A Complete Guide to Paired-End RNA-seq Alignment with STAR: From Basics to Advanced Optimization
This article provides a comprehensive, step-by-step protocol for aligning paired-end RNA-seq reads using the Spliced Transcripts Alignment to a Reference (STAR) software. Tailored for researchers and scientists in drug development and biomedical research, it covers foundational concepts, detailed methodology, critical troubleshooting, and validation techniques. Readers will learn to perform both standard and advanced 2-pass mapping, optimize parameters for specific experimental goals like somatic mutation or fusion transcript detection, and interpret alignment outputs to ensure data quality for downstream differential expression and splicing analysis.
A Complete Guide to Paired-End RNA-seq Alignment with STAR: From Basics to Advanced Optimization
Abstract
This article provides a comprehensive, step-by-step protocol for aligning paired-end RNA-seq reads using the Spliced Transcripts Alignment to a Reference (STAR) software. Tailored for researchers and scientists in drug development and biomedical research, it covers foundational concepts, detailed methodology, critical troubleshooting, and validation techniques. Readers will learn to perform both standard and advanced 2-pass mapping, optimize parameters for specific experimental goals like somatic mutation or fusion transcript detection, and interpret alignment outputs to ensure data quality for downstream differential expression and splicing analysis.
Understanding STAR: Why It's the Gold Standard for Spliced RNA-seq Alignment
The alignment of high-throughput sequencing reads to a reference genome represents a critical step in the analysis of both RNA-seq and DNA-seq data. This process enables crucial downstream analyses including gene discovery, gene quantification, splice variant analysis, and the identification of chimeric fusion genes [1]. RNA-seq data alignment presents unique computational challenges, primary among them being the accurate mapping of reads that span non-contiguous genomic regions—a consequence of the splicing mechanism that joins exons together in eukaryotic transcriptomes [2]. Traditional DNA-seq aligners, which assume sequence contiguity, are ill-suited for this task, necessitating the development of specialized "splice-aware" aligners.
Spliced Transcripts Alignment to a Reference (STAR) is a highly accurate and ultra-fast splice-aware aligner specifically designed to address the mapping challenges inherent to RNA-seq data [1] [2]. First introduced in 2012, STAR employs a novel alignment algorithm based on sequential maximum mappable seed search in uncompressed suffix arrays, followed by seed clustering and stitching procedures [2]. This strategy allows STAR to outperform other splice-aware RNA-seq aligners such as HISAT2 and TopHat2 in terms of mapping rate and speed, though it typically requires more substantial computational memory resources [1]. A key advantage of STAR is its high precision in identifying both canonical and non-canonical splice junctions, alongside its capability to detect chimeric transcripts and accurately map long reads generated by third-generation sequencing technologies [1] [2].
STAR Algorithm and Computational Strategy
Core Algorithmic Principles
The STAR algorithm operates through a two-step process that fundamentally differs from approaches used by earlier aligners. Rather than extending DNA-seq alignment methods, STAR was designed from the ground up to align non-contiguous sequences directly to the reference genome [2]. This core design principle enables its exceptional performance in handling spliced transcripts.
The first phase, seed searching, involves identifying the longest sequences from reads that exactly match one or more locations on the reference genome. For each read, STAR searches for the Maximal Mappable Prefix (MMP)—the longest substring starting from read position i that matches exactly one or more substrings of the reference genome G [2]. When a read contains a splice junction, it cannot be mapped contiguously; thus the first MMP maps to a donor splice site, and the algorithm sequentially searches the unmapped portion of the read to find the next MMP, which maps to an acceptor splice site [3] [2]. This sequential application of MMP search exclusively to unmapped read portions contributes significantly to STAR's computational efficiency.
The second phase, clustering, stitching, and scoring, involves reconstructing complete alignments by stitching together individually mapped seeds. Seeds are clustered based on proximity to selected "anchor" seeds—those with limited genomic mapping locations [2]. A dynamic programming algorithm then stitches seed pairs together within user-defined genomic windows, allowing for mismatches but only one insertion or deletion per seed pair [2]. This approach naturally accommodates paired-end reads by clustering and stitching seeds from both mates concurrently, treating them as pieces of the same sequence, which enhances alignment sensitivity [2].
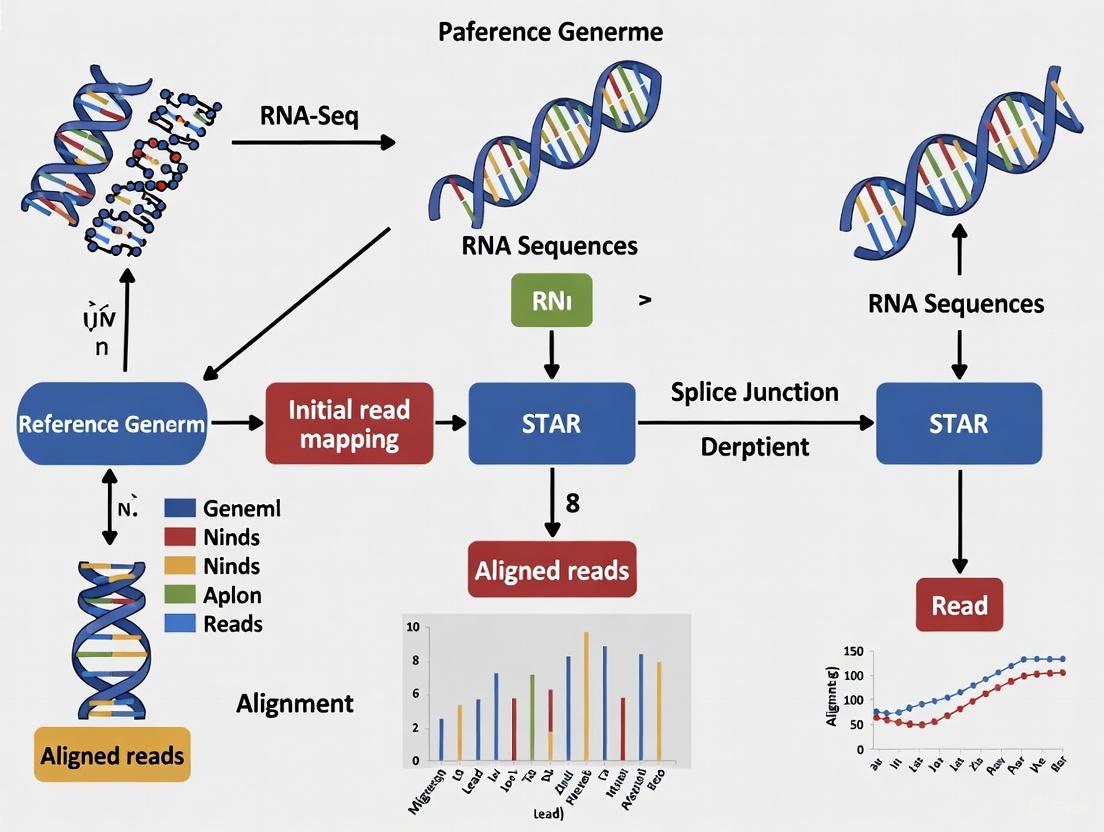
Visualizing the STAR Alignment Workflow
The following diagram illustrates the complete STAR alignment workflow, from initial read processing to final output generation:
Key Algorithmic Advantages
STAR's algorithmic design provides several distinct advantages over traditional approaches. The use of uncompressed suffix arrays enables rapid searching with logarithmic scaling relative to reference genome size, allowing efficient processing even against large genomes [2]. The sequential MMP approach represents a more natural method for identifying splice junction locations compared to arbitrary read-splitting methods used in other aligners [2]. Furthermore, STAR detects splice junctions in a single alignment pass without requiring preliminary contiguous alignment or pre-existing junction databases, enabling unbiased de novo discovery of both canonical and non-canonical splice junctions [2].
The two-step mapping process also provides robustness against sequencing artifacts. When MMP extension fails due to poor sequence quality or adapter contamination, STAR can identify and soft-clip these problematic regions [3] [2]. This capability ensures that alignment quality remains high even with imperfect sequencing data.
Experimental Protocols and Implementation
Computational Requirements and Installation
STAR requires significant computational resources, particularly memory, for optimal performance. The following table outlines typical system requirements:
Table 1: STAR Computational Requirements for Human Genome Alignment
| Resource Type | Minimum Recommended | Optimal for Human Genome |
|---|---|---|
| RAM | 16 GB | 32 GB or higher |
| Processors | 4 cores | 8-12 cores |
| Storage | 100 GB free space | 500 GB free space |
| Operating System | Linux or Mac OS | Linux or Mac OS |
Installation can be accomplished either by downloading pre-compiled binaries or compiling from source [1]. For Linux systems, the following commands install STAR and add its binaries to the system path:
Genome Index Generation Protocol
STAR requires a genome index before read alignment can commence. The index generation protocol requires both a reference genome sequence in FASTA format and gene annotation in GTF or GFF3 format [1] [3].
Protocol Steps:
- Create a directory for genome indices:
mkdir /path/to/genome_indices - Execute the genomeGenerate run mode with appropriate parameters
- Verify index generation by checking for the presence of output files in the genome directory
The following command provides a template for genome index generation:
Table 2: Critical Parameters for Genome Index Generation
| Parameter | Description | Recommended Value |
|---|---|---|
--runThreadN |
Number of parallel threads to use | 6-12 (based on available cores) |
--genomeDir |
Path to directory for storing genome indices | User-defined directory path |
--genomeFastaFiles |
Reference genome file in FASTA format | Genome sequence file |
--sjdbGTFfile |
Gene annotation file in GTF format | Annotation file matching genome |
--sjdbOverhang |
Length of genomic sequence around annotated junctions | Read length minus 1 |
For read lengths of 150 bp, the --sjdbOverhang parameter should be set to 149, though the default value of 100 often produces similar results [1] [3]. When using GFF3 annotation files instead of GTF, an additional parameter --sjdbGTFtagExonParentTranscript Parent must be included to define parent-child relationships [1].
Read Alignment Protocol
With genome indices prepared, RNA-seq reads can be aligned using STAR's standard mapping mode. The protocol differs slightly for single-end versus paired-end reads.
For single-end reads:
For paired-end reads:
For compressed FASTQ files (.fastq.gz), include the parameter --readFilesCommand zcat or --readFilesCommand gunzip -c to enable on-the-fly decompression [1].
Table 3: Essential Parameters for Read Alignment
| Parameter | Description | Importance |
|---|---|---|
--readFilesIn |
Input read file(s) | Mandatory; specifies sequence data |
--genomeDir |
Path to genome indices | Mandatory; specifies reference |
--outSAMtype BAM SortedByCoordinate |
Output sorted BAM format | Enables efficient downstream processing |
--outFileNamePrefix |
Output file name prefix | Organizes output files by sample |
--quantMode |
Gene-level quantification | Optional: outputs read counts per gene |
For studies focused on novel splice junction discovery or differential splicing analysis, a 2-pass mapping approach is recommended. This involves re-building genome indices using splice junctions identified in an initial mapping step, then repeating alignment with these enhanced indices [1].
Output Files and Interpretation
STAR Output File Types
A successful STAR alignment generates multiple output files, each containing different types of information about the alignment results.
Table 4: STAR Output Files and Their Contents
| Output File | Contents | Downstream Applications |
|---|---|---|
*Aligned.sortedByCoord.out.bam |
Coordinate-sorted alignments | Visualization, variant calling, transcript assembly |
*Log.final.out |
Summary alignment statistics | Quality control, experimental assessment |
*SJ.out.tab |
Filtered splice junction information | Splice junction analysis, novel junction discovery |
*ReadsPerGene.out.tab |
Read counts per gene | Differential expression analysis |
*Log.progress.out |
Alignment progress statistics | Performance monitoring, optimization |
The BAM file containing sorted alignments represents the primary output for most downstream analyses, including transcript assembly with StringTie and differential expression analysis with tools like DESeq2 [4]. The splice junction file (*SJ.out.tab) contains information about all detected junctions, including genomic coordinates, strand information, and annotation status [1].
Interpretation of Alignment Statistics
The *Log.final.out file provides comprehensive alignment statistics essential for quality assessment. The following table illustrates typical alignment rates from a successful experiment:
Table 5: Example STAR Alignment Statistics from Arabidopsis thaliana Data
| Metric | Value | Interpretation |
|---|---|---|
| Number of input reads | 11,889,751 | Total sequencing depth |
| Uniquely mapped reads | 90.48% | High-quality alignment rate |
| Average mapped length | 200.74 bp | Read length after trimming |
| Mismatch rate per base | 0.65% | Acceptable error rate |
| Multi-mapping reads | 6.55% | Reads mapping to multiple loci |
| Splice junctions detected | 7,206,200 | Total splice events |
A uniquely mapped reads percentage above 70-80% generally indicates successful alignment, though this varies by organism and RNA-seq library quality [1] [3]. The splice junction classification provides insights into splicing biology, with canonical GT/AG junctions typically representing the majority of splicing events [1].
Performance Comparison with Alternative Aligners
STAR versus Pseudoaligners
STAR is frequently compared with pseudoalignment tools like Kallisto, particularly in the context of single-cell and bulk RNA-seq analysis. The following table summarizes key differences:
Table 6: Feature Comparison Between STAR and Kallisto
| Feature | STAR | Kallisto |
|---|---|---|
| Algorithm type | Splice-aware alignment to genome | Pseudoalignment to transcriptome |
| Accuracy | Higher gene detection rates [5] | Faster with slightly lower detection |
| Computational speed | Fast but memory-intensive | Extremely fast and lightweight |
| Memory usage | High (28GB reported) [6] | Low (3.6GB reported) [6] |
| Novel junction detection | Excellent for canonical and non-canonical | Limited to annotated transcripts |
| Input requirements | Genome sequence + annotations | Transcriptome sequence |
Research comparing these tools on single-cell RNA-seq data from Drop-seq, Fluidigm, and 10x Genomics platforms has demonstrated that STAR generally produces higher gene counts and greater alignment rates (62.40% versus 35.11% in Drop-seq data) [5] [6]. Additionally, STAR shows better correlation with RNA-FISH validation data, particularly for the Gini index, which measures expression distribution across cells [5].
Performance Optimization Strategies
Recent research has identified several strategies for optimizing STAR performance in high-throughput computing environments:
Genome Version Selection: Using newer Ensembl genome releases (e.g., release 111 versus 108) can dramatically reduce index size (85 GB to 29.5 GB) and decrease execution time by more than 12-fold while maintaining nearly identical mapping rates [7].
Early Stopping: Implementing an "early stopping" approach that terminates alignment if less than 30% of the initial read subset maps successfully can reduce total computation time by approximately 19.5% by filtering out low-quality samples early in the process [7].
Cloud-Native Architecture: Implementing STAR in scalable cloud environments using pre-computed indices and spot instances can significantly reduce computational costs while maintaining alignment accuracy for large-scale transcriptomic studies [7].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the STAR alignment protocol requires several key computational reagents and resources. The following table details these essential components:
Table 7: Essential Research Reagents and Computational Materials for STAR Alignment
| Resource Type | Specific Examples | Function in Protocol |
|---|---|---|
| Reference Genome | GRCh38 (human), GRCm39 (mouse), Sorghum_bicolor (plant) | Genomic coordinate system for read alignment |
| Gene Annotation | GTF files from Ensembl, GENCODE, or RefSeq | Defines known gene models and splice junctions |
| Sequence Reads | FASTQ files (single or paired-end) | Raw sequencing data for alignment |
| Alignment Software | STAR (version 2.7.10b or newer) | Core alignment algorithm execution |
| Sequence Manipulation Tools | SAMtools, BEDTools, FASTQC | BAM file processing and quality control |
| Downstream Analysis Packages | StringTie, DESeq2, Ballgown | Transcript assembly and differential expression |
| Rubelloside B | Rubelloside B, MF:C42H66O14, MW:795.0 g/mol | Chemical Reagent |
| Dihydroniloticin | Dihydroniloticin, MF:C30H50O3, MW:458.7 g/mol | Chemical Reagent |
The quality and version compatibility of these resources significantly impact alignment success. Using matched versions of genome sequences and gene annotations from the same source (e.g., both from Ensembl release 111) prevents coordinate mismatches and ensures optimal junction annotation [7]. Regular updates to genome assemblies and annotations can improve alignment accuracy and reduce computational requirements.
Advanced Applications and Integration in Research Pipelines
Specialized Applications
STAR supports several advanced analysis modes that extend its utility beyond basic read alignment:
Two-Pass Mapping: For novel splice junction discovery, a two-pass approach first identifies junctions from initial alignment, then incorporates these junctions into the reference for a second alignment round, significantly improving sensitivity for unannotated splicing events [1].
Chimeric Alignment: STAR can detect chimeric (fusion) transcripts by identifying alignments where read segments map to different genomic loci, enabling discovery of gene fusions with important implications in cancer research [2].
Long Read Alignment: Although designed for short reads, STAR can accurately map long reads from third-generation sequencing technologies (PacBio, Oxford Nanopore), making it suitable for full-length transcriptome analysis [2].
Integration with Downstream Analysis Workflows
STAR aligns seamlessly into comprehensive RNA-seq analysis pipelines. A typical integrated workflow might include:
- Quality Control: FastQC for raw read assessment
- Alignment: STAR for genome mapping
- Transcript Assembly: StringTie for transcript model construction [4]
- Quantification: featureCounts or STAR's built-in quantMode for gene-level counts
- Differential Expression: DESeq2 or Ballgown for identifying expression changes [4]
This integration is exemplified in the nf-core/RNA-seq pipeline, which incorporates STAR as a primary aligner alongside comprehensive quality control metrics [8]. The output of STAR—particularly the coordinate-sorted BAM files and junction information—serves as input for diverse downstream applications including variant calling, differential splicing analysis, and visualization in genome browsers.
The following diagram illustrates STAR's role in a comprehensive RNA-seq analysis workflow:
STAR's position as a central component in modern RNA-seq analysis workflows underscores its importance in contemporary genomics research. Its balance of speed, accuracy, and versatility makes it particularly valuable for large-scale transcriptomic studies, including those conducted by major consortia such as ENCODE [2]. As RNA-seq technologies continue to evolve, with increasing read lengths and throughput, STAR's unique algorithmic approach positions it to remain a cornerstone of transcriptome analysis for the foreseeable future.
The Spliced Transcripts Alignment to a Reference (STAR) software package represents a foundational tool in modern transcriptomics, specifically engineered to address the unique computational challenges of RNA-seq data analysis. In the context of paired-end RNA-seq alignment protocol research, STAR's algorithm provides a critical solution for mapping sequences derived from non-contiguous genomic regions, a task essential for accurate transcriptome reconstruction [2]. The strategic importance of STAR lies in its dual capacity for ultra-fast processing and high-sensitivity detection of complex RNA arrangements, enabling researchers to comprehensively analyze the full complexity of cellular transcriptomes. This capability is particularly valuable for drug development professionals investigating disease-specific transcriptional alterations, where discovering novel splice variants and fusion transcripts can reveal potential therapeutic targets.
STAR's performance advantages stem from its novel alignment strategy, which fundamentally differs from earlier approaches that treated RNA-seq alignment as an extension of DNA read mapping [2]. By directly addressing the non-contiguous nature of RNA transcripts, STAR achieves unprecedented mapping speed while maintaining accuracy—processing 550 million 2×76 bp paired-end reads per hour on a standard 12-core server, outperforming other aligners by more than a factor of 50 [2] [3]. This scalability is crucial for large consortia efforts and clinical studies where processing thousands of samples efficiently is paramount. Furthermore, STAR's ability to detect both canonical and non-canonical splices, chimeric (fusion) transcripts, and circular RNAs provides researchers with a comprehensive toolset for exploratory transcriptome analysis in both basic research and clinical applications [9].
Technical Breakdown of Key Advantages
Superior Mapping Rate and Speed
STAR achieves its exceptional mapping performance through an innovative two-step algorithm that maximizes both speed and accuracy. The first phase involves sequential searching for Maximal Mappable Prefixes (MMPs), defined as the longest subsequences from reads that exactly match one or more locations on the reference genome [2] [3]. This approach represents a natural way of identifying precise splice junction locations without prior knowledge of junction loci or properties. The sequential application of MMP search exclusively to unmapped portions of reads distinguishes STAR from other algorithms and underlies its significant speed advantage [2].
The technical implementation utilizes uncompressed suffix arrays (SAs), which provide logarithmic scaling of search time with reference genome length, enabling rapid searching against large genomes [2]. In benchmark comparisons, STAR demonstrates both high mapping sensitivity and precision, with typical mapping rates of 92-95% for human RNA-seq data [9] [3]. The algorithm's efficiency is further enhanced through parallel processing capabilities, allowing researchers to leverage multi-core computing architectures to accelerate analysis timelines—a critical advantage in time-sensitive drug discovery pipelines.
Table 1: Comparative Mapping Performance Metrics
| Performance Metric | STAR Performance | Typical Range for Other Aligners |
|---|---|---|
| Mapping Speed | 550 million PE reads/hour (12-core server) | <10 million PE reads/hour [2] |
| Mapping Rate | 92.2% (example from human RNA-seq) | 70-90% varies by aligner [9] [10] |
| Unique Mapping Rate | 92.2% | 80-95% varies by aligner [9] |
| Multi-Mapping Rate | 6.0% | 5-15% varies by aligner [9] |
| Unmapped Reads | 1.7% | 5-20% varies by aligner [9] |
Comprehensive Splice Junction Discovery
STAR excels in splice junction detection through its ability to identify both annotated and novel splice junctions without prior annotation. The algorithm accomplishes this by stitching separately mapped seeds across intronic regions, with the maximum intron size determined by user-defined genomic windows during the clustering phase [2]. This approach allows STAR to discover novel splice junctions de novo, a capability verified through experimental validation where 1960 novel intergenic splice junctions were confirmed with an 80-90% success rate using Roche 454 sequencing of RT-PCR amplicons [2].
The splice detection sensitivity is further enhanced when using paired-end reads, as seeds from both mates are clustered and stitched concurrently, with each paired-end read represented as a single sequence [2]. This principled utilization of paired-end information reflects the biological reality that mates are fragments of the same RNA molecule, increasing detection sensitivity particularly for junctions where only one mate provides strong alignment evidence. For optimal junction detection, STAR can utilize gene annotations in GTF format during genome indexing, significantly improving identification and accurate mapping across known splice junctions [9]. When annotations are unavailable, STAR's two-pass mapping method enables sensitive novel junction discovery by leveraging splice junctions detected in an initial mapping round to inform a second, more accurate alignment phase [9].
Splice Junction Discovery Workflow: STAR's two-phase algorithm for identifying splice junctions from RNA-seq reads.
Advanced Chimeric RNA Detection
STAR possesses unique capabilities for detecting chimeric RNA molecules, including fusion transcripts that result from chromosomal rearrangements—a phenomenon of particular interest in cancer research and biomarker discovery. The algorithm identifies chimeric alignments by searching for multiple genomic windows that collectively cover the entire read sequence, with different portions mapping to distal genomic loci, different chromosomes, or different strands [2]. This approach allows STAR to detect both internal chimeric junctions within single reads and chimeras where the junction falls between paired-end mates in the unsequenced portion of the RNA molecule [2].
The chimeric detection functionality enables researchers to identify fusion transcripts without additional specialized tools, streamlining analytical workflows. STAR can pinpoint precise locations of chimeric junctions in the genome, providing structural information essential for understanding the molecular consequences of genomic rearrangements [2]. This capability was demonstrated in the detection of BCR-ABL fusion transcripts in K562 erythroleukemia cells, a clinically relevant fusion in hematological malignancies [2]. For comprehensive chimeric detection, STAR includes dedicated options that can be enabled during alignment to systematically report chimeric alignments in separate output files [9].
Application Notes for Experimental Design
Recommended Protocols for Optimal Performance
To maximize STAR's performance advantages in paired-end RNA-seq studies, researchers should follow established best practices for experimental design and computational resource allocation. The quality of input RNA significantly impacts mapping outcomes, with recommendations for assessing RNA integrity number (RIN) and selecting appropriate RNA extraction protocols based on sample type [10]. For eukaryotic samples, researchers must choose between poly(A) selection and ribosomal RNA depletion, with poly(A) selection preferred for high-quality samples and rRNA depletion necessary for degraded samples or bacterial transcriptomes [10].
Library preparation considerations include the use of strand-specific protocols (such as the dUTP method) to preserve information about the transcribed strand, which is particularly valuable for identifying antisense transcripts and accurately quantifying overlapping genes [10]. Paired-end sequencing is strongly recommended over single-end approaches for comprehensive transcriptome characterization, as the additional information significantly enhances splice junction detection and novel isoform discovery [10]. Sequencing depth should be determined based on research objectives, with typical recommendations ranging from 20-50 million reads per sample for standard differential expression studies to 100 million reads or more for comprehensive isoform-level analysis [10].
Table 2: STAR Alignment Parameters for Different Research Applications
| Research Application | Critical STAR Parameters | Recommended Settings | Performance Considerations |
|---|---|---|---|
| Standard Gene Expression | --outSAMtype BAM SortedByCoordinate --quantMode GeneCounts |
Sorted BAM, read counts per gene | Fast processing, lower memory |
| Novel Isoform Discovery | --twopassMode Basic --outSAMstrandField intronMotif |
Two-pass mapping, strand information | 2x computation time, higher sensitivity |
| Fusion Transcript Detection | --chimSegmentMin 15 --chimJunctionOverhangMin 15 |
Chimeric alignment enabled | Additional computation, specialized output |
| Clinical RNA-seq | --outFilterScoreMinOverLread 0.3 --outFilterMatchNminOverLread 0.1 |
Strict filtering, quality metrics | Balanced sensitivity/specificity |
| Long Read Analysis | --seedSearchStartLmax 20 --seedPerReadNmax 100000 |
Modified seed parameters | Adapted for emerging technologies |
Computational Requirements and Resource Planning
STAR's exceptional performance requires substantial computational resources, particularly during the genome indexing phase. Memory requirements scale with reference genome size, with approximately 10× genome size bytes needed—for example, ~30 GB for the human genome [9]. The developers recommend 32 GB of RAM for human genome alignments to ensure stable operation [9]. Disk space requirements are also significant, with >100 GB of free space recommended for storing output files, including alignment files, splice junction lists, and logging information [9].
For processing efficiency, STAR supports multi-threaded execution, with the number of threads specified using the --runThreadN parameter [9]. Optimal performance is achieved when this parameter matches the number of physical processor cores available, though some systems with efficient hyper-threading may benefit from increasing threads up to twice the number of physical cores [9]. In high-performance computing environments, researchers should request appropriate resources through job schedulers, typically specifying 6-12 cores and 32-64 GB of RAM for human transcriptome alignment [3].
Detailed Methodologies and Protocols
Genome Index Generation Protocol
The initial step in any STAR analysis involves generating genome indices, which must be completed before read alignment. This process requires the reference genome sequence in FASTA format and gene annotations in GTF format. The following protocol outlines the critical steps for creating optimized indices:
Necessary Resources:
- Reference genome FASTA file
- Gene annotation GTF file
- Computer with Unix/Linux/Mac OS X operating system
- Sufficient memory (≥ 30 GB for human genome)
- Adequate disk space (≥ 100 GB)
Step-by-Step Procedure:
- Create and navigate to a directory for genome indices:
- Execute the genome generation command:
The --sjdbOverhang parameter should be set to the maximum read length minus 1, with 100 being a standard value that works well in most cases [3]. For paired-end reads, this parameter should reflect the length of the read that aligns across the splice junction, which is typically the read length minus 1 [9]. The genome indexing process is computationally intensive but only needs to be performed once for each combination of reference genome and annotation set.
Basic Paired-End Read Alignment Protocol
Once genome indices are prepared, researchers can perform the actual read alignment. This protocol describes the essential mapping workflow for paired-end RNA-seq data:
Input Files Requirements:
- Paired FASTQ files (read 1 and read 2)
- Genome indices generated as described in Protocol 4.1
Alignment Command:
Critical Parameter Explanations:
--runThreadN 12: Utilizes 12 computational threads for parallel processing--readFilesCommand zcat: Specifies decompression for gzipped input files--outSAMtype BAM SortedByCoordinate: Outputs alignment in coordinate-sorted BAM format--outSAMunmapped Within: Includes unmapped reads in the output BAM file--outSAMattributes Standard: Includes standard alignment attributes in output
This basic protocol generates a sorted BAM file suitable for downstream analysis, including transcript quantification and visualization. During execution, STAR provides progress updates through both real-time console output and the Log.progress.out file, which displays mapping statistics updated at regular intervals [9].
Two-Pass Mapping for Novel Junction Discovery
For applications requiring maximum sensitivity for novel splice junction detection, the two-pass mapping method provides enhanced performance:
Two-Pass Execution:
The two-pass approach first identifies splice junctions from the data itself, then incorporates these junctions into the genome index for a second, more sensitive alignment round [9]. This method is particularly valuable when analyzing samples with potentially novel splicing patterns, such as disease tissues or uncharacterized biological conditions. The additional computational requirements are substantial (approximately double the alignment time) but yield significantly improved sensitivity for detecting rare or novel splicing events.
STAR Paired-End RNA-seq Analysis Workflow: Complete workflow from raw sequencing data to analysis-ready outputs.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Computational Tools for STAR RNA-seq Analysis
| Category | Item/Reagent | Specification/Version | Critical Function |
|---|---|---|---|
| Biological Materials | Quartet Reference RNA Samples | Multi-center study validated [11] | Cross-laboratory standardization and quality control |
| ERCC RNA Spike-In Controls | 92 synthetic RNAs [11] | Technical performance monitoring and quantification calibration | |
| High-Quality Total RNA | RIN >8.0, minimal degradation [10] | Optimal library preparation input material | |
| Library Preparation | Strand-Specific Kit | dUTP second strand method [10] | Preservation of strand information in sequencing reads |
| Poly(A) Selection Beads | mRNA enrichment protocol [10] | Focus on messenger RNA population | |
| Ribo-Depletion Reagents | rRNA removal protocol [10] | Comprehensive transcriptome including non-polyadenylated RNAs | |
| Computational Resources | Reference Genome | GRCh38/hg38 or current version | Alignment coordinate system and annotation basis |
| Gene Annotations | GTF format (e.g., Ensembl, GENCODE) | Splice junction guidance and feature quantification | |
| High-Performance Computing | 12+ cores, 32+ GB RAM [9] | Execution of memory-intensive alignment algorithms | |
| Software Tools | STAR Aligner | Version 2.4.1a or later [9] | Primary alignment engine with splice-aware capability |
| SAMtools | Version 1.17 or later [12] | BAM file processing and indexing utilities | |
| FastQC | Version 0.12.1 or later [12] | Raw read quality assessment and troubleshooting | |
| Trimmomatic or Cutadapt | Current versions [12] | Adapter trimming and read quality processing | |
| Fupenzic acid | Fupenzic acid, CAS:119725-20-1, MF:C30H44O5, MW:484.7 g/mol | Chemical Reagent | Bench Chemicals |
| Isohyenanchin | Isohyenanchin | High-purity Isohyenanchin for laboratory research. Investigate its mechanism and applications. For Research Use Only. Not for diagnostic or therapeutic use. | Bench Chemicals |
STAR's combination of superior mapping rates, comprehensive splice junction discovery, and advanced chimeric RNA detection establishes it as a foundational technology in modern transcriptomics research. The algorithms and protocols detailed in this application note provide researchers with a robust framework for extracting maximum biological insight from paired-end RNA-seq data, particularly in contexts where novel transcript discovery and detection of complex RNA arrangements are research priorities.
As RNA-seq applications continue to evolve toward clinical implementation, quality assessment using appropriate reference materials like the Quartet samples becomes increasingly important, particularly for detecting subtle differential expression with diagnostic significance [11]. The ongoing development of long-read sequencing technologies presents new opportunities and challenges for alignment algorithms, and STAR's capacity to handle sequences of any length with moderate error rates positions it well for these emerging applications [9] [2].
For drug development professionals and clinical researchers, STAR's precision in identifying fusion transcripts and alternatively spliced isoforms provides critical capabilities for biomarker discovery and therapeutic target identification. By implementing the optimized protocols and quality control measures described in this application note, researchers can ensure the reliability and reproducibility of their RNA-seq analyses, ultimately accelerating the translation of transcriptomic discoveries into clinical applications.
Within the framework of a broader thesis on paired-end RNA-seq alignment, the selection of the STAR aligner (Spliced Transcripts Alignment to a Reference) is often dictated by its superior speed and accuracy in handling spliced transcripts [13]. However, its significant computational demands necessitate careful planning to ensure successful and efficient analysis. This application note details the critical hardware preparations and experimental protocols required to execute a STAR-based RNA-seq workflow, specifically tailored for researchers and scientists in drug development who need to process large-scale genomic data. Proper resource allocation is not merely an administrative task; it is a fundamental prerequisite that underpins the validity and reliability of downstream differential expression and transcriptomic analysis.
Quantitative Hardware Requirements
The alignment of RNA-seq reads to a reference genome is one of the most computationally intensive steps in the entire bioinformatics pipeline. The requirements below are specifically curated for aligning human or other similarly sized mammalian genomes, which are the most common use cases in therapeutic and drug discovery research.
The following table synthesizes hardware recommendations for running the STAR aligner under different operational scales, from minimal viable operation to optimal performance for large-scale batch processing.
Table 1: Detailed hardware requirements for STAR RNA-seq alignment
| Component | Minimum Requirement | Recommended for Standard Use | Large-Scale/Batch Processing |
|---|---|---|---|
| RAM (Memory) | 16 GB for mammalian genomes [14] | 32 GB - 38 GB for human genome [15] [16] [14] | 48 GB - 64 GB+ [15] [17] |
| Processor (CPU) | 8-core processor [18] | 16-24 cores [18] | 8 vCPU per task/instance, modern CPU architecture recommended [17] |
| Storage | 500 GB - 1 TB SSD [15] [18] | 1 TB+ high-performance (NVMe) SSD [15] | Large-scale block storage (EBS), 550 GB+ per instance with high IOPS [17] |
| Key Considerations | Risk of failure due to insufficient memory. | Balance of cost and performance for most single-sample analyses. | Essential for parallel processing of multiple samples; older CPU models can significantly slow processing [17]. |
Analysis of Resource Utilization
- Memory (RAM): The primary driver of STAR's high memory usage is the need to load the entire reference genome index into memory. For the human genome, this index is approximately 30 GB in size [17]. The recommended 32-38 GB provides the necessary headroom for the operating system and other concurrent processes, ensuring stable alignment without memory allocation errors. Attempting to run STAR with less than the minimum required RAM will almost certainly result in job failure [14].
- Processor (CPU): STAR is designed to leverage multiple CPU cores effectively. While it can run on fewer cores, performance scales with increased core count, significantly reducing alignment time. It is critical to note that CPU architecture impacts performance; a benchmark study noted that older-generation CPUs available in some serverless configurations led to slower alignment times compared to modern EC2 instances [17].
- Storage: The use of Solid-State Drives (SSD) is strongly recommended over traditional hard drives due to the high I/O demands of reading FASTQ files and writing SAM/BAM outputs [15]. For a dataset of 100 human samples with ~21 million reads each, total storage needs will easily exceed 1 TB when accounting for raw data, intermediate files, and final results.
Experimental Protocols for Resource Management
This protocol assumes prior installation of STAR and preparation of the reference genome index.
- Hardware Allocation: Provision a computational node with at least 32 GB of RAM and 8 CPU cores. Ensure a minimum of 500 GB of free space on a high-speed (SSD) storage volume.
- Input File Preparation: Confirm that paired-end FASTQ files are available. STAR can directly read gzipped files (e.g.,
sample_1.fastq.gz,sample_2.fastq.gz), which saves disk space and I/O time [19]. - STAR Execution Command: Execute a standard alignment command. The
--runThreadNparameter should be set to the number of available CPU cores to maximize parallelization. - Monitoring and Validation: Monitor the system's resource usage (e.g., using
toporhtop) during the initial phase of alignment to verify that memory consumption is within expected limits. Check the generatedLog.final.outfile for alignment statistics and any reported errors.
Protocol: Reprocessing RNA-seq Data Without Realignment
For workflows that involve recalculating gene counts or re-running downstream QC without altering the alignment itself, a BAM reprocessing workflow can save substantial time and computational resources [16]. This is highly relevant for refining analyses in long-term research projects.
- Prerequisite: An initial pipeline run (e.g., using nf-core/rnaseq) must be executed with the
--save_align_intermedsflag, which preserves the genomic and transcriptomic BAM files [16]. - Samplesheet Generation: The pipeline will auto-generate a new samplesheet (e.g.,
samplesheet_with_bams.csv) that includes paths to the previously generated BAM files. - Reprocessing Execution: Launch the pipeline in reprocessing mode, specifying the
--skip_alignmentflag and providing the generated samplesheet. - Outcome: The pipeline will bypass the resource-intensive STAR alignment step and use the existing BAM files for all downstream quantification and quality control steps, drastically reducing compute time and resource consumption [16].
Workflow Visualization
The following diagram illustrates the complete STAR-based RNA-seq analysis workflow, highlighting the critical pathway and key decision points for resource management.
Diagram 1: STAR RNA-seq analysis workflow
This table details the key computational "reagents" and infrastructure components required for a successful STAR alignment project.
Table 2: Essential computational materials and resources
| Resource / Tool | Function & Application in the Protocol |
|---|---|
| STAR Aligner [14] | The core software for performing spliced alignment of RNA-seq reads to a reference genome. Its high speed comes with significant RAM requirements. |
| Reference Genome Index [17] | A pre-computed data structure from a reference genome (e.g., human GRCh38). For STAR, this is a ~30 GB file that must be loaded into RAM, defining the memory requirement. |
| High-Performance Computing (HPC) or Cloud Platform [15] [17] | Local computing clusters or cloud services (AWS EC2/ECS) are typically required to meet the memory and CPU demands for mammalian genomes, as standard desktop computers are often insufficient. |
| nf-core/rnaseq Pipeline [16] | A robust, community-maintained Nextflow pipeline that encapsulates the entire RNA-seq analysis, including STAR alignment, quantification, and QC, ensuring reproducibility. |
| Elastic Block Storage (EBS) / SSDs [17] | High-input/output (IOPS) block storage, crucial for handling the large volumes of data involved in reading FASTQ files and writing alignment outputs efficiently. |
RNA sequencing (RNA-seq) has become a foundational tool in modern molecular biology and drug development, enabling comprehensive, genome-wide quantification of transcriptomes [20]. Unlike earlier methods like microarrays, RNA-seq provides finer resolution for dynamic expression changes and allows for the discovery of novel RNA species and splicing events [21]. A critical first step in the analysis of RNA-seq data is the accurate and efficient alignment of millions of short sequencing reads to a reference genome. The Spliced Transcripts Alignment to a Reference (STAR) software package performs this task with high levels of accuracy and speed, and is uniquely capable of detecting not only annotated and novel splice junctions but also more complex RNA sequence arrangements, such as chimeric and circular RNA [22]. This application note details the core two-step workflow of STAR—genome generation and read mapping—providing researchers and drug development professionals with detailed protocols for performing paired-end RNA-seq alignment within the context of advanced genomic research.
The STAR workflow is fundamentally structured around two primary stages. The first stage involves generating a genome index from a reference genome and annotation file. This index is not a simple lookup table; it is a highly efficient, pre-processed structure that stores the genomic sequences along with crucial junction information, enabling STAR to perform ultra-fast searching during the alignment phase. The second stage uses this custom-generated index to map the RNA-seq reads (typically in FASTQ format) to the reference genome. The process results in aligned read files (BAM) and various output files that can be used for downstream analyses such as differential gene expression, novel isoform reconstruction, and signal visualization [22].
The following diagram illustrates the logical sequence and data flow between these two core processes:
Step 1: Protocol for Genome Index Generation
The creation of a STAR genome index is a one-time per genome/annotation release pre-processing step. A well-constructed index is crucial for the speed and accuracy of the subsequent mapping.
Experimental Methodology
This protocol requires a reference genome sequence in FASTA format and a gene annotation file in GTF format. The genome is processed into a suffix array, and the annotations are used to create a database of known splice junctions, which STAR uses to guide the alignment of reads across intron boundaries.
Input Files:
genome.fa: Reference genome sequences.annotation.gtf: Gene annotations.
Computational Command:
Key Parameters for Index Generation
The --sjdbOverhang parameter should be set to the read length minus 1. For paired-end reads, this is based on the length of one read. The following table summarizes critical parameters and their functions.
Table 1: Key Parameters for STAR Genome Index Generation
| Parameter | Function | Typical Setting for Paired-End |
|---|---|---|
--runMode genomeGenerate |
Directs STAR to operate in genome index generation mode. | Mandatory |
--genomeDir |
Specifies the directory where the genome index will be stored. | User-defined path |
--genomeFastaFiles |
Provides the path to the reference genome FASTA file(s). | Path to genome.fa |
--sjdbGTFfile |
Provides the gene annotation file to generate splice junction database. | Path to annotation.gtf |
--sjdbOverhang |
Specifies the length of the genomic sequence around annotated junctions. | Read length - 1 (e.g., 99 for 100bp reads) |
Step 2: Protocol for Mapping RNA-seq Reads
Once the genome index is built, it can be used to map the RNA-seq reads from each sample. This protocol is designed for paired-end reads, which provide higher mapping accuracy and better resolution of splice junctions compared to single-end reads.
Experimental Methodology
This protocol takes paired-end FASTQ files and the previously generated genome index as input. STAR performs a two-pass alignment process by default, which significantly improves the detection of novel splice junctions.
Input Files:
sample_R1.fastq: Forward reads.sample_R2.fastq: Reverse reads.- Genome index (from Step 1).
Computational Command:
Key Parameters for Read Mapping
The following parameters control the mapping behavior and output format. Adjusting the number of threads (--runThreadN) can significantly speed up the process on multi-core systems.
Table 2: Key Parameters for STAR Read Mapping
| Parameter | Function | Typical Setting |
|---|---|---|
--genomeDir |
Path to the genome index directory created in Step 1. | Mandatory |
--readFilesIn |
Specifies the paths to the FASTQ files (forward, then reverse). | sample_R1.fastq sample_R2.fastq |
--runThreadN |
Number of parallel threads to use for the alignment. | Dependent on available CPU cores |
--outSAMtype |
Specifies the format of the alignment output. | BAM SortedByCoordinate |
--quantMode |
Directs STAR to output counts for gene expression analysis. | GeneCounts |
The Scientist's Toolkit: Research Reagent Solutions
Successful execution of the STAR workflow is predicated on the quality of its inputs. The following table details the essential "research reagents"—the data and software components—required for the featured protocol.
Table 3: Essential Research Reagents and Materials for the STAR Workflow
| Item | Function / Role in the Workflow | Technical Specifications & Notes |
|---|---|---|
| Reference Genome (FASTA) | The canonical DNA sequence for the organism serves as the alignment template. | Source: Ensembl, UCSC, or NCBI. Must match the annotation file's version. |
| Gene Annotation (GTF) | Provides the coordinates of known genes, transcripts, and exon-intron boundaries. | Critical for guiding splice-aware alignment and quantifying gene-level counts. |
| RNA-seq Reads (FASTQ) | The raw data representing fragments of the transcriptome from the experimental sample. | For paired-end sequencing, two files (R1 and R2) are required per sample [20]. |
| STAR Software | The aligner software that executes the two-step genome generation and mapping workflow. | Open source, available for Unix, Linux, or Mac OS X systems [22]. |
| High-Performance Computing (HPC) | Computational environment to run the analysis. | STAR is memory-intensive; genome generation for human requires ~32GB RAM [22]. |
| Rubiarbonol B | Rubiarbonol B, MF:C30H50O3, MW:458.7 g/mol | Chemical Reagent |
| Coronarin B | Coronarin B, CAS:119188-38-4, MF:C20H30O4, MW:334.4 g/mol | Chemical Reagent |
Integration in the Broader RNA-seq Analysis Context
The STAR alignment protocol is a central component in a larger RNA-seq data analysis pipeline. The outputs from STAR feed directly into multiple downstream applications crucial for biomedical research and drug development.
The following diagram maps STAR's role within the end-to-end analytical workflow, from raw data to biological insight:
As shown, the process begins with raw FASTQ files, which must first undergo quality control (QC) and adapter trimming using tools like FastQC and Trimmomatic to identify technical errors and remove low-quality sequences [20]. The cleaned reads are then passed to STAR for alignment. The resulting BAM files can be used for visualization of sequencing coverage and splice junctions, while the generated count matrix becomes the direct input for statistical analysis of differential gene expression (DGE) using specialized tools like DESeq2 or edgeR [20]. This entire workflow enables researchers to move from raw sequencing data to biologically interpretable results, such as identifying biomarker candidates for disease or targets for therapeutic intervention.
Step-by-Step Protocol: From Genome Indexing to Read Alignment
The initial and most critical computational step in a paired-end RNA-seq alignment protocol using the STAR (Spliced Transcripts Alignment to a Reference) aligner is the generation of a genome index. This process, executed with the --runMode genomeGenerate command, is not a simple formatting step but a fundamental preprocessing operation that constructs a comprehensive map of the reference genome. This map enables STAR's ultra-fast alignment algorithm, which is based on a strategy of searching for Maximal Mappable Prefixes (MMPs) and subsequently clustering, stitching, and scoring these seeds to generate accurate spliced alignments [3] [2]. For RNA-seq data, this index must be "splice-aware," meaning it incorporates known gene annotations to dramatically improve the accuracy of identifying non-contiguous alignments that span exon-intron boundaries [9] [10]. A properly constructed index is the cornerstone of the entire analysis, influencing the speed, sensitivity, and precision of all downstream results, from gene expression quantification to the discovery of novel splice variants and fusion transcripts [9] [2].
Building a genome index is a resource-intensive process. The primary consideration is RAM memory, as STAR loads the entire genome and its indices into memory during the build process. For complex genomes like human, this requires a substantial amount of memory.
- RAM Requirements: A minimum of 10 x the genome size in bytes is recommended. For the human genome (~3 GigaBases), this translates to approximately 30 GigaBytes of RAM, with 32 GB often recommended for safe operation [9]. In practice, failures in index generation, particularly for large genomes, are frequently due to insufficient RAM.
- Storage and Processing: The process requires sufficient disk space for the resulting index files, which are typically large, and benefits from multiple CPU cores to speed up computation [3] [9]. The table below summarizes the requirements for different genome examples.
Table 1: Example Genome Index Resource Requirements
| Organism | Genome Size (Approx.) | Recommended RAM | Index Directory Size (Approx.) |
|---|---|---|---|
| Human (chr1 only) | ~300 Mbp [3] | 16 GB [3] | Not Specified |
| Human (full) | ~3 Gbp [9] | 30-32 GB [9] | ~30 GB [17] |
| Drosophila | Not Specified | Not Specified | Not Specified |
Detailed Experimental Protocol
Input Data Preparation
The genome indexing process requires two primary input files.
- Reference Genome Sequence: A reference genome file in FASTA format (e.g.,
Homo_sapiens.GRCh38.dna.primary_assembly.fa). The file must be uncompressed for STAR to process it [23]. If downloaded in a compressed format (e.g.,.gz), usegunziporzcatto decompress it. - Gene Annotation File: A gene annotation file in GTF or GFF format (e.g.,
Homo_sapiens.GRCh38.92.gtf). This file provides the coordinates of known exons, introns, and genes, which STAR uses to create a database of splice junctions [3] [9]. This file must also be uncompressed.
Command-Line Execution
The following protocol outlines the step-by-step process for generating the genome index.
- Create an Output Directory: First, create a dedicated directory to store the genome indices. It is best practice to use scratch space with large temporary storage capacity if available [3].
- Execute the Genome Generate Command: Run the STAR command with the
genomeGenerateparameters. The following example uses a SLURM job submission script for use on a high-performance computing (HPC) cluster. - Clean Up: After successful index generation, remember to remove the uncompressed FASTA and GTF files if they were created temporarily, to save disk space [23].
The workflow for this entire process is summarized in the following diagram:
Critical Parameters and Optimization
Understanding the key parameters is essential for generating an optimal genome index.
Table 2: Critical Parameters for STAR Genome Index Generation
| Parameter | Function and Explanation | Recommended Setting |
|---|---|---|
--runThreadN |
Number of CPU cores to use for parallel processing. | Match to available physical cores (e.g., 6) [3]. |
--runMode |
Specifies the operation mode; must be set to genomeGenerate. |
genomeGenerate |
--genomeDir |
Path to the directory where the genome indices will be stored. | User-defined directory path. |
--genomeFastaFiles |
Path(s) to the reference genome FASTA file(s). | Path to your .fa file(s). |
--sjdbGTFfile |
Path to the annotation file in GTF format. | Path to your .gtf file [3] [9]. |
--sjdbOverhang |
Specifies the length of the genomic sequence around annotated junctions to be indexed. | Read length minus 1 [3] [23]. For 100bp PE, use 99 [3]. For 51bp PE, use 50 [23]. If unsure, a default of 100 is often suitable [3]. |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents and Computational Tools
| Item | Function in Protocol | Specification Notes |
|---|---|---|
| Reference Genome (FASTA) | The DNA sequence against which RNA-seq reads are aligned. | Ensure version compatibility with the GTF annotation file (e.g., GRCh38). |
| Gene Annotation (GTF) | Provides coordinates of known genes and splice sites for splice-aware indexing. | Use a version that matches the reference genome. Sources include Ensembl, GENCODE [23]. |
| STAR Aligner | The software package that performs the genome indexing and subsequent read alignment. | Download the latest release from the official GitHub repository [9]. |
| High-Performance Computing (HPC) Environment | Provides the necessary computational power, memory, and storage for index generation. | Requires Unix/Linux OS, ~30 GB RAM for human genome, and sufficient disk space [9]. |
| Methyl 2-amino-2-(2-chlorophenyl)acetate | Methyl 2-amino-2-(2-chlorophenyl)acetate, CAS:141109-13-9, MF:C9H10ClNO2, MW:199.63 g/mol | Chemical Reagent |
| Koumine N-oxide | Koumine N-oxide, MF:C20H22N2O2, MW:322.4 g/mol | Chemical Reagent |
Troubleshooting and Validation
A successful index generation run will conclude with a log message such as "..... finished successfully" [23]. The primary output is the directory specified by --genomeDir, which will be populated with numerous files (e.g., Genome, SA, SAindex, chromosome-specific files, and junction information files). It is important to note that many of these files are for STAR's internal use and do not require direct interpretation by the researcher [24]. The most reliable validation of the index is its subsequent successful use in a mapping job. If the alignment step runs without errors and produces expected mapping statistics, the index has been built correctly.
Essential Parameters for Genome Indexing ('--sjdbOverhang', '--genomeDir')
In paired-end RNA-seq analysis, the STAR (Spliced Transcripts Alignment to a Reference) aligner employs a sophisticated two-step process where genome indexing is the critical first step that enables efficient subsequent read mapping [3]. The indexing process creates a pre-computed reference structure that allows STAR to rapidly identify potential mapping locations for sequencing reads, accounting for challenges specific to RNA-seq data, particularly splicing events where exons can be separated by large introns [3]. Proper configuration of the --sjdbOverhang and --genomeDir parameters during this indexing phase is fundamental to the success of the entire alignment workflow, directly impacting mapping sensitivity, accuracy, and computational efficiency [23] [25].
Parameter Deep Dive: --genomeDir and --sjdbOverhang
The --genomeDir Parameter
The --genomeDir parameter specifies the path to the directory where STAR will store and access the generated genome indices. This directory must be created before running the genome generation step and requires sufficient storage space for the index files, which are typically several tens of gigabytes for mammalian genomes [3] [14].
Key considerations for --genomeDir:
- Storage Location: For computational efficiency, particularly with large datasets, place this directory on storage with high I/O throughput [26].
- Memory Mapping: During alignment, STAR loads portions of the index from this directory into memory, making accessible storage crucial for runtime performance [26].
- Path Specification: The same
--genomeDirpath must be provided during both the genome generation and alignment steps [3].
The --sjdbOverhang Parameter
The --sjdbOverhang parameter defines the length of the genomic sequence around annotated splice junctions to be included in the genome index. This "overhang" allows STAR to effectively map reads that cross splice junctions, with the optimal value being read length minus 1 [3] [23].
Calculation and Default Behavior:
The parameter significantly affects splice junction detection. For diverse read lengths, the recommended approach is setting --sjdbOverhang to max(ReadLength)-1 [23] [27]. If unspecified during genome generation with annotations, STAR applies a default value of 100 [25].
Table 1: sjdbOverhang Configuration for Common Read Lengths
| Read Length | Recommended sjdbOverhang | Considerations |
|---|---|---|
| 51 bp | 50 | Optimal for maximum sensitivity [23] |
| 76 bp | 75 | Standard for shorter paired-end reads |
| 101 bp | 100 | Common default value [25] |
| 151 bp | 150 | Suitable for longer paired-end reads [27] |
| Variable lengths | max(ReadLength)-1 | Recommended for multiple read lengths [27] |
Critical Implementation Constraint: The --sjdbOverhang value must be identical during genome generation and alignment when using pre-indexed annotations. Mismatched values will cause fatal errors [25] [27]. For flexibility with varying read lengths, build the genome index without annotations, then supply the GTF file and --sjdbOverhang during alignment for "on-the-fly" junction inclusion [25].
Experimental Protocol for Genome Indexing
Computational Requirements and Setup
STAR genome indexing is computationally intensive, particularly for mammalian genomes. The following resources are recommended:
Table 2: Computational Requirements for Mammalian Genomes
| Resource | Minimum Requirement | Recommended |
|---|---|---|
| RAM | 16 GB | 32 GB or more [14] |
| CPU Cores | 4-6 | 8+ for faster execution [3] |
| Storage | Sufficient for ~30GB index | High I/O throughput storage [26] |
Software Installation: STAR can be compiled from source or installed via package managers:
Step-by-Step Indexing Protocol
Step 1: Prepare Reference Files
- Obtain genome sequence (FASTA) and annotation (GTF) files from Ensembl or GENCODE
- Ensure chromosome naming consistency between FASTA and GTF files
- Decompress files if necessary:
zcat Homo_sapiens.GRCh38.dna.chromosome.10.fa.gz > genome.fa[23]
Step 2: Create Genome Directory
Step 3: Execute Genome Generation
Critical Parameters Explained:
--runThreadN 8: Number of parallel threads to use [3]--runMode genomeGenerate: Specifies genome indexing mode [3] [23]--genomeDir /path/to/STAR_index: Output directory for indices [3]--genomeFastaFiles /path/to/genome.fa: Input genome sequence [3] [23]--sjdbGTFfile /path/to/annotations.gtf: Gene annotation file [3] [23]--sjdbOverhang 99: Optimal for 100bp reads (100-1=99) [3]
Quality Assessment and Verification
After completion, verify successful index generation by checking for these key files in the --genomeDir directory:
Genome: Binary genome sequenceSA: Suffix arraysjdbInfo.txt: Splice junction database information
Check the log file for successful completion message: "finished successfully" [23].
Integration with Downstream Alignment
Workflow Integration
The genome indexing step directly enables the subsequent read alignment process. Once indexing is complete, the same --genomeDir path is provided to STAR during alignment:
Visualization of the STAR Alignment Workflow
The following diagram illustrates the complete STAR alignment workflow, highlighting the critical role of genome indexing with proper parameter configuration:
Research Reagent Solutions
Table 3: Essential Computational Resources for STAR Genome Indexing
| Resource Type | Specific Solution | Function in Workflow |
|---|---|---|
| Reference Genome | ENSEMBL (Homosapiens.GRCh38.dna.primaryassembly.fa) | Genomic sequence for read alignment [3] |
| Gene Annotation | GENCODE (gencode.v29.annotation.gtf) | Gene models for splice junction identification [23] |
| Compute Infrastructure | AWS EC2 instances (memory-optimized) | High-RAM computational environment [26] |
| Container Platform | Docker/Singularity | Environment reproducibility for alignment [26] |
| Quality Control | RSeQC, Qualimap | Post-alignment quality assessment [28] |
Troubleshooting and Optimization
Common Implementation Issues
- sjdbOverhang Mismatch Error: If encountering "present --sjdbOverhang=XX is not equal to the value at the genome generation step =YY", ensure consistent values or rebuild the index [25] [27].
- Insufficient Memory: For mammalian genomes, allocate at least 32GB RAM to prevent crashes during indexing [14].
- Storage Considerations: Genome indices require significant storage (~30GB for human); ensure adequate space in
--genomeDirlocation [26].
Performance Optimization Strategies
- Parallel Processing: Use 6-8 threads (
--runThreadN) to reduce indexing time [3]. - Reference Optimization: For large-scale processing, consider distributing pre-built indices to compute nodes to avoid redundant indexing [26].
- Cloud Optimization: On AWS, memory-optimized instances (r-series) provide cost-efficient indexing performance [26].
The alignment of high-throughput sequencing reads to a reference genome is a critical step in RNA-seq data analysis [1]. For paired-end RNA-seq data, this process involves determining the precise genomic origins of millions of short sequence read pairs derived from fragmented RNA transcripts. The Spliced Transcripts Alignment to a Reference (STAR) software package addresses the unique challenges of RNA-seq mapping through a novel algorithm that enables ultra-fast and accurate alignment of spliced sequences [2] [29]. STAR's ability to detect both annotated and novel splice junctions, combined with its capacity to identify complex RNA arrangements such as chimeric and circular RNAs, makes it particularly suitable for comprehensive transcriptome studies in biomedical and drug development research [9].
Unlike DNA-seq alignment, RNA-seq alignment must account for non-contiguous genomic regions resulting from RNA splicing. Traditional aligners designed for DNA sequences often fail to identify splice junctions accurately, leading to substantial information loss. STAR employs an uncompressed suffix array-based searching method followed by a clustering/stitching procedure that allows it to precisely map reads across intron boundaries without relying exclusively on existing transcript annotations [2]. This capability is especially valuable for drug development professionals investigating novel transcripts or splicing variants that may serve as therapeutic targets or biomarkers.
Theoretical Foundation of the STAR Algorithm
The STAR alignment algorithm operates through two principal phases: seed searching followed by clustering, stitching, and scoring [2]. During the seed search phase, STAR identifies the Maximal Mappable Prefix (MMP)—the longest substring from the read start that matches exactly to one or more genomic locations. When a read contains a splice junction, the initial MMP maps to the donor splice site, and the algorithm repeats the search on the unmapped portion, which typically maps to an acceptor splice site. This sequential MMP search provides a natural mechanism for pinpointing splice junction locations without prior knowledge of junction databases.
In the second phase, STAR clusters the aligned seeds by genomic proximity and stitches them together using a dynamic programming approach that allows for mismatches and small indels [2]. For paired-end reads, seeds from both mates are processed concurrently, significantly enhancing alignment sensitivity. This approach treats paired-end reads as fragments of the same RNA molecule, allowing accurate alignment even when only one mate contains a reliable anchor sequence.
Table 1: Key Algorithmic Features of STAR for Paired-End RNA-seq Alignment
| Algorithmic Feature | Description | Advantage for Paired-End RNA-seq |
|---|---|---|
| Maximal Mappable Prefix (MMP) Search | Identifies longest exact matches between read segments and reference genome | Enables detection of novel splice junctions without prior annotation |
| Sequential MMP Application | Repeatedly searches unmapped portions of reads after each MMP identification | Naturally identifies precise splice junction locations in a single alignment pass |
| Seed Clustering & Stitching | Groups aligned seeds by genomic proximity and connects them via dynamic programming | Allows comprehensive alignment of reads spanning multiple exons with small indels/mismatches |
| Concurrent Mate Processing | Processes both paired-end mates together during clustering/stitching | Increases sensitivity; one reliable mate can guide alignment of the entire fragment |
| Two-Pass Mapping Mode | Uses splice junctions discovered in first pass to inform second alignment pass | Enhances accuracy for novel junction detection without increasing false positive rate |
Computational Requirements and Resource Planning
Effective utilization of STAR requires careful consideration of computational resources, particularly for large-scale studies in pharmaceutical development environments. STAR's memory footprint is substantial due to its use of uncompressed suffix arrays, which trade memory usage for significant speed advantages [2] [9]. For the human genome (approximately 3 gigabases), STAR requires ~30GB of RAM, making 32GB a recommended minimum [9]. Smaller genomes such as mouse or rat require proportionally less memory, while plant genomes with high repetitive content may need additional resources.
STAR efficiently utilizes multiple processor cores, with alignment speed scaling nearly linearly with core count up to physical processor limits [9]. A modest 12-core server can align approximately 550 million 2×76 bp paired-end reads per hour [29], though hyper-threading can sometimes provide additional speed improvements. Disk space requirements are substantial, with output files for large datasets often exceeding 100GB, particularly when storing intermediate files and multiple alignment formats [9].
Table 2: Computational Requirements for STAR with Different Genome Sizes
| Resource Type | Human Genome (3GB) | Mouse Genome (2.7GB) | Arabidopsis (120MB) |
|---|---|---|---|
| Recommended RAM | 32GB | 28GB | 8GB |
| Disk Space for Index | ~30GB | ~27GB | ~4GB |
| Typical Alignment Speed | 550M reads/hour (12 cores) | 600M reads/hour (12 cores) | >1B reads/hour (12 cores) |
| Output File Space | 100-500GB | 90-450GB | 20-100GB |
Experimental Protocol: Genome Index Generation
Preparation of Reference Files
Before generating genome indices, researchers must obtain reference genome sequences and annotation files. The reference genome should be in FASTA format (uncompressed), while annotations can be in GTF or GFF3 format [1]. For GFF3 files, additional parameters are required to define parent-child relationships between exons and transcripts [1]. For drug development studies, using comprehensive annotations from sources such as GENCODE (human) or Ensembl is recommended, as these include protein-coding genes, non-coding RNAs, and pseudogenes that may be relevant to disease mechanisms.
Index Generation Command
The following protocol generates a genome index for the human GRCh38 assembly:
Critical Parameters:
--genomeDir: Directory where genome indices will be stored [1]--genomeFastaFiles: Reference genome FASTA file(s) [1]--sjdbGTFfile: Gene annotation file in GTF format [1]--sjdbOverhang: Length of genomic sequence around annotated junctions [9]; should be set to read length minus 1 [1]--runThreadN: Number of parallel threads to use [1]--genomeSAindexNbases: Length of the suffix array index; must be scaled down for small genomes [30]--genomeChrBinNbits: Scaling parameter for genomes with many contigs [30]
For non-model organisms or those with limited annotations, the --sjdbGTFfile parameter can be omitted, though this may reduce junction detection accuracy. In such cases, two-pass mapping is strongly recommended [9].
Figure 1: Genome Index Generation Workflow
Experimental Protocol: Mapping Paired-End Reads
Basic Mapping Protocol
The fundamental protocol for aligning paired-end RNA-seq reads assumes prior genome index generation and quality-checked FASTQ files. The example below processes paired-end reads from a typical drug treatment experiment:
Critical Mapping Parameters:
--readFilesIn: Specifies paired-end read files (read1 then read2) [1]--readFilesCommand: Uncompression command for compressed inputs (zcatfor .gz,bzcatfor .bz2) [1]--outSAMtype: Output format;BAM SortedByCoordinateenables direct use in downstream analysis [1]--outFileNamePrefix: Output file naming convention [1]--outFilterType BySJout: Reduces spurious alignments using junction information [31]--outSAMstrandField intronMotif: Adds strand information for transcript assembly [31]
Two-Pass Mapping for Novel Junction Discovery
For studies investigating novel splicing events or working with non-model organisms where annotation is limited, two-pass mapping significantly improves sensitivity:
Two-pass mapping does not substantially increase the number of detected novel junctions but improves the alignment of reads containing these junctions [31]. This approach is particularly valuable in cancer research or toxicology studies where alternative splicing may reveal disease mechanisms or drug effects.
Figure 2: Two-Pass Mapping Workflow
Output Interpretation and Quality Assessment
Output Files and Their Applications
STAR generates multiple output files that serve different purposes in downstream analysis:
- Aligned Reads (BAM):
*Aligned.sortedByCoord.out.bamcontains coordinate-sorted alignments suitable for variant calling, visualization, and read counting [1] - Junction File:
*SJ.out.tablists detected splice junctions with genomic coordinates and strand information [1] - Alignment Statistics:
*Log.final.outprovides comprehensive mapping summary statistics [1] - Progress Log:
*Log.progress.outenables real-time monitoring of large mapping jobs [9]
Interpretation of Alignment Metrics
The Log.final.out file contains critical quality metrics that researchers should evaluate before proceeding to differential expression analysis:
Table 3: Key Alignment Metrics and Their Interpretation
| Metric | Optimal Range | Biological/Technical Interpretation |
|---|---|---|
| Uniquely Mapped Reads | >70% for complex genomes | Lower percentages may indicate contamination, poor RNA quality, or excessive PCR duplicates |
| Splice Junction Detection | High percentage annotated for well-annotated organisms | Increased novel junctions may indicate alternative splicing or insufficient annotation |
| Canonical Splice Sites | >95% of junctions | Non-canonical sites may indicate technical artifacts or biologically relevant non-canonical splicing |
| Mismatch Rate | <1% with reference genomes | Elevated rates may indicate poor sequencing quality, genetic variation, or incorrect reference |
| Multi-Mapping Reads | Variable by genome complexity | Higher in repetitive regions; may be filtered or handled probabilistically in quantification |
| Insertion/Deletion Rates | <0.1% | Elevated rates may indicate sequencing errors or alignment artifacts |
Table 4: Essential Resources for Paired-End RNA-seq Alignment with STAR
| Resource Category | Specific Example | Function in Protocol |
|---|---|---|
| Reference Genome | GRCh38 (human), GRCm39 (mouse) | Provides genomic coordinate system for alignment and annotation |
| Gene Annotation | GENCODE, Ensembl GTF | Defines known gene models and splice junctions for guided alignment |
| Quality Control Tools | FastQC, fastp, Trim Galore | Assesses read quality and performs adapter trimming prior to alignment [32] |
| Alignment Software | STAR (v2.7.9a or newer) | Performs splice-aware alignment of RNA-seq reads [33] |
| Sequence Visualization | IGV, UCSC Genome Browser | Enables visual inspection of aligned reads and splicing patterns |
| Downstream Analysis | featureCounts, HTSeq, RSEM | Quantifies reads per gene for differential expression analysis [34] |
| Computational Environment | High-performance computing cluster | Provides necessary memory and processing resources for large datasets |
Advanced Applications in Drug Development Research
STAR's alignment capabilities enable several advanced applications particularly relevant to pharmaceutical research and development. The detection of chimeric (fusion) transcripts can identify oncogenic drivers in cancer studies [2], while comprehensive splice junction analysis can reveal alternative splicing patterns induced by drug treatments or disease states. For quantitative transcriptomics, STAR alignments can be processed by tools like RSEM or Cufflinks to estimate transcript abundances [9], providing crucial data for biomarker discovery and mechanism of action studies.
In large-scale drug screening applications, STAR's speed becomes particularly valuable. The ability to process hundreds of millions of reads per hour on modest computing infrastructure enables rapid turnaround from sequencing to expression analysis [29]. This throughput advantage facilitates time-course studies of drug response or large cohort analyses where dozens or hundreds of samples require processing.
For specialized applications in immuno-oncology or host-pathogen interactions, researchers can employ modified alignment strategies that combine human and pathogen references in a single index. This approach enables simultaneous quantification of host response genes and pathogen transcriptomes from infection models, providing comprehensive insights into drug effects on integrated biological systems.
Within the broader research on paired-end RNA-seq alignment protocols, the Spliced Transcripts Alignment to a Reference (STAR) aligner has established itself as a cornerstone tool due to its high accuracy and speed [3] [9]. Its unique strategy of performing seed searching followed by clustering, stitching, and scoring allows it to efficiently handle the challenges of RNA-seq data mapping, particularly the accurate alignment of reads across splice junctions [3]. This application note provides detailed, practical protocols for both basic and advanced mapping scenarios, enabling researchers and drug development professionals to reliably generate the alignment data necessary for downstream expression and differential analysis.
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details the key materials and software required to perform RNA-seq alignment with STAR.
Table 1: Essential Research Reagents and Computational Tools for STAR Alignment
| Item | Function / Description |
|---|---|
| Reference Genome (FASTA) | The DNA sequence of the target organism against which reads are aligned (e.g., Homo_sapiens.GRCh38.dna.primary_assembly.fa). |
| Gene Annotation (GTF/GFF) | File containing genomic coordinates of known genes, transcripts, and exons. Used by STAR to inform spliced alignment (e.g., Homo_sapiens.GRCh38.92.gtf) [3] [9]. |
| STAR Aligner | The core software package that performs ultra-fast, splice-aware alignment of RNA-seq reads [9]. |
| RNA-seq Reads (FASTQ) | The input sequence files, which can be single-end or paired-end (two files per sample for the latter). |
| SAMtools | A suite of utilities used for post-processing alignments, such as converting SAM to BAM, sorting, and indexing [12] [23]. |
| Brachynoside heptaacetate | Brachynoside heptaacetate, MF:C45H54O22, MW:946.9 g/mol |
| 7-Oxohinokinin | 7-Oxohinokinin |
Core STAR Alignment Strategy and Workflow
The efficiency of the STAR algorithm stems from its two-step mapping process. First, it searches for the Maximal Mappable Prefixes (MMPs)—the longest segments of the read that exactly match the reference genome [3]. These segments, or "seeds," are identified sequentially from the unmapped portions of the read. In the second stage, STAR clusters these seeds based on proximity, stitches them together into a complete alignment, and scores them, allowing it to construct complex spliced alignments spanning multiple exons [3].
The following diagram illustrates the logical workflow and decision points in a STAR-based RNA-seq analysis, from data preparation to final output.
Experimental Protocols and Commands
Protocol 1: Basic Mapping for a Single Sample
This is the foundational protocol for aligning a single RNA-seq sample to a reference genome, producing a coordinate-sorted BAM file ready for downstream analysis [3] [9].
Methodology:
- Genome Index Generation: The reference genome and annotations must first be converted into a STAR-specific index. This is a one-time requirement for a given genome and annotation combination.
- Read Alignment: The sequencing reads are mapped against the pre-built genome index. STAR performs splice-aware alignment, utilizing the provided gene annotations to guide the mapping across introns.
Basic Single-Sample Alignment Command:
Code 1: Command for aligning a single-end FASTQ file. For paired-end reads, specify both files separated by a space in --readFilesIn (e.g., read_1.fastq read_2.fastq) [3] [9].
Key Options:
--genomeDir: Path to the directory containing the genome indices.--runThreadN: Number of CPU threads to use for alignment.--readFilesIn: Path(s) to the input FASTQ file(s).--outSAMtype BAM SortedByCoordinate: Directs STAR to output a coordinate-sorted BAM file, which is the standard for most downstream applications [3] [23].--readFilesCommand zcat: Required if input FASTQ files are compressed (e.g.,.fastq.gz) [9].
Protocol 2: Advanced 2-Pass Mapping for Novel Junction Discovery
For discoveries that go beyond known annotations, such as novel splice junctions or isoforms, the 2-pass mapping strategy is recommended [9]. This method increases the sensitivity of novel junction detection by leveraging information from all samples in an experiment.
Methodology:
- First Pass: Align all samples individually. During this process, STAR collects data on novel splice junctions detected in each sample, outputting them to a file (
SJ.out.tab). - Second Pass: STAR is run again on all samples, but this time it incorporates the novel junctions discovered in the first pass across the entire dataset into its mapping model, leading to more comprehensive and accurate alignments.
Protocol 3: Efficient Mapping of Multiple Samples
Processing multiple samples is a common requirement in experimental designs for drug development. This can be achieved efficiently using a shell loop to process samples sequentially or in parallel, ensuring separate output files for each sample [35] [36].
Methodology: A shell script is used to iterate over a list of sample identifiers. For each sample, the corresponding paired-end FASTQ files are defined, and a dedicated STAR command is executed. This method provides clear organization and is easier to debug and manage than attempting to process all samples in a single STAR command, which merges results [36].
Shell Script for Multiple Sample Alignment:
Code 2: A bash loop for processing multiple paired-end samples. Each sample produces a separate BAM file and set of output files [35].
The following diagram visualizes the control flow of this multiple sample processing strategy.
Table 2: Summary of Basic and Advanced STAR Alignment Commands
| Protocol Scenario | Core Command / Script Structure | Key Output Files |
|---|---|---|
| Single Sample (Paired-end) | STAR --genomeDir /index --readFilesIn R1.fastq R2.fastq --outSAMtype BAM SortedByCoordinate --outFileNamePrefix sample_ [3] [9] |
sample_Aligned.sortedByCoord.out.bam: Sorted alignments.sample_Log.final.out: Mapping statistics.sample_SJ.out.tab: Detected splice junctions. |
| Multiple Samples (Loop) | for base in sample1 sample2; do STAR --readFilesIn ${base}_1.fq ${base}_2.fq --outFileNamePrefix ${base}_ ... ; done [35] |
A set of the above output files for each sample1 and sample2. |
| Quantification during Mapping | STAR ... --quantMode GeneCounts [23] |
sample_ReadsPerGene.out.tab: Raw counts per gene for unstranded (col 2), forward (col 3), and reverse (col 4) strands [23]. |
The protocols detailed herein provide a robust framework for implementing the STAR aligner in both standard and complex research scenarios. The basic single-sample command forms the essential building block, while the multiple-sample loop script offers an efficient and organized approach for larger-scale studies. Furthermore, the advanced 2-pass method ensures maximal discovery of novel transcriptomic features. Mastery of these commands empowers researchers to reliably transform raw RNA-seq data into high-quality alignments, forming a solid foundation for all subsequent bioinformatic analysis in the drug development pipeline.
Within the broader methodology of paired-end RNA-seq alignment using STAR, efficient handling of compressed sequencing files is a critical, yet often overlooked, technical step. This application note details the use of the --readFilesCommand zcat parameter, a best-practice solution for direct, on-the-fly decompression during alignment. We provide a standardized protocol, quantitative performance data, and integrated workflow visualizations to guide researchers and drug development professionals in implementing robust, resource-efficient RNA-seq analyses, thereby ensuring data integrity from raw sequences to aligned reads.
RNA sequencing (RNA-seq) has become a cornerstone of modern genomics, enabling transcriptome-wide analysis of gene expression, splicing, and genetic variation. The Spliced Transcripts Alignment to a Reference (STAR) aligner is widely recognized for its speed and accuracy in handling the complexities of RNA-seq data, including spliced alignment across exon-intron boundaries [23]. A typical RNA-seq experiment generates substantial data volumes, with raw sequencing files (FASTQ) often compressed using gzip to conserve storage space. Consequently, a routine analytical challenge involves managing these compressed files without introducing unnecessary I/O overhead or pipeline complexity. The --readFilesCommand zcat parameter within STAR provides an elegant solution, allowing the aligner to directly stream decompressed data from compressed .fastq.gz or .fq.gz inputs into the alignment engine, bypassing the need for manual decompression and intermediate disk storage [37] [23]. This note formalizes the application of this command within a comprehensive paired-end RNA-seq alignment protocol.
Research Reagent and Computational Solutions
The following table catalogues the essential reagents, software, and data resources required to execute the STAR alignment protocol with compressed input files.
Table 1: Essential Research Reagents and Computational Resources for RNA-seq Alignment with STAR
| Category | Item | Description and Function |
|---|---|---|
| Software | STAR Aligner [37] [23] [38] | A specialized aligner for RNA-seq data that performs spliced alignment to a reference genome. |
| Software | zcat / gunzip -c [37] [23] |
A standard Unix/Linux command used for decompressing .gz files to standard output without deleting the original file. |
| Data | Compressed FASTQ Files [37] [39] | The raw paired-end sequencing data files (e.g., sample_R1.fastq.gz, sample_R2.fastq.gz). |
| Reference Data | Genome Index [37] [23] [39] | A pre-built STAR index of the reference genome, which is required for the alignment step. |
| Reference Data | Annotation File (GTF/GFF) [37] [23] [40] | A file in GTF or GFF format containing known gene models and transcript structures for the reference genome. |
Core Protocol: Integrating--readFilesCommand zcatinto STAR Alignment
Principle and Rationale
The --readFilesCommand parameter instructs STAR on how to preprocess the files specified in --readFilesIn. By setting it to zcat, STAR executes the zcat command (or the equivalent gunzip -c) for each compressed input file, reading the decompressed content directly from the standard output stream [37] [23]. This method is memory-efficient and eliminates the need to create large, temporary decompressed files on disk, which is particularly advantageous in high-performance computing (HPC) environments with limited storage quotas or during the processing of large-scale datasets.
Detailed Command-Line Methodology
The following steps outline the complete procedure for aligning paired-end RNA-seq data from compressed FASTQ files.
Step 1: Define Key Variables and Paths Organize your workflow by first defining shell variables for paths to critical resources. This enhances command readability, reduces errors, and facilitates scripting.
Step 2: Execute STAR with --readFilesCommand zcat
Construct and execute the STAR command for paired-end reads. The following example incorporates common parameters for a comprehensive analysis [37] [35] [23].
Explanation of Key Parameters:
--runThreadN 8: Utilizes 8 CPU threads for parallel processing to accelerate alignment [37].--readFilesIn: Specifies the pair of compressed FASTQ files for the sample [35].--readFilesCommand zcat: The core command enabling direct decompression [37] [23].--outSAMtype BAM SortedByCoordinate: Outputs alignments as a BAM file sorted by genomic coordinates, which is the required input for many downstream tools [37] [23].--quantMode GeneCounts: Directs STAR to output read counts per gene, leveraging the provided GTF annotation file [37] [23].
Data Presentation and Performance Metrics
The integration of --readFilesCommand zcat impacts workflow efficiency primarily by reducing disk I/O. The table below summarizes a comparative analysis of file handling strategies.
Table 2: Quantitative Comparison of File Handling Methods in STAR RNA-seq Alignment
| Parameter | Manual Decompression (Then STAR) | STAR with --readFilesCommand zcat |
|---|---|---|
| Intermediate Disk Usage | High (2x compressed file size) | None |
| I/O Overhead | High (read compressed, write uncompressed, read uncompressed) | Low (read compressed, stream to STAR) |
| Pipeline Complexity | Higher (requires separate decompression step and cleanup) | Lower (single, integrated command) |
| Required Command | gunzip -c sample_R1.fastq.gz > sample_R1.fastq followed by --readFilesIn sample_R1.fastq |
--readFilesIn sample_R1.fastq.gz --readFilesCommand zcat |
| Handling of Paired-end Files | Requires decompressing two files separately | Simultaneous streaming of both files |
Integrated Workflow Visualization
The following diagram illustrates the logical flow and comparative advantage of using the --readFilesCommand zcat parameter within the end-to-end STAR alignment workflow for paired-end RNA-seq data.
Diagram 1: Workflow comparison of file handling methods in STAR RNA-seq alignment. The optimized method with --readFilesCommand zcat eliminates intermediate disk-intensive steps.
Discussion and Best Practices
The --readFilesCommand zcat parameter represents a best practice for handling compressed RNA-seq data in STAR workflows. Its primary advantage is the significant reduction in disk I/O, which translates to faster processing times and lower storage wear in production environments. This approach is not limited to zcat; for other compression formats, such as those ending in .bz2, the command can be substituted with bzcat [38].
For large-scale studies involving multiple samples, this command can be seamlessly incorporated into a scripting framework to automate alignment. A for loop that iterates over sample base names, constructs the respective filenames for paired-end reads, and executes the integrated STAR command ensures reproducibility and efficiency [37] [35].
In conclusion, the proper use of --readFilesCommand zcat is a simple yet powerful technique that optimizes the initial and crucial alignment step in RNA-seq analysis. By integrating this protocol, researchers in both academic and drug development settings can enhance the robustness and scalability of their genomic pipelines, ensuring that data processing is both accurate and resource-conscious.
This application note provides a detailed protocol for configuring the output of the Spliced Transcripts Alignment to a Reference (STAR) aligner to generate sorted BAM files optimized for downstream RNA-seq analysis. Properly sorted BAM files, where read alignments are ordered by genomic coordinates, constitute a critical prerequisite for numerous bioinformatics tools including transcript quantifiers, variant callers, and genome browsers. This guide details the specific parameters and computational strategies for generating analysis-ready alignments within the context of paired-end RNA-seq studies, enabling researchers to ensure data compatibility and analytical efficiency throughout their experimental pipeline.
In RNA-seq analysis, the alignment of sequencing reads represents a pivotal step where raw sequence data is mapped to a reference genome. The STAR aligner was specifically designed to address the unique challenges of RNA-seq data, particularly the accurate alignment of reads across splice junctions [2]. However, the raw output of any aligner requires further processing to be functionally useful for downstream applications.
A BAM file is the binary, compressed version of a Sequence Alignment/Map (SAM) file, storing aligned sequencing reads along with their mapping qualities and genomic coordinates [41]. While STAR can generate BAM files directly, the default ordering of reads is often not optimal for subsequent analysis tools. Sorting these alignments by genomic coordinate—arranging them by chromosome and base position—creates a structured file that enables rapid random access, efficient indexing, and compatibility with downstream bioinformatics tools [42] [43]. This process transforms a collection of alignments into an organized dataset ready for quantitative analysis.
The Critical Role of Sorted BAM Files in Downstream Analysis
Foundational Requirements for Downstream Tools
Most established bioinformatics tools for RNA-seq analysis explicitly require coordinate-sorted BAM files as input. This requirement exists because sorted data allows programs to process genomic regions systematically without loading entire files into memory.
- Variant Calling: Detection of single nucleotide polymorphisms (SNVs) and other genetic variants relies on sorted BAM files to comprehensively assess evidence at each genomic locus [42].
- Transcript Quantification: Tools like HTSeq and Cufflinks require sorted alignments to accurately assign reads to genomic features while handling overlapping regions [44].
- Visualization: Genome browsers such as IGV (Integrative Genomics Viewer) depend on sorted BAM files and their accompanying indexes (.bai) for efficient visualization and rapid navigation across genomic regions [41] [43].
Performance and Analytical Advantages
The computational advantages of sorted BAM files extend beyond simple compatibility. Indexing, a process that creates a separate index file enabling rapid look-up of alignments in specific genomic regions, can only be performed on coordinate-sorted BAM files [42] [43]. This index-file combination allows analytical tools and visualizers to quickly access data from a specific genomic locus without processing the entire file, dramatically improving computational efficiency, especially with large datasets.
Complete STAR Alignment Protocol for Sorted BAM Output
Preliminary Step: Genome Index Generation
Before conducting alignments, STAR requires a genome index to be generated. This one-time process creates a searchable reference structure that enables STAR's ultrafast alignment performance.
Protocol:
- Obtain reference genome sequences (in FASTA format) and annotation files (in GTF format).
- Unzip the reference files if they are compressed.
- Execute the STAR genome generation command with appropriate parameters:
Table 1: Key Parameters for STAR Genome Index Generation
| Parameter | Typical Setting | Explanation |
|---|---|---|
--runMode genomeGenerate |
N/A | Specifies genome index generation mode. |
--genomeDir |
User-defined path | Directory to store the generated index. |
--sjdbOverhang |
ReadLength - 1 | Optimal value depends on read length; crucial for splice junction detection [23]. |
--runThreadN |
Variable | Number of CPU threads to use for parallelization. |
Core Protocol: Alignment with Sorted BAM Output
The following protocol details the alignment step for paired-end reads, specifically configured to output a coordinate-sorted BAM file.
Protocol:
- Navigate to your working directory containing the FASTQ files.
- Create an output directory for alignment results:
mkdir alignments - Execute the STAR alignment command with parameters configured for sorted BAM output:
Table 2: Essential STAR Parameters for Generating Sorted BAM Files
| Parameter | Setting | Function |
|---|---|---|
--readFilesIn |
read1.fastq read2.fastq |
Specifies paired-end read files [35] [23]. |
--readFilesCommand |
zcat |
Enables reading of compressed (.gz) input files. |
--outSAMtype |
BAM SortedByCoordinate |
Critical parameter that directs STAR to output a coordinate-sorted BAM file [23]. |
--quantMode |
GeneCounts |
Optional but recommended: simultaneously generates read counts per gene. |
--outFileNamePrefix |
alignments/sample_ |
Defines directory and prefix for all output files. |
Output File Verification and Handling
Upon successful completion, STAR generates several output files in the specified directory. The key file is Aligned.sortedByCoord.out.bam, which contains the coordinate-sorted alignments. To verify the successful sorting of the BAM file, use samtools:
The output should display SO:coordinate, confirming the file is sorted by genomic coordinate. To enable compatibility with genome browsers and various analysis tools, generate an index for the sorted BAM file:
This command creates an index file named sample_Aligned.sortedByCoord.out.bam.bai [43].
Experimental Workflow and Data Flow
The following diagram illustrates the complete experimental workflow from raw sequencing data to analysis-ready sorted BAM files, highlighting the integration between STAR and downstream tools.
Diagram 1: Complete workflow for generating and utilizing sorted BAM files from paired-end RNA-seq data.
The Scientist's Toolkit: Essential Research Reagents and Computational Tools
Table 3: Research Reagent Solutions for RNA-seq Alignment and Analysis
| Resource Type | Specific Tool/Resource | Function in Workflow |
|---|---|---|
| Alignment Software | STAR Aligner | Performs ultrafast spliced alignment of RNA-seq reads to reference genome [2] [23]. |
| File Manipulation Tools | Samtools | Provides utilities for sorting, indexing, and viewing BAM files; essential for post-processing [42] [41] [43]. |
| Reference Genome | ENSEMBL GRCh38 | Standardized human reference genome sequence in FASTA format provides mapping context. |
| Gene Annotation | GENCODE (v29+) | Comprehensive gene annotation in GTF format defines gene models for read quantification [23]. |
| Quantification Tools | HTSeq, featureCounts | Count reads overlapping genomic features using coordinate-sorted BAM files as input [44]. |
| Visualization Software | IGV, GenomeBrowse | Enables visual inspection of alignments in sorted BAM files across genomic regions [41] [45]. |
| Toddalosin | Toddalosin|For Research | High-purity Toddalosin for research. Explore its anti-inflammatory and anticancer mechanisms. This product is For Research Use Only. Not for human or veterinary use. |
Configuring the STAR aligner to generate coordinate-sorted BAM files through the --outSAMtype BAM SortedByCoordinate parameter is a fundamental yet powerful step in establishing a robust RNA-seq analysis pipeline. This protocol provides researchers with the specific commands and conceptual understanding needed to produce analysis-ready alignment files that ensure compatibility with the broad ecosystem of downstream bioinformatics tools. By implementing this optimized output configuration, scientists can significantly enhance the efficiency, reproducibility, and reliability of their transcriptomic studies, from initial quality assessment through final visualization and interpretation.
The foundational step in RNA sequencing (RNA-seq) analysis involves aligning short sequence reads to a reference genome, a process that presents unique challenges compared to DNA sequence alignment. RNA sequences are often spliced, meaning they are derived from non-contiguous genomic regions (exons) separated by introns [9]. While standard alignment tools can identify reads originating from known, annotated splice junctions, the discovery of novel splice junctions remains computationally difficult. Traditional single-pass alignment methods implicitly favor known junctions by requiring stronger alignment evidence for novel ones, creating a quantification bias that can obscure important biological discoveries [46].
Two-pass alignment has emerged as a powerful strategy to overcome this limitation. This approach separates the processes of splice junction discovery and read quantification, thereby increasing sensitivity to novel splicing events without compromising alignment accuracy [46] [47]. Initially made feasible by advances in ultra-fast aligners like STAR (Spliced Transcripts Alignment to a Reference), two-pass alignment is particularly valuable for studies focusing on differential splicing analysis, variant discovery, and comprehensive transcriptome characterization [9] [46]. This protocol details the implementation and advantages of two-pass mapping within the context of paired-end RNA-seq alignment using STAR, providing researchers with a robust framework for enhancing novel junction detection.
Theoretical Foundation: How Two-Pass Mapping Enhances Sensitivity
The core principle of two-pass alignment involves performing an initial mapping pass to discover splice junctions across the entire dataset, followed by a second pass that utilizes these discovered junctions as supplementary annotation to guide more sensitive alignment [46]. In standard single-pass mode, aligners like STAR use only pre-existing gene annotation files (GTF/GFF) to identify known splice junctions. While this approach is efficient, it inherently biases quantification against unannotated junctions, as reads spanning novel junctions require more stringent evidence to align [46].
The two-pass method addresses this limitation through a sophisticated realignment process. In the first pass, reads are aligned to the reference genome with standard parameters, during which a comprehensive set of splice junctions is collected, including both annotated and novel candidates [47]. In the second pass, these newly discovered junctions are incorporated into the alignment process, either by regenerating the genome indices or by directly supplying the junctions during mapping. This allows reads to be realigned with reference to the complete set of junctions discovered in the first pass, significantly improving the alignment rate for reads spanning novel splice sites [46] [47].
Performance profiling across diverse RNA-seq datasets demonstrates that two-pass alignment improves quantification of at least 94% of simulated novel splice junctions, providing as much as 1.7-fold deeper median read depth over these junctions compared to single-pass methods [46]. This enhancement works primarily by permitting alignment of sequence reads with shorter overhangs across splice junctions, thereby increasing sensitivity without substantially compromising specificity when appropriate filtering is applied [46].
Methodological Approaches: Implementing Two-Pass Mapping
Comparative Analysis of Two-Pass Implementation Strategies
Researchers can implement two-pass mapping using STAR through two distinct methodologies, each with specific advantages and considerations. The table below summarizes the key characteristics of both approaches:
Table 1: Comparison of STAR Two-Pass Mapping Implementation Strategies
| Feature | Traditional Two-Pass with Index Rebuilding | Direct Two-Pass Mapping |
|---|---|---|
| Workflow Complexity | Multi-step process requiring separate genome index generation | Simplified single-step process |
| Computational Overhead | Higher; requires storing and processing multiple genome indices | Lower; uses original genome indices |
| Sample Integration | Enables uniform junction discovery across multiple samples [48] | Processes each sample independently |
| Implementation Control | Allows for careful filtering of novel junctions prior to second pass [47] | Uses all junctions discovered in first pass automatically |
| Recommended Use Case | Studies with multiple samples requiring consistent junction annotation | Individual sample analysis or rapid prototyping |
Two-Pass Mapping Workflow
The following diagram illustrates the logical workflow for both implementation strategies of two-pass mapping with STAR:
Two-Pass Mapping Implementation Workflow
Detailed Protocol: Traditional Two-Pass with Index Rebuilding
This approach provides the highest quality results, particularly for studies involving multiple samples, as it enables uniform novel splicing detection across all samples [48].
Step 1: Initial Genome Index Generation Before beginning two-pass mapping, generate standard STAR genome indices using your reference genome and annotation file:
The --sjdbOverhang parameter should be set to the read length minus 1. For paired-end reads, this typically corresponds to the length of one read minus 1 [9].
Step 2: First Pass Alignment - Junction Discovery Perform initial alignment on all samples to discover splice junctions:
Repeat this step for each sample in your dataset. The critical output for two-pass mapping is the SJ.out.tab file generated for each sample.
Step 3: Junction Filtering and Consolidation Combine and filter junction files from all samples to remove likely false positives:
Filtering parameters can be adjusted based on experimental needs. The fifth column in SJ.out.tab indicates canonical splicing (1=non-canonical, 0=non-canonical), and the seventh column indicates the number of uniquely mapping reads [47].
Step 4: Genome Index Regeneration with Novel Junctions Regenerate genome indices incorporating the filtered novel junctions:
The --sjdbFileChrStartEnd parameter is used to incorporate the filtered junction file into the new genome indices [47].
Step 5: Second Pass Alignment - Sensitive Mapping Realign all reads using the new genome indices containing both annotated and novel junctions:
This second pass alignment will produce the final BAM files with enhanced sensitivity for novel junctions, suitable for all downstream analyses including differential expression and splicing analysis.
Alternative Protocol: Direct Two-Pass Mapping
For individual samples or rapid analysis, STAR offers a streamlined two-pass mode that doesn't require explicit index regeneration:
The --twopassMode Basic parameter instructs STAR to perform both passes automatically [48]. This approach is computationally more efficient but processes each sample independently, which might lead to less consistent junction discovery across multiple samples compared to the traditional method [48].
Performance Characterization and Quality Assessment
Quantitative Performance Metrics
Extensive benchmarking across diverse RNA-seq datasets reveals consistent improvements in novel junction quantification with two-pass alignment. The table below summarizes performance gains observed across various biological contexts:
Table 2: Performance Improvements with Two-Pass Alignment Across Sample Types
| Sample Type | Read Length | Junctions Improved | Median Read Depth Ratio | Expected Read Depth Ratio |
|---|---|---|---|---|
| Lung Adenocarcinoma Tissue | 48 nt | 99% | 1.68× | 1.75× |
| Lung Normal Tissue | 48 nt | 98% | 1.71× | 1.75× |
| Reference RNA (UHRR) | 75 nt | 94-97% | 1.25-1.26× | 1.35× |
| Lung Cancer Cell Lines | 101 nt | 97% | 1.19-1.21× | 1.19-1.23× |
| Arabidopsis Tissues | 101 nt | 95-97% | 1.12× | 1.12× |
Data sourced from performance profiling across twelve publicly available RNA-seq datasets [46]. The "Median Read Depth Ratio" represents the fold-change in read depth over novel splice junctions comparing two-pass to single-pass alignment, while "Junctions Improved" indicates the percentage of junctions showing quantification benefits.
Technical Validation and Error Profiling
While two-pass alignment significantly improves sensitivity, it may potentially introduce alignment errors. However, these are readily identifiable through simple classification methods [46]. The primary mechanism of improvement involves permitting alignment of reads with shorter spanning lengths across splice junctions (as few as 3-8 nucleotides compared to more stringent requirements in single-pass mode) [46].
Quality assessment should include:
- Junction saturation analysis using RSeQC to determine if sequencing depth adequately captures splice junction diversity [49]
- Read distribution metrics to verify appropriate distribution across genomic features [49]
- Junction annotation comparing detected junctions to known reference annotations [49]
- Duplicate read analysis to distinguish technical versus biological duplication [49]
Successful implementation of two-pass mapping requires specific computational resources and software tools. The following table details the essential components:
Table 3: Essential Research Reagent Solutions for STAR Two-Pass Mapping
| Resource Category | Specific Tool/Requirement | Function/Purpose | Implementation Notes |
|---|---|---|---|
| Alignment Software | STAR (v2.7.10b or newer) | Spliced alignment of RNA-seq reads | Ultra-fast universal RNA-seq aligner; supports two-pass mode natively [12] [47] |
| Reference Genome | Species-specific FASTA file | Genomic coordinate system for alignment | Human: GRCh38; Arabidopsis: TAIR10; ensure compatibility with annotation [46] |
| Gene Annotation | GTF/GFF3 file | Annotation of known gene structures | GENCODE-Basic recommended for human; essential for guided alignment [46] |
| Computing Resources | 32+ GB RAM (human genome) | Memory for alignment processes | Minimum 10× genome size; 30GB for human genome recommended [9] |
| Computing Resources | Multi-core processors | Parallel processing of alignment | Significantly increases mapping throughput [9] |
| Quality Control | RSeQC, Qualimap | Alignment quality assessment | Provides junction saturation, read distribution metrics [49] |
| Junction Filtering | Custom awk scripts | Removal of false positive junctions | Filter by canonical splicing, unique read support [47] |
Two-pass alignment with STAR represents a significant methodological advancement for novel splice junction discovery in RNA-seq experiments. By separating the processes of junction discovery and quantification, this approach provides substantially improved sensitivity for detecting unannotated splicing events while maintaining high alignment accuracy. The documented 1.7-fold improvement in read depth over novel junctions across diverse biological contexts makes this protocol particularly valuable for studies investigating alternative splicing, transcriptome diversity, and rare splicing variants [46].
The two implementation strategies offer flexibility for different experimental designs: the traditional index-rebuilding approach provides the highest quality for multi-sample studies, while the direct mapping method offers computational efficiency for individual samples [47] [48]. As RNA-seq applications continue to evolve toward single-cell analyses and long-read sequencing, the principles of iterative alignment refinement embodied in two-pass mapping will remain relevant for maximizing biological discovery from transcriptomic data.
Researchers should consider two-pass alignment as a standard approach when novel junction discovery is a primary study objective, particularly in clinical contexts where comprehensive transcriptome characterization is essential for understanding disease mechanisms and identifying therapeutic targets.
Beyond Defaults: Optimizing Parameters and Solving Common Problems
Optimizing '--sjdbOverhang' for Your Read Length
In the analysis of paired-end RNA-seq data, the accurate alignment of reads that span splice junctions is a critical step. The STAR (Spliced Transcripts Alignment to a Reference) aligner addresses this challenge through its splice junction database (sjdb) system. The --sjdbOverhang parameter is a central configuration point for this system, specifically used during the genome indexing step. Its purpose is to define the length of the genomic sequence on each side of an annotated junction that STAR will include in its indices [50] [9]. Proper configuration of this parameter ensures that the aligner can effectively map the portions of a read that lie on either side of a splice site. Setting this parameter correctly is foundational to the sensitivity and accuracy of the entire alignment process, impacting downstream analyses such as novel isoform discovery and differential gene expression.
Conceptual Foundation and Technical Specifications
The Role of sjdbOverhang in Genome Indexing
The --sjdbOverhang parameter is exclusively utilized during the genomeGenerate run mode. When a gene annotation file (GTF/GFF) is provided, STAR extracts all known splice junctions. For each junction, it then takes N bases from the donor (exon) side and N bases from the acceptor (exon) side and splices these sequences together to create an artificial "junction" sequence in the genome index [51]. The value N is specified by the --sjdbOverhang option. This process effectively enriches the reference genome with junctional sequences, creating a bridge that allows STAR to recognize and align reads that cross splice boundaries with high fidelity. It is crucial to distinguish this from the --alignSJDBoverhangMin parameter, which is used later, during the read mapping step, to define the minimum number of bases a read must have on each side of a junction for the alignment to be considered valid [50].
Determining the Ideal sjdbOverhang Value
The established rule for determining the optimal --sjdbOverhang value is mate_length - 1 [50] [51] [3]. For example, in a 2x100 bp paired-end experiment, the mate length is 100 bp, making the ideal sjdbOverhang value 99. This logic accounts for the most extreme but plausible scenario where a read spans a junction with a single base on one side and 99 bases on the other. Setting the parameter to this value ensures the genome index contains junction sequences long enough to accommodate such an alignment. The following table summarizes the recommended values for common sequencing read lengths:
Table 1: Recommended --sjdbOverhang Values for Common Read Lengths
| Read Length (bp) | Ideal --sjdbOverhang |
Standard Practice |
|---|---|---|
| 50 | 49 [23] | 49 |
| 75 | 74 [50] | 100 [51] |
| 100 | 99 [3] | 100 [51] [9] |
| 101 | 100 [27] | 100 |
| 150 | 149 | 100 [51] or 149 [27] |
For datasets with a single, fixed read length, applying the mate_length - 1 rule is straightforward. As the developer Alexander Dobin notes, for longer reads (e.g., 100 bp and above), using a generic value of 100 often works with minimal performance loss and is a widely adopted standard for simplicity [51] [9]. However, for shorter reads (e.g., <50 bp), using the ideal value is strongly recommended to maintain sensitivity [51].
Experimental Protocols and Practical Application
Protocol 1: Standard Genome Indexing for a Fixed Read Length
This protocol is designed for experiments with a single, uniform read length, such as a newly generated dataset.
Necessary Resources:
- Reference Genome: FASTA file for the target organism.
- Gene Annotations: GTF or GFF file, preferably from a trusted source like Ensembl or GENCODE.
- Computational Resources: A server with sufficient RAM (e.g., ~32 GB for human) and multiple CPU cores.
Procedure:
- Calculate
sjdbOverhang: Determine your read length. If your data is 100 bp paired-end, the value is 99. - Execute the Indexing Command:
- Validation: Upon successful completion, the specified
--genomeDirwill contain the binary genome indices, including the splice junction databases.
Protocol 2: Handling Multiple or Variable Read Lengths
A common challenge in meta-analyses is integrating datasets with different read lengths. The strategy depends on the length variation.
Strategy A: Uniformly Long Reads
- If all datasets have read lengths ≥100 bp (e.g., 100 bp, 125 bp, 150 bp), a single index with
--sjdbOverhang 100is sufficient and highly practical [51]. This approach avoids the need to maintain multiple indices.
Strategy B: Mix Including Short Reads
- If any dataset has reads shorter than 100 bp (e.g., 50 bp, 75 bp), the optimal solution is to build separate indices tailored to the shorter read lengths. For a 75 bp dataset, an index with
--sjdbOverhang 74should be generated [50] [23].
Strategy C: Variable Lengths within a Dataset
- For reads of varying lengths from a single sample (e.g., after quality trimming), the parameter should be set to
max(ReadLength)-1[3] [23]. If the maximum read length is 150 bp, use--sjdbOverhang 149. If the maximum length is very long, the default of 100 remains a safe, efficient choice [51].
Table 2: Decision Matrix for Multi-Dataset Analysis
| Scenario | Example Read Lengths | Recommended Strategy | --sjdbOverhang Value |
|---|---|---|---|
| All long reads | 100, 101, 150 | Single index with default long value | 100 |
| Includes short reads | 50, 100, 150 | Multiple indices | 49 (for 50 bp data), 100 (for others) |
| Single sample, variable length | 70-150 bp after trimming | Single index based on max length | 149 [3] |
The following workflow diagram synthesizes the decision process for selecting and implementing the correct --sjdbOverhang value:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Computational Tools for STAR Alignment
| Item | Function in Protocol | Specification |
|---|---|---|
| Reference Genome (FASTA) | The reference sequence against which reads are aligned. | Species-specific (e.g., GRCh38 for human); obtain from Ensembl, GENCODE, or UCSC. |
| Gene Annotation (GTF) | Provides known transcript structures and splice sites for building the sjdb. | Match to genome build (e.g., Ensembl v109 for GRCh38); crucial for sensitivity. |
| STAR Aligner | The software package that performs the spliced alignment of RNA-seq reads. | Use the latest stable version from GitHub [9]. |
| High-Performance Computing (HPC) Node | Executes the computationally intensive genome generation and alignment steps. | Minimum 32 GB RAM for human genome; multiple CPU cores (e.g., 8-16) for speed. |
| RNA-seq Read Files (FASTQ) | The input data containing the sequenced RNA fragments. | Paired-end files (R1, R2); know the read length and quality profile. |
Troubleshooting and Optimization
Common Errors and Solutions
A frequent fatal error occurs when the --sjdbOverhang value specified during the alignment step does not match the value used to build the genome index [52] [27]. The solution is to ensure consistency by either using the same value or omitting the parameter during alignment to allow STAR to use the value stored in the pre-built index.
Advanced Optimization
For users seeking to maximize alignment sensitivity, particularly for novel junctions, consider the interaction between --sjdbOverhang and --seedSearchStartLmax. The latter controls the maximum length of the alignment "seeds" during the mapping step. The developer recommends that --sjdbOverhang should be at least as large as --seedSearchStartLmax - 1 [51]. If you decrease --seedSearchStartLmax below its default of 50 to increase sensitivity for shorter blocks, ensure your --sjdbOverhang is adjusted accordingly. For ultimate accuracy with a single dataset, building an index with the ideal mate_length - 1 value is the gold standard.
When to Use Two-Pass Mode ('--twopassMode Basic') for Sensitivity
The alignment of RNA-seq reads to a reference genome presents a unique computational challenge due to the discontinuous nature of transcriptional data, primarily caused by splicing. For most standard analyses involving well-annotated organisms, single-pass alignment with STAR is sufficient. However, in scenarios prioritizing the discovery and accurate quantification of novel splice junctions, the two-pass alignment method provides a significant enhancement in sensitivity. This application note details the implementation, benefits, and specific use cases for the --twopassMode Basic option within the STAR aligner, providing a structured protocol for researchers pursuing sensitive spliced alignment.
RNA-seq alignment requires mapping sequencing reads across splice junctions, where reads span exon-intron boundaries. Aligners like STAR use a reference genome and often a set of annotated splice junctions (from a GTF file) to guide this process. A inherent limitation of single-pass alignment is its systematic bias against reads that span novel (unannotated) splice junctions. To align a read across a junction, the aligner must find a match for each segment of the read on two different genomic loci. For annotated junctions, this process is facilitated by the existing annotation. For novel junctions, the aligner must perform a more extensive search, often requiring more evidence (i.e., a longer overhang) to consider the alignment valid, which can lead to reduced sensitivity [46].
Two-pass alignment addresses this bias by separating the processes of junction discovery and read quantification. In the first pass, STAR performs a standard alignment with high stringency to identify a comprehensive set of splice junctions, including novel ones, specific to the sample. In the second pass, these newly discovered junctions are incorporated as "annotated" junctions, allowing STAR to use a lower stringency threshold when aligning reads to them. This method effectively levels the playing field, granting novel junctions the same alignment advantages as known ones and thereby increasing the sensitivity of their detection and quantification [9] [46].
Key Applications and Quantitative Benefits of Two-Pass Mode
The decision to use two-pass alignment should be guided by the primary goals of the research. The following table summarizes the primary use cases where --twopassMode Basic is recommended, alongside scenarios where it may be unnecessary.
Table 1: Guidelines for Implementing STAR Two-Pass Alignment
| Recommended For | Not Recommended For | Rationale |
|---|---|---|
| Novel Splice Junction Discovery [47] [46] | Differential Gene Expression (Gene-level) [53] | Maximizes sensitivity for unannotated splicing events; gene-level counts are largely unaffected. |
| Differential Splicing Analysis [54] [55] | Routine alignment in well-annotated genomes where novel splicing is not a focus. | Accurate junction quantification is critical for tools like rMATS and MAJIQ. |
| Variant Discovery from RNA-seq [56] | Studies with severe computational constraints (time/memory). | Improved alignment around junctions aids in variant calling. |
| Transcriptome Assembly [54] | Provides more input data for assemblers like StringTie or Cufflinks. | |
| Studies with limited or poor annotations [53] | Partially compensates for missing annotation. |
Empirical evidence strongly supports the use of two-pass alignment for its intended purposes. An independent analysis across a variety of RNA-seq datasets—including human cancer samples, cell lines, and Arabidopsis thaliana—demonstrated consistent and significant improvements.
Table 2: Quantitative Performance Gains from Two-Pass Alignment Across Diverse RNA-seq Samples [46]
| Sample Description | Read Length | Junctions Improved | Median Read Depth Ratio (2-pass / 1-pass) |
|---|---|---|---|
| Lung Adenocarcinoma Tissue | 48 nt | 99% | 1.68x |
| Lung Normal Tissue | 48 nt | 98% | 1.71x |
| Universal Human Reference RNA (UHRR) | 75 nt | 94% | 1.25x |
| Lung Cancer Cell Line (A549) | 101 nt | 97% | 1.21x |
| Arabidopsis Flower Buds | 101 nt | 97% | 1.12x |
The data shows that two-pass alignment improved the quantification of the vast majority (94-99%) of novel splice junctions. The "Median Read Depth Ratio" indicates a substantial increase in the number of reads aligning to these junctions, with gains of up to 1.7-fold, directly translating to higher sensitivity and more robust statistical power in downstream analyses [46].
The following table lists key materials and software required to implement the two-pass alignment protocol described in this note.
Table 3: Essential Research Reagents and Computational Tools for STAR Two-Pass Alignment
| Item | Function / Explanation | Example / Note |
|---|---|---|
| STAR Aligner | Ultra-fast universal RNA-seq aligner capable of spliced alignment and two-pass mode. | Latest version recommended from GitHub [9]. |
| Reference Genome | A reference genome sequence in FASTA format. | e.g., GRCh38 for human; TAIR10 for A. thaliana. |
| Annotation File | Gene annotation in GTF format for initial genome indexing and first-pass alignment. | e.g., from Ensembl or GENCODE. Not strictly required but highly recommended [9]. |
| High-Performance Computing Node | Computer with sufficient RAM and multiple CPU cores. | RAM: ~30GB for human genome. CPUs: Multiple cores (e.g., 12) for parallel execution [9]. |
| RNA-seq Reads | Input sequencing data in FASTQ format. | Can be single-end or paired-end. Gzip-compressed files are supported. |
Splice Junction File (SJ.out.tab) |
Output from the first pass of STAR, listing all detected splice junctions. | Used as input to build the enhanced genome index for the second pass [47]. |
Experimental Protocols and Implementation
The two-pass alignment workflow with STAR can be executed via two primary methods: (1) re-generating the genome indices using the discovered junctions, or (2) providing the junction file directly during the second mapping step. The first method is described in the protocol below and visualized in the subsequent diagram.
Detailed Two-Pass Protocol with Genome Index Regeneration
Step 1: First-Pass Alignment to Discover Junctions
The initial step is a standard STAR alignment run. Its key purpose is to generate a comprehensive list of splice junctions for the sample, saved in the SJ.out.tab file.
Step 2: Filtering the Discovered Splice Junctions
Before using the SJ.out.tab file, it is good practice to filter out low-quality junctions to reduce potential false positives. A typical filter keeps only canonical junctions (column 5 != 0) supported by a minimum number of uniquely mapping reads (column 7 > 2), and can optionally exclude already annotated junctions (column 6 == 0) [47].
Step 3: Re-generating the Genome Index with New Junctions The filtered splice junctions are fed back into the genome generation step of STAR. This creates a new, sample-aware genome index.
Step 4: Second-Pass Alignment with the Enhanced Index Finally, the original reads are realigned against the new genome index. STAR now treats the sample-specific junctions as known, improving the alignment sensitivity.
The final output, sample1_pass2_Aligned.sortedByCoord.out.bam, is the alignment file with enhanced sensitivity for novel splice junctions, ready for downstream analysis.
Workflow Visualization
The following diagram illustrates the logical flow and data products of the two-pass alignment protocol.
The --twopassMode Basic feature in STAR is a powerful strategy for enhancing the sensitivity of RNA-seq analyses that depend on the comprehensive detection of splicing events. While not required for all studies—such as basic differential gene expression—it has become a recommended standard for sensitive variant discovery [56], differential splicing [55], and de novo transcript assembly [54]. The protocol, which involves a straightforward two-step alignment process, effectively mitigates the inherent bias against novel junctions in single-pass alignment, leading to more accurate and complete molecular profiles. Integrating this method into relevant RNA-seq workflows empowers researchers to more fully exploit the information contained in their sequencing data.
In the analysis of paired-end RNA-seq data using the STAR aligner, the detection of fusion transcripts (or chimeric transcripts) is a critical capability for cancer genomics and studies of genomic rearrangements. A chimeric alignment in STAR occurs when parts of a single read (or read pair) map to two different genomic loci, which may be on different chromosomes, different strands, or are too far apart on the same chromosome to represent a conventional splice junction [57]. The --chimSegmentMin parameter is a fundamental setting that directly controls the sensitivity and specificity of this fusion detection process by defining the minimum length of each mapped segment that can be considered part of a valid chimeric alignment [57]. Understanding and properly configuring this parameter, along with its related settings, is essential for researchers aiming to discover novel gene fusions with clinical and therapeutic significance in oncological research and drug development.
Theoretical Foundation of Chimeric Segmentation
Defining Chimeric Segments
In STAR's algorithmic framework, each chimeric alignment consists of two "segments," where each segment is itself non-chimerically aligned, but the segments are chimeric relative to each other [57]. These segments represent the portions of a read (or read pair) that map to different genomic locations. The --chimSegmentMin parameter controls the minimum mapped length (in bases) that each of these two segments must achieve to be considered a valid part of a chimeric alignment [57]. For example, with 2x75 base pair reads and --chimSegmentMin 20, a chimeric alignment with 130 bases on one chromosome and 20 bases on the other would be output, while a configuration with 135 and 15 bases would not, as the 15-base segment falls below the threshold [57].
Relationship to Other Key Parameters
The --chimSegmentMin parameter functions in concert with several other STAR parameters to define what constitutes a valid chimeric junction:
--chimJunctionOverhangMin: Defines the minimum overhang length for a chimeric junction [58] [59]. While--chimSegmentMincontrols the total segment length,--chimJunctionOverhangMinspecifically addresses the junction-flanking sequences.--chimMainSegmentMultNmax: Controls how many multi-mapping segments are allowed for the main (longer) chimeric segment [59].--chimOutType: Determines the output format for chimeric alignments, with options such asJunctions SeparateSAMold WithinBAM SoftClipbeing commonly used in production pipelines [59].
Table 1: Key Parameters for Chimeric Detection in STAR
| Parameter | Default Value | Function | Recommended Setting for Fusions |
|---|---|---|---|
--chimSegmentMin |
0 (disabled) | Minimum length of each chimeric segment | 15-20 bases |
--chimJunctionOverhangMin |
20 | Minimum overhang for chimeric junctions | 15-20 bases |
--chimMainSegmentMultNmax |
1 | Maximum multi-mapping sites for main segment | 1 |
--chimOutType |
- | Output format for chimeras | Junctions WithinBAM SoftClip |
Configuration Strategies for Different Applications
Fusion Detection in Cancer Research
For identifying somatic gene fusions in tumor RNA-seq data, the --chimSegmentMin parameter should be set to balance sensitivity and specificity. The GDC mRNA Analysis Pipeline, which processes cancer RNA-seq data for projects like The Cancer Genome Atlas (TCGA), utilizes --chimSegmentMin 15 in conjunction with --chimJunctionOverhangMin 15 [59]. This configuration is designed to detect chimeric events with moderate stringency, helping to identify potential driver fusions while reducing false positives. When working with standard 2x101 bp paired-end reads, setting --chimSegmentMin to 15-20 bases typically provides optimal results, as it ensures that each chimeric segment has sufficient length for confident mapping while allowing detection of fusions that may occur near read ends.
Circular RNA Detection
The nf-core/circrna pipeline, designed specifically for circular RNA detection, utilizes --chimSegmentMin 10 as part of its standardized workflow [60]. This lower threshold is appropriate for circular RNA detection because back-splice junctions characteristic of circRNAs may result in shorter chimeric segments. The pipeline combines this with --chimJunctionOverhangMin 10 and specific settings for --alignMatesGapMax 200 to optimize for circular RNA candidates [60]. This example demonstrates how the parameter must be tuned based on the specific biological phenomenon being investigated.
Two-Pass Alignment for Novel Junction Discovery
For comprehensive fusion detection, particularly when seeking novel junctions not present in standard annotations, a two-pass mapping method is recommended [59] [61]. In this approach:
- First Pass: STAR aligns reads with
--twopassMode Basicto detect novel splice junctions, including potential chimeric junctions. - Second Pass: The genome index is regenerated incorporating junctions discovered in the first pass, then all reads are realigned with chimeric detection enabled using appropriate
--chimSegmentMinvalues.
This method significantly improves sensitivity for detecting previously unannotated fusion events by providing STAR with a more complete junction database before the final alignment.
Experimental Protocol for Fusion Detection
Sample Preparation and Sequencing Considerations
For optimal fusion detection using STAR with --chimSegmentMin:
- Utilize paired-end sequencing with read lengths of at least 2x75 bp, though 2x101 bp or longer is preferred
- Ensure stranded RNA-seq library preparation to resolve ambiguous alignments involving overlapping genes
- Sequence to sufficient depth (>50 million read pairs for tumor samples) to detect low-frequency fusion events
- Include positive control samples with known fusion events when possible to validate parameter settings
STAR Alignment with Chimeric Detection
The following protocol details the complete procedure for fusion detection in human RNA-seq data:
Step 1: Genome Index Generation
Step 2: Two-Pass Alignment with Chimeric Detection
Table 2: Critical Parameter Settings for Fusion Detection
| Parameter Category | Parameter | Setting | Rationale |
|---|---|---|---|
| Chimeric Detection | --chimSegmentMin |
15 | Ensures segments have minimal length for confident mapping |
| Chimeric Detection | --chimJunctionOverhangMin |
15 | Balanced sensitivity for junction overhangs |
| Chimeric Detection | --chimOutType |
Junctions WithinBAM SoftClip | Multiple output formats for downstream analysis |
| Alignment Sensitivity | --alignIntronMax |
1000000 | Allows detection of fusions with large genomic distances |
| Alignment Sensitivity | --outFilterMultimapNmax |
20 | Controls for highly repetitive regions |
| Two-Pass Mode | --twopassMode |
Basic | Enhances novel junction discovery |
Output Interpretation and Validation
STAR generates several output files relevant to chimeric detection:
Chimeric.out.junction: Tab-delimited file detailing each chimeric junction with genomic coordinates, junction type, and supporting read information [57]Chimeric.out.sam(or within the main BAM withWithinBAMoption): SAM-formatted alignments of chimeric readsSJ.out.tab: File containing all detected splice junctions, including chimeric ones
To filter high-confidence chimeric junctions from the output:
This filtering removes junctions involving the mitochondrial genome, non-canonical splice sites, and those with excessive repeat overlap, helping to prioritize likely biologically relevant fusions [57].
Visualization of Chimeric Alignment Detection
The following diagram illustrates how the --chimSegmentMin parameter functions within STAR's chimeric detection workflow:
Chimeric Read Processing with chimSegmentMin Filter - This workflow illustrates how STAR processes potential chimeric alignments and applies the --chimSegmentMin threshold to determine valid fusion events.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Fusion Detection
| Reagent/Resource | Function | Example/Source |
|---|---|---|
| STAR Aligner | Spliced transcript alignment with fusion detection | GitHub Repository [58] |
| Reference Genome | Genomic coordinate system for alignment | GRCh38 from GENCODE [59] |
| Gene Annotations | Splice junction database for alignment guidance | GENCODE v36 [59] |
| Positive Control Samples | Validation of fusion detection sensitivity | Cell lines with known fusions (e.g., K562 with BCR-ABL) [57] |
| STAR-Fusion | Downstream analysis of chimeric outputs | Broad Institute tool for fusion interpretation [58] |
| nf-core/circrna | Specialized pipeline for circular RNA analysis | nf-core workflow [60] |
Troubleshooting and Optimization Guidelines
Parameter Optimization Strategies
When optimizing --chimSegmentMin for specific research contexts:
- For high-sensitivity detection in clinical screening: Decrease to 10-12 bases, but expect higher false positive rates
- For high-specificity requirements in validation studies: Increase to 20-25 bases to ensure only high-confidence fusions
- For long-read sequencing technologies: Adjust proportionally to read length (e.g., 5-10% of total read length)
- Always use matched values for
--chimSegmentMinand--chimJunctionOverhangMin(e.g., both 15) unless specific biological hypotheses justify asymmetry
Common Issues and Solutions
- No chimeric output: Ensure
--chimSegmentMinis set to a positive value and--chimOutTypeis appropriately specified [57] [61] - Excessive false positives: Increase
--chimSegmentMinand implement more stringent post-filtering based on junction type and repeat overlap [57] - Memory issues during alignment: Use
--genomeLoad NoSharedMemoryand ensure sufficient RAM is available [59] - Low sensitivity for novel fusions: Implement two-pass mode and consider increasing
--alignIntronMaxto detect fusions with large genomic distances [59]
The --chimSegmentMin parameter represents a critical control point in the STAR RNA-seq alignment workflow for fusion transcript detection. When configured appropriately for the specific biological question and sequencing technology, this parameter enables researchers to balance sensitivity and specificity in chimeric alignment identification. The recommended value of 15 bases, used in conjunction with related parameters like --chimJunctionOverhangMin within a two-pass alignment framework, provides a robust foundation for fusion detection in cancer research and drug development applications. As sequencing technologies evolve and clinical applications expand, continued optimization of these key parameters will further enhance our ability to detect biologically and therapeutically relevant gene fusions across diverse research contexts.
Troubleshooting Low Mapping Rates and High Unmapped Reads
In paired-end RNA-seq experiments, a high proportion of unmapped reads can compromise downstream analyses and lead to inaccurate biological interpretations. The STAR (Spliced Transcripts Alignment to a Reference) aligner, while highly accurate and efficient, is particularly sensitive to specific experimental and computational parameters. This protocol synthesizes a systematic approach to diagnose and resolve the prevalent issue of low mapping rates, drawing from empirical evidence and established bioinformatics practices. The guidance is framed within a broader research context on optimizing RNA-seq alignment protocols for reliable transcriptomic analysis in drug development and biomedical research.
Initial Diagnosis and Root Cause Analysis
A structured initial assessment is critical for efficiently identifying the source of poor mapping performance. The following workflow provides a logical sequence for diagnosis.
Verify Reference Genome and Annotation Compatibility
Ensure the reference genome and annotation files are appropriate for your data. Mismatched or incomplete references are a primary cause of low mapping rates [62]. For total RNA-seq, using a genome that includes all scaffolds and non-chromosomal sequences is essential, as ribosomal RNA (rRNA) genes are often not placed on main chromosomes [63]. Confirm that the species and genome assembly version of your reference match your samples. For non-model organisms, high sample divergence from the reference genome can also cause mapping failures [64].
Assess Sequence Quality and Adapter Content
Systematically evaluate raw sequencing data. Use FastQC to visualize quality scores and detect overrepresented sequences, which can indicate adapter contamination or other issues [65]. Perform adapter trimming with tools like CutAdapt or Trimmomatic. While one user found that trimming did not significantly improve their overall mapping stats, it remains a critical step to remove technical sequences that prevent proper alignment [64]. Also, check for an abundance of short fragments, which can result from RNA degradation and lead to reads being classified as "unmapped: too short" [63].
Test for Library Contamination
rRNA contamination due to inefficient depletion during library preparation is a very common cause of low unique mapping rates [66] [65]. A high fraction of reads originating from multi-copy genes like rRNAs will either map to multiple loci and be filtered out or fail to map if the rRNA sequences are absent from the reference genome.
- rRNA BLAST Search: Extract unmapped and multi-mapped reads, convert them to FASTA format, and use BLAST to search against a database of rRNA sequences from a closely related species [65].
- Use Dedicated Tools: Tools like SortMeRNA can directly filter rRNA sequences from your FASTQ files [65].
- Check for Other Contaminants: Consider using Kraken to screen for bacterial or viral contamination, especially for samples derived from environmental or non-sterile sources [66].
Key Experimental Protocols for Troubleshooting
Protocol: BLAST Analysis of Unmapped Reads
This protocol helps identify the biological source of reads that failed to align [64] [65].
- Extract Unmapped Reads: Use SAMtools to extract unmapped reads from the BAM file.
- Convert to FASTA: Use bedtools to convert the BAM file to FASTA format.
- Run BLAST Search: BLAST the FASTA file against a custom database (e.g., rRNA sequences from NCBI) or the non-redundant nucleotide (nt) database remotely.
Protocol: Genome Index Generation with Optimized Parameters
Proper genome indexing is foundational for successful alignment. This protocol outlines key parameters, especially --sjdbOverhang.
Critical Parameter:
--sjdbOverhang: This specifies the length of the genomic sequence around annotated junctions. The ideal value is read length - 1 [3] [1]. For example, for 150bp paired-end reads, use--sjdbOverhang 149. The default is 100, which works well in most cases but optimizing it can improve junction detection [3].
Protocol: Two-Pass Alignment for Novel Junction Detection
For experiments focused on discovering novel splice variants, a two-pass method is recommended [1].
- First Pass Mapping: Perform a standard alignment. This run will generate a file of novel splice junctions (
SJ.out.tab). - Incorporate New Junctions: Use the junctions discovered in the first pass to create a new genome index, or include them directly in the second mapping step.
- Second Pass Mapping: Re-map all reads using the updated genome index or by feeding the
SJ.out.tabfile from the first pass to achieve more comprehensive alignment of spliced reads [1].
Parameter Optimization and The Scientist's Toolkit
Key Research Reagent Solutions
The following table details essential computational "reagents" required for effective troubleshooting.
| Research Reagent | Function & Purpose | Specification Notes |
|---|---|---|
| Reference Genome (FASTA) | Spliced alignment scaffold; provides genomic context for mapping. | Must include all scaffolds and contigs; verify species/assembly match. |
| Gene Annotation (GTF/GFF3) | Defines known exon-intron structures; critical for splice-aware alignment. | Ensure compatibility with the FASTA genome version. |
| rRNA Sequence Database | Reference for identifying ribosomal RNA contamination in unmapped reads. | Compile from sources like NCBI or Ensembl for target species. |
| Adapter Sequence File | Defines oligonucleotide sequences for trimming during pre-processing. | Must match the specific library preparation kit used (e.g., Illumina). |
| STAR Genome Index | Pre-computed data structure for ultra-fast sequence search and alignment. | Must be built with the corresponding FASTA and GTF files. |
Optimization of STAR Alignment Parameters
Empirical testing has shown that adjusting specific STAR parameters can significantly recover unmapped reads. The table below summarizes critical parameters and their effects based on case studies [64].
| Parameter | Default Value | Tested/Optimized Value | Effect on Mapping |
|---|---|---|---|
--outFilterScoreMinOverLread |
0.66 | 0 | Relaxes overall alignment score threshold. |
--outFilterMatchNminOverLread |
0.66 | 0 | Relaxes the threshold for the number of matched bases relative to read length. |
--outFilterMatchNmin |
0 | 20 or 30 | Sets a minimum absolute number of matched bases [64]. |
--alignIntronMin |
21 | 10 | Allows detection of shorter introns. |
--alignIntronMax |
0 | 100000 | Reduces the maximum intron size from default (can help in smaller genomes). |
--outFilterMultimapNmax |
10 | 50 or higher | Increases the number of allowed multi-mapping locations, useful for gene families and rRNAs [63]. |
--seedSearchStartLmax |
50 | 12-15 | Can improve sensitivity for finding alignment seeds in shorter reads. |
Trade-off Consideration: Relaxing parameters like --outFilterMatchNmin can increase the percentage of uniquely mapped reads but often comes with a trade-off, such as a higher mismatch rate per base and a greater proportion of reads mapped to multiple loci [64]. It is crucial to find a balance that maximizes informative alignments without introducing excessive noise.
Resolving low mapping rates in STAR RNA-seq alignment requires an integrated troubleshooting strategy that spans the entire workflow, from wet-lab library preparation to computational parameter optimization. The most effective approach involves systematically verifying the reference genome and annotation, rigorously checking for and removing contaminants like rRNA, and fine-tuning key alignment parameters with a clear understanding of the associated trade-offs. By implementing the diagnostic workflows and experimental protocols detailed in this document, researchers can significantly improve alignment efficiency and data quality, thereby ensuring the reliability of their downstream transcriptomic analyses in drug development and biomedical research.
Within the framework of a comprehensive thesis on paired-end RNA-seq alignment, rigorous quality control (QC) is a critical step to ensure the reliability of downstream analyses. The STAR (Spliced Transcripts Alignment to a Reference) aligner is a widely used, ultrafast tool designed to address the unique challenges of RNA-seq data mapping, such as the presence of splice junctions [2]. Upon completing a mapping run, STAR generates a Log.final.out file, which provides a crucial summary of the alignment performance. This application note details the methodology for interpreting this log file, transforming its raw metrics into actionable insights for researchers, scientists, and drug development professionals engaged in transcriptomic studies.
The STAR Aligner and Its Final Log Output
STAR's algorithm operates through a two-step process: a seed search for maximal mappable prefixes and a subsequent clustering, stitching, and scoring phase to generate full alignments [67] [2]. This design allows it to efficiently handle spliced reads. The alignment process culminates in the Log.final.out file, a consolidated report distinct from the running logs (Log.out and Log.progress.out). This final log is the primary resource for a high-level overview of the mapping outcomes for a given sample [68].
Table 1: Key Inputs for the STAR Alignment Workflow
| Item | Function in the Protocol |
|---|---|
| Reference Genome (FASTA) | The genomic sequence against which reads are aligned. Provides the coordinate system [69] [67]. |
| Gene Annotation (GTF) | A file containing genomic coordinates of known genes and transcripts. Used during index generation to improve mapping accuracy, especially for splice junctions [69] [67]. |
| STAR Genome Index | A pre-built index of the reference genome and annotations, enabling rapid alignment. Must be generated before the read alignment step [68] [69]. |
| RNA-seq Reads (FASTQ) | The input sequencing data. For paired-end analysis, both read pair files (R1 and R2) must be provided [67]. |
The following workflow outlines the key stages of a paired-end RNA-seq analysis using STAR, highlighting the generation and role of the Log.final.out file in the quality control process.
A Section-by-Section Guide to 'Log.final.out'
The Log.final.out file is structured into clear sections. The most critical metrics for a primary quality assessment are found in the "Mapping statistics" table. It is essential to remember that STAR, like other mappers, estimates the most likely genomic position for each read but does not know its true origin; therefore, these metrics are estimates of mapping performance rather than absolute measures of precision and recall [70].
Table 2: Key Metrics in the 'Log.final.out' File for Quality Control
| Metric Category | Specific Metric | Interpretation and Benchmark |
|---|---|---|
| Read Input | Number of input reads |
Total reads processed from the FASTQ file(s). Verify it matches expectations. |
| Uniquely Mapped Reads | Uniquely mapped reads % |
A primary QC indicator. The percentage of reads mapped to a single genomic location. High rates (e.g., >70-80% for human) are typically desirable [68] [70]. |
| Multi-Mapped Reads | % of reads mapped to multiple loci |
Reads aligned to several locations. High percentages may indicate repetitive sequences. |
| Unmapped Reads | % of reads unmapped: too short |
Reads trimmed short, potentially due to adapters or poor quality. |
% of reads unmapped: other |
Reads failing to map for other reasons. Investigate if this value is high. | |
| Splicing Events | Number of splices: Total |
Total number of splice junctions detected in all aligned reads. |
Number of splices: Annotated (sjdb) |
Splice junctions that match the provided annotation (GTF file). | |
Number of splices: GT/AG |
The number of canonical splice sites. Expect this to be the dominant category. | |
| Mismatch and Deletion Rates | Mismatch rate per base, % |
Frequency of base mismatches in the alignment. |
Deletion rate per base / Insertion rate per base |
Frequency of small insertions or deletions. |
Integrating Log File Interpretation into a Robust QC Protocol
Interpreting the Log.final.out should not be done in isolation. A robust QC protocol integrates these metrics with other tools and checks to form a complete picture of data quality.
Cross-Validation with Other STAR Outputs: The
Log.final.outprovides a summary, but other STAR outputs offer deeper insights. TheSJ.out.tabfile contains all high-confidence splice junctions, which can be used to verify novel splice discovery or check for non-canonical splices [68] [69]. Visualizing the sorted BAM file in a genome browser like IGV allows for manual inspection of read alignments in specific genomic regions, providing a qualitative check against the quantitative metrics [70].Tool-Based Quality Assessment: For a more automated and comprehensive assessment of the alignment, use dedicated QC tools. For instance, Qualimap can compute various quality metrics from the BAM file, such as coverage biases, DNA or rRNA contamination, and 5'-3' biases, which are not directly available in the STAR log [68]. This step is highly recommended for a thorough QC.
Troubleshooting Common Issues: The log file is the first place to look when an alignment underperforms.
- Low Uniquely Mapped Read Percentage: This can be caused by high levels of ribosomal RNA, contamination, or low RNA quality. Check the
% of reads unmapped: otherand consider using tools like FastQC on the raw reads to assess initial quality. - Unexpected Splice Junction Counts: A low number of annotated splices might suggest that an inappropriate or outdated gene annotation file was used during the genome indexing step [69]. Ensure the GTF file is from a reputable source and matches the genome build.
- Low Uniquely Mapped Read Percentage: This can be caused by high levels of ribosomal RNA, contamination, or low RNA quality. Check the
For researchers relying on RNA-seq data, from basic science to drug development, the STAR Log.final.out file is an indispensable gateway to assessing data quality. By systematically interpreting its metrics within a broader analytical framework—which includes using the correct research reagents, following a structured protocol, and leveraging complementary tools like Qualimap—scientists can confidently evaluate their alignment data. This rigorous approach ensures that the foundational data for all subsequent differential expression and transcriptome analysis is sound, ultimately supporting the generation of reliable and biologically meaningful conclusions.
Ensuring Accuracy: Validating Results and Comparing Methodological Choices
Within the context of paired-end RNA-seq alignment protocol research, selecting an optimal aligner is a foundational decision that profoundly impacts the quality of all subsequent biological interpretations. The Spliced Transcripts Alignment to a Reference (STAR) aligner has emerged as a prominently used tool, renowned for its high-speed performance and precision. This application note provides a systematic evaluation of STAR against other common aligners, drawing upon recent benchmarking studies to quantify performance across critical metrics such as alignment accuracy, splice junction detection, and computational efficiency. The data and protocols summarized herein are intended to guide researchers, scientists, and drug development professionals in making informed decisions for their transcriptomics pipelines, particularly when working with paired-end reads.
Performance Benchmarking and Quantitative Comparison
Base-Level and Junction-Level Alignment Accuracy
Recent independent benchmarking studies provide quantitative evidence of STAR's performance. A 2024 study using simulated Arabidopsis thaliana data assessed alignment accuracy at both the base level and the junction level. At the base-level assessment, STAR demonstrated superior performance, achieving over 90% overall accuracy under varying test conditions. However, at the more challenging junction base-level assessment, which evaluates the accurate mapping of reads across splice junctions, SubRead emerged as the most accurate tool, with over 80% accuracy under most conditions [71]. This indicates that while STAR excels overall, the choice of aligner for studies heavily focused on alternative splicing may warrant specific consideration.
Performance in Clinical and Complex Sample Types
The performance of an aligner can be significantly affected by sample quality, as is often the case with clinically derived specimens. A 2019 study comparing HISAT2 and STAR using RNA-seq data from Formalin-Fixed Paraffin-Embedded (FFPE) breast cancer samples identified notable differences. The study reported that STAR generated more precise alignments, a critical advantage for the analysis of early neoplasia samples. In contrast, HISAT2 was more prone to misaligning reads to retrogene genomic loci [72]. This finding is particularly relevant for precision medicine and clinical research, where FFPE samples are a primary resource, and alignment precision is paramount for accurate biomarker discovery.
Alignment Runtime and Computational Efficiency
Computational efficiency, including runtime and memory usage, is a practical concern for many research groups. A 2021 comparison of several aligners using an RNA-seq dataset from Erysiphe necator highlighted that HISAT2 was approximately three times faster than the next fastest aligner in terms of runtime [73]. While STAR is engineered for high-speed mapping through its efficient algorithm, its runtime is generally longer than that of HISAT2. It is important to note that STAR's design prioritizes a balance between speed and alignment sensitivity, especially for spliced alignments, but it is also more memory-intensive compared to FM-index-based aligners [3] [2] [73].
Table 1: Summary of Key Performance Metrics for RNA-seq Aligners
| Aligner | Base-Level Accuracy | Junction-Level Accuracy | Alignment Precision (FFPE) | Relative Runtime | Primary Strength |
|---|---|---|---|---|---|
| STAR | >90% [71] | Varies [71] | High [72] | Medium [73] | Balanced speed and sensitivity |
| HISAT2 | Not Specified | Not Specified | Lower (retrogene misalignment) [72] | Fastest [73] | Computational speed |
| SubRead | Not Specified | >80% [71] | Not Specified | Not Specified | Junction accuracy |
| BWA | Not Specified | Not Specified | Not Specified | Not Specified | General performance on long transcripts [73] |
Experimental Protocols for Benchmarking Aligners
To ensure the reproducibility and reliability of aligner comparisons, a standardized experimental workflow is essential. The following protocol, synthesized from the cited literature, outlines a robust methodology for benchmarking RNA-seq aligners.
Sample Preparation and Sequencing
- Sample Type: The use of paired-end sequencing is strongly recommended. As confirmed in a community discussion, longer paired-end reads (e.g., 150bp) provide greater specificity and alignment accuracy compared to shorter or single-end reads [74].
- Spike-in Controls: For absolute quantification and assessment of technical performance, include spike-in RNAs with known concentrations, such as Sequins, ERCC, or SIRV mixes [75].
- Biological and Technical Replicates: Incorporate multiple replicates to account for biological and technical variability. For instance, the SG-NEx project sequenced each cell line with at least three high-quality replicates [75].
Computational Workflow for Alignment Comparison
The core analysis involves processing the raw sequencing data through different aligners and comparing their outputs against a ground truth or reference standard.
Diagram 1: Experimental Workflow for Benchmarking RNA-seq Aligners
Protocol Steps:
Data Input and Quality Control (QC):
- Input: Raw paired-end sequencing reads in FASTQ format.
- QC and Trimming: Use tools like
Trimmomatic,Cutadapt, orBBDukto remove adapter sequences and low-quality bases. Parameters typically include a Phred quality score threshold of >20 and a minimum read length of 50bp after trimming [76].
Genome Indexing:
- Obtain the reference genome sequence (e.g., GRCh38 for human) and the corresponding annotation file (GTF format).
- Each aligner requires building a specific genome index.
- Example STAR Indexing Command:
The
--sjdbOverhangparameter should be set to the read length minus 1 [3].
Read Alignment:
- Execute each aligner (STAR, HISAT2, etc.) on the trimmed reads using identical computational resources (core count, memory) for a fair comparison.
- Example STAR Alignment Command: This command produces a coordinate-sorted BAM file ready for downstream analysis [3].
Metric Calculation and Accuracy Assessment:
- Alignment Rate: The percentage of input reads successfully mapped to the reference.
- Junction Detection: The number of annotated splice junctions detected and the accuracy of their boundaries.
- Comparison to Ground Truth: When using simulated data (e.g., generated with
Polyester[71] [77] orRSEM[77]), accuracy can be directly measured by comparing alignments to known read origins using metrics like Mean Absolute Relative Difference (MARD) and Pearson correlation [77]. For experimental data, consistency with spike-in controls and qRT-PCR validation can serve as benchmarks [76].
Successful execution of an RNA-seq alignment study requires a suite of computational tools and genomic resources. The following table details key components used in the benchmark experiments cited in this note.
Table 2: Key Research Reagent Solutions for RNA-seq Alignment Analysis
| Item Name | Function / Purpose | Specifications / Notes |
|---|---|---|
| Reference Genome | Linear sequence to which reads are aligned. | Use a version-matched genome FASTA and GTF annotation file (e.g., from Gencode or ENSEMBL). Critical for accurate splice-aware alignment [3] [77]. |
| Spike-in Control RNAs | Internal controls for technical performance assessment. | Synthetic RNAs (e.g., ERCC, Sequin, SIRV) spiked into samples at known concentrations to evaluate quantification accuracy and sensitivity [75]. |
| STAR Aligner | Spliced alignment of RNA-seq reads to a reference genome. | Known for high speed and precision. Requires significant RAM (e.g., 32GB for mammalian genomes). Best for alignment-sensitive applications [72] [2] [14]. |
| HISAT2 Aligner | Highly efficient spliced read alignment. | Uses a memory-efficient FM-index. Often faster than STAR, making it suitable for resource-constrained environments [72] [73]. |
| Quality Control Tools | Assess raw read quality and perform adapter trimming. | Tools like FastQC, Trimmomatic, and Cutadapt are used to preprocess data, which improves overall mapping rates [76]. |
| Simulation Software | Generates RNA-seq data with a known ground truth. | Tools like Polyester and RSEM simulate reads from a transcriptome, enabling precise calculation of alignment accuracy [71] [77]. |
| Quantification Tools | Assign reads to genes/transcripts and estimate abundance. | Tools like featureCounts, Salmon, or Kallisto use the aligned BAM files (or reads directly) to generate count or TPM tables for differential expression analysis [72] [77]. |
Visualization of STAR's Algorithmic Advantage
STAR's performance stems from its unique two-step alignment algorithm, which is fundamentally different from the FM-index-based strategies used by many other aligners.
Diagram 2: STAR's Two-Step Alignment Algorithm
Algorithm Explanation:
Step 1: Seed Searching STAR uses an uncompressed suffix array (SA) to rapidly find the longest possible exact matches between the read and the reference genome, known as Maximal Mappable Prefixes (MMPs). It starts from the beginning of the read, finds the first MMP, then repeats the search on the unmapped portion of the read. This sequential search is highly efficient and allows STAR to detect splice junctions de novo without prior annotation [2].
Step 2: Clustering, Stitching, and Scoring In the second step, the separately mapped "seeds" (MMPs) are clustered together based on their proximity in the genome. A clustering and stitching algorithm then connects these seeds, allowing for a single genomic gap (an intron) between them. The final stitched alignment is scored based on the number of mismatches, insertions, and deletions [2]. This two-step process is key to STAR's high sensitivity in identifying spliced alignments.
The systematic comparisons outlined in this note demonstrate that STAR is a robust and high-performing choice for paired-end RNA-seq alignment, particularly in contexts demanding high precision and sensitive junction detection. Its performance in analyzing complex and clinically relevant FFPE samples makes it especially suitable for biomedical and drug development research. However, the benchmarks also show that no single aligner is universally superior across all metrics. For projects where computational speed is the primary constraint and the organism has a well-annotated transcriptome, HISAT2 presents a compelling alternative. Ultimately, the choice of aligner should be guided by the specific biological questions, sample types, and computational resources available to the researcher.
Validating with Spike-In RNAs and qRT-PCR for Expression Correlation
Within the framework of paired-end RNA-seq alignment research using the STAR protocol, the independent verification of expression findings is a critical step to ensure data reliability. While RNA-seq is a powerful tool for transcriptome-wide discovery, technical variations can influence results [78]. This application note details a complementary validation approach using spike-in RNAs and quantitative RT-PCR (qRT-PCR). Spike-in controls serve as external benchmarks to monitor technical performance across samples and sequencing runs, while qRT-PCR provides a highly sensitive and specific orthogonal method to confirm the expression levels of key genes identified by RNA-seq [79] [78]. This document provides detailed protocols for integrating these validation techniques into a STAR-based RNA-seq workflow, ensuring robust and reproducible gene expression data for critical decision-making in research and drug development.
Background and Principles
The transition from a discovery-based tool like RNA-seq to a validated, reliable dataset requires understanding the principles of analytical validation. RNA-seq data, especially when derived from a precise aligner like STAR (Spliced Transcripts Alignment to a Reference), provides a comprehensive view of the transcriptome [2] [80]. However, its accuracy must be evaluated against key performance metrics.
STAR utilizes a novel RNA-seq alignment algorithm that performs sequential maximum mappable seed search in uncompressed suffix arrays, followed by seed clustering and stitching. This design allows for fast and accurate alignment, including the detection of canonical and non-canonical splice junctions, which is crucial for comprehensive transcriptome analysis [2].
For validation, the "fit-for-purpose" concept is key, where the level of assay validation is sufficient to support its specific context of use [79]. The core performance characteristics for any quantitative method like qRT-PCR include:
- Analytical accuracy/trueness: Closeness of measured value to the true value.
- Analytical precision: Closeness of repeated measurements to each other.
- Analytical sensitivity: The minimum detectable concentration of an analyte.
- Analytical specificity: The ability to distinguish the target from non-target analytes [79].
Spike-in RNAs, which are exogenous synthetic RNA sequences added to samples in known quantities, provide a means to track these metrics throughout the entire workflow, from library preparation to sequencing and alignment, thereby controlling for technical noise [78].
The Relationship Between RNA-seq and qRT-PCR
While RNA-seq and qRT-PCR are both used for gene expression analysis, they serve different primary purposes in the research pipeline. RNA-seq is a hypothesis-generating tool ideal for unbiased, genome-wide discovery of differentially expressed genes and novel transcripts [81] [78]. In contrast, qRT-PCR is a hypothesis-testing tool, offering high sensitivity, specificity, and reproducibility for validating the expression of a limited number of pre-selected targets [78] [82].
Studies have shown a generally high correlation between results obtained from RNA-seq and qRT-PCR. A comprehensive analysis by Everaert et al. (as cited in [78]) revealed that depending on the analysis workflow, 15-20% of genes might be 'non-concordant,' with the vast majority of these showing low fold changes (<2) or very low expression levels. This underscores that for genes with strong, biologically relevant expression changes, RNA-seq is highly reliable. Therefore, qRT-PCR validation is most critical when a research conclusion hinges on the expression of a small number of genes, particularly if those genes are lowly expressed or show modest fold-changes [78].
Workflow Integration and Experimental Design
The successful integration of spike-ins and qRT-PCR validation into an RNA-seq workflow requires careful planning. The following diagram illustrates the complete experimental journey from sample preparation to validated results.
Critical Considerations for Experimental Design
To ensure meaningful validation, the experimental design must minimize variability and bias.
- Minimize Batch Effects: Technical batch effects are a major source of non-biological variation. All steps, including RNA isolation, library preparation, and sequencing runs for compared samples, should be performed simultaneously or in a randomized and balanced manner [81].
- Biological Replication: The study must be powered with an adequate number of biological replicates (e.g., cells or tissues from different individuals) rather than technical replicates. This is essential for capturing biological variance and achieving statistical robustness in downstream differential expression analysis [81] [78].
- RNA Quality: Use high-quality RNA with an RNA Integrity Number (RIN) > 7.0 to ensure reliable results for both RNA-seq and downstream qRT-PCR [81].
- Selection of Genes for qRT-PCR Validation: Choose genes based on RNA-seq results that are critical to the biological story. This includes genes with high statistical significance, large fold-changes, and those relevant to the pathways of interest. It is also prudent to include genes with varying expression levels and fold-changes to broadly confirm the RNA-seq data [78].
Protocols
Protocol 1: Using Spike-In RNA Controls in RNA-seq
Spike-in controls, such as the External RNA Controls Consortium (ERCC) mixes, are used to monitor technical performance.
I. Materials and Reagents
- Spike-in RNA Mix: A commercially available set of synthetic RNA transcripts at known, varying concentrations (e.g., ERCC Spike-In Mix).
- Nuclease-free Water.
- RNA Isolation Kits.
II. Procedure
- Spike-In Dilution: Prepare a working dilution of the spike-in mix according to the manufacturer's instructions. The dilution factor should be calculated to result in an appropriate concentration relative to your total RNA input.
- Addition to Sample: Add a fixed volume of the diluted spike-in mix to each cell lysate or purified total RNA sample at the very beginning of the RNA isolation procedure. Adding at the lysis stage ensures the spike-ins control for the entire RNA extraction and library preparation process.
- RNA-seq Library Preparation: Proceed with standard RNA-seq library preparation protocols, including poly-A selection or rRNA depletion. The spike-in RNAs will be co-processed with your endogenous RNA.
- Sequencing and Alignment: Sequence the libraries and align the reads using STAR. The spike-in sequences must be included in the reference genome/transcriptome index used for alignment.
- Data Analysis: Quantify the reads mapped to each spike-in transcript. The measured abundance should correlate with the known input amount. Deviations from the expected correlation can indicate issues with library complexity, amplification bias, or sequencing saturation.
Protocol 2: qRT-PCR Validation of RNA-seq Results
This protocol describes the steps for validating RNA-seq findings using qRT-PCR, including the critical process of reference gene selection.
I. Materials and Reagents
- RNA samples: The same biological replicates used for RNA-seq.
- Reverse Transcription Kit: Includes reverse transcriptase, buffers, and random hexamers/oligo-dT primers.
- qPCR Master Mix: Contains DNA polymerase, dNTPs, MgClâ‚‚, and reaction buffers.
- Primer Pairs: Validated, gene-specific primers for target and reference genes.
- Multi-well Plates and Sealing Films.
- Real-time PCR Instrument.
II. Procedure
Step 1: cDNA Synthesis
- For each RNA sample, synthesize first-strand cDNA using a reverse transcription kit.
- Use a fixed amount of total RNA (e.g., 500 ng - 1 µg) per reaction to maintain consistency.
- Include a no-reverse transcriptase (-RT) control for each sample to check for genomic DNA contamination.
Step 2: Selection of Reference Genes
- The selection of stable reference genes is critical for accurate qRT-PCR normalization. Traditional housekeeping genes (e.g., ACTB, GAPDH) can be variable under different experimental conditions [82].
- Software-assisted selection: Use tools like Gene Selector for Validation (GSV) to identify the most stable reference genes directly from your RNA-seq dataset [82]. GSV applies filters to select genes with high and stable expression (e.g., TPM > 0 in all samples, low coefficient of variation < 0.2) across your specific biological conditions.
- Validate the stability of at least two reference genes for your experiment.
Step 3: qPCR Reaction Setup and Cycling
- Design qPCR reactions in triplicate for each sample, target gene, and reference gene.
- A typical 20 µL reaction might contain: 10 µL of 2x qPCR Master Mix, 1 µL of forward and reverse primer mix, 2 µL of cDNA template, and 7 µL of nuclease-free water.
- Run the plate on the real-time PCR instrument using a standard cycling protocol (e.g., 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min).
Step 4: Data Analysis
- Calculate the average Cq (quantification cycle) for each replicate.
- Normalize the Cq of the target genes to the geometric mean of the Cq values from the validated reference genes (∆Cq).
- Calculate the relative expression levels (e.g., using the 2^–∆∆Cq method) between experimental groups.
- Correlate the fold-change values obtained from qRT-PCR with those from the STAR-aligned RNA-seq data. A strong positive correlation (e.g., R² > 0.8) validates the RNA-seq findings.
Data Presentation and Analysis
Presenting quantitative data in a clear, structured format is essential for effective communication. The following tables summarize key performance characteristics and reagent solutions used in the validation workflow.
Table 1: Key Performance Characteristics for qRT-PCR Assay Validation [79]
| Performance Characteristic | Definition | Acceptance Criterion Example |
|---|---|---|
| Analytical Accuracy | Closeness of measured value to the true value. | Slope of standard curve: 100% ± 10% |
| Analytical Precision | Closeness of repeated measurements (Repeatability & Reproducibility). | Intra-assay CV < 5%; Inter-assay CV < 10% |
| Analytical Sensitivity (LOD) | The minimum detectable concentration of the analyte. | Cq value < 35 in dilution series |
| Analytical Specificity | Ability to distinguish the target from non-target sequences. | No amplification in NTC and -RT controls |
| Dynamic Range | The range of concentrations over which the assay is accurate and precise. | 5-6 orders of magnitude |
Table 2: Research Reagent Solutions for Validation Workflows
| Reagent / Solution | Function / Application | Key Considerations |
|---|---|---|
| Spike-In RNA Controls (e.g., ERCC) | Exogenous RNA standards added to samples to monitor technical variance throughout RNA-seq workflow. | Add at lysis stage; use a mix covering a wide dynamic range; include in STAR reference index [78]. |
| STAR Aligner | Spliced Transcripts Alignment to a Reference; aligns RNA-seq reads to the genome, detecting splice junctions and chimeric transcripts. | High speed and sensitivity; requires a reference genome and gene annotation file (GTF) [2] [12]. |
| Reference Genes | Endogenous genes used for normalization in qRT-PCR to correct for sample-to-sample variation. | Stability must be validated for each experimental condition; tools like GSV can select candidates from RNA-seq data [82]. |
| qPCR Master Mix | Optimized buffer containing polymerase, dNTPs, and salts for efficient and specific cDNA amplification. | Choose mixes with high fidelity and specific fluorophores (e.g., SYBR Green or probe-based) [79]. |
Troubleshooting and Common Pitfalls
Even with a robust protocol, challenges can arise. The table below outlines common issues and recommended solutions.
Table 3: Troubleshooting Guide for Validation Experiments
| Problem | Potential Cause | Solution |
|---|---|---|
| Poor correlation between RNA-seq and qRT-PCR fold-changes. | 1. Unstable reference genes used for qRT-PCR.2. Saturation of RNA-seq counts for highly expressed genes.3. Lowly expressed genes with high variability. | 1. Re-evaluate reference gene stability using software like GeNorm or NormFinder [82].2. Inspect read distribution; consider using a quantification method less prone to saturation.3. Focus validation on genes with moderate to high expression and significant p-values [78]. |
| High variability in spike-in recoveries. | 1. Inconsistent addition of spike-in mix across samples.2. Degradation of the spike-in RNA stock. | 1. Create a master mix of spike-ins for all samples and use a calibrated pipette for dispensing.2. Aliquot spike-in stock and avoid multiple freeze-thaw cycles. |
| Low sequencing library complexity in RNA-seq. | 1. Insufficient RNA input.2. Over-amplification during PCR. | 1. Use a higher RNA input or a library prep kit designed for low inputs.2. Reduce the number of PCR cycles; use a protocol with bead-based cleanup to remove small fragments and primers. |
Assessing 5'-End Specificity and TSS Peak Calling for Promoter Analysis
Within the framework of paired-end RNA-seq alignment research using the STAR protocol, the precise identification of transcription start sites (TSSs) represents a critical step for elucidating gene regulation mechanisms. Accurate promoter analysis depends heavily on the specific capture of a transcript's 5'-end and subsequent robust computational identification of TSS peaks [83]. While standard RNA-seq protocols provide comprehensive transcriptome coverage, they often fail to resolve the precise boundaries of 5' ends due to biases introduced during library preparation [84]. Specialized 5' end RNA-seq methods have therefore been developed to address this gap, enabling researchers to study condition-specific promoter usage, alternative TSSs, and regulatory elements such as enhancers [83]. This protocol outlines a systematic approach for assessing 5'-end specificity and performing TSS peak calling, integrating these specialized methodologies with the STAR aligner to generate a comprehensive pipeline for promoter analysis in drug development and basic research.
Comparative Analysis of 5'-End RNA-Seq Methods
Several specialized methods exist for identifying the 5' ends of transcripts, each employing distinct molecular strategies to enrich for sequences containing TSS information. The most prominent methods include CAGE (Cap Analysis Gene Expression), RAMPAGE (RNA Annotation and Mapping of Promoters for Analysis of Gene Expression), STRT (Single-Cell Tagged Reverse Transcription), NanoCAGE, Oligo Capping (TSS-Seq), and GRO-cap [83]. These techniques primarily differ in their initial RNA processing steps: CAGE captures the 5' cap of mRNAs through cap-trapping; RAMPAGE uses a template-switching mechanism; STRT and NanoCAGE are optimized for low-input samples; Oligo Capping replaces the cap with a synthetic oligonucleotide; while GRO-cap identifies actively transcribing RNA polymerases [83] [85].
Performance Metrics and Method Selection
A comprehensive comparative analysis of these methods revealed significant differences in their performance characteristics. When evaluated using standardized metrics including 5'-end specificity, sensitivity, precision, and quantification accuracy, CAGE emerged as the best-performing method for mRNA TSS identification, while GRO-cap was superior for detecting enhancer RNAs (eRNAs) [83].
Table 1: Performance Comparison of 5'-End RNA-Seq Methods for mRNA
| Method | 5'-End Specificity | Sensitivity | Precision | Quantification Error |
|---|---|---|---|---|
| CAGE | Highest | High | Highest | Lowest (1.1%) |
| RAMPAGE | High | High | Moderate | Moderate (2.0%) |
| GRO-cap | Moderate | Highest | Moderate | Not Reported |
| STRT | Moderate | Low | High | Not Reported |
| NanoCAGE-XL | Low | Low | High | Not Reported |
| Oligo Capping | Low | Low | Low | Not Reported |
The evaluation utilized synthetic spike-in RNA assays to distinguish technical performance from biological phenomena or annotation issues [83]. CAGE demonstrated superior 5' end coverage of both cellular transcripts and spike-in controls, coupled with the lowest quantification error, making it particularly suitable for quantifying differences in TSS usage across samples [83].
Experimental Protocol for 5'-End Specificity Assessment
Library Preparation with CAGE
The CAGE method provides an optimal balance of specificity and quantification accuracy for promoter analysis. The following protocol details library construction and processing:
RNA Input and Quality Control: Use high-quality total RNA with RNA Integrity Number (RIN) ≥ 7 [10]. The CAGE protocol typically requires 100-500 ng of total RNA as starting material [83].
Cap Trapping:
- Reverse transcribe RNA using random primers and reverse transcriptase.
- Specifically biotinylate the 5' cap structure of cDNA molecules using cap-catching chemistry.
- Capture biotinylated cDNAs using streptavidin beads, effectively enriching for full-length transcripts containing authentic 5' ends [83].
Library Construction for Sequencing:
- Ligate sequencing adapters to the captured cDNA fragments.
- Amplify the library with a minimal number of PCR cycles (typically 10-12) to maintain representation accuracy.
- Include Unique Molecular Identifiers (UMIs) during adapter ligation to correct for PCR duplicates in downstream analysis [85].
Sequencing Parameters: Sequence libraries on Illumina platforms using paired-end sequencing (minimum 50 bp read length, 30-60 million reads per sample for standard differential expression analysis) [10] [86].
Spike-In RNA Assay for Technical Validation
To control for technical variability and accurately assess 5'-end specificity, incorporate synthetic spike-in RNAs:
Spike-in Selection: Use ERCC (External RNA Controls Consortium) spike-in mixes [83]. These artificial transcripts with known concentrations and sequences provide an internal standard for evaluating method performance.
Spike-in Addition: Add a defined quantity of ERCC spike-ins to the total RNA sample prior to library preparation, following manufacturer recommendations for dilution ratios.
Performance Assessment: After sequencing, calculate the 5' end coverage and quantification accuracy for each spike-in transcript. Compare the observed read distribution and abundance to the expected values to quantify technical variability [83].
TSS Peak Calling and Analysis Pipeline
STAR Alignment for 5'-End RNA-Seq Data
The STAR aligner is particularly suited for 5' end RNA-seq data due to its ability to handle spliced alignments and accurately map reads across exon junctions [9] [3].
Genome Index Generation:
Note: The
--sjdbOverhangparameter should be set to read length minus 1 [3].Read Alignment:
This produces coordinate-sorted BAM files suitable for downstream peak calling [3].
Alignment Quality Control:
TSS Peak Calling with Paraclu
Following alignment, specialized peak-calling algorithms identify precise TSS locations from the enriched 5' end reads:
Input Preparation: Extract the 5' most position of properly paired, high-quality aligned reads from BAM files. For paired-end data, use only read 1 5' ends as they originate from the 5' end of transcripts [83].
Peak Calling with Paraclu:
- Use the Paraclu algorithm with parameters optimized for sensitivity and precision [83].
- Adjust minimum tag count thresholds based on sequencing depth (typically 10-20 tags per peak for 20 million reads).
- Cluster nearby TSS signals into representative peaks representing distinct promoter regions.
Peak Filtering:
- Implement additional filtering steps to distinguish true TSS peaks from background noise [83].
- Remove peaks falling within annotated rRNA genes and other structural RNAs.
- Filter out peaks with low complexity sequences that may represent technical artifacts.
Figure 1: TSS Peak Calling and Promoter Analysis Workflow
Annotation and Validation of TSS Peaks
Genomic Annotation:
- Annotate identified TSS peaks relative to known gene models using reference annotations (e.g., UCSC, ENSEMBL, or Gencode) [83].
- Classify peaks as annotated (matching known TSSs) or unannotated (potential novel promoters).
Validation:
- Support unannotated TSS peaks with evidence from other genomic methods (e.g., chromatin marks, conservation) [83].
- Calculate precision and sensitivity metrics by comparing against validated TSS databases.
- For CAGE data, expect approximately 24% of reads to map to regions not close to annotated 5' ends (beyond 10% of transcript length), representing background, incomplete annotation, or biological phenomena like RNA recapping [83].
Research Reagent Solutions
Table 2: Essential Research Reagents for 5'-End RNA-Seq Studies
| Reagent/Category | Specific Examples | Function and Application Notes |
|---|---|---|
| 5' End Library Prep Kits | CAGE Kit, STRT-N Kit | Specialized reagents for 5' end capture and library construction; select based on RNA input requirements and application [83] [85]. |
| Spike-in RNA Controls | ERCC RNA Spike-In Mix | Artificial RNA transcripts with known sequences and abundances; enable technical performance assessment and normalization [83]. |
| RNA Quality Assessment | Bioanalyzer RNA Kit, RIN Algorithm | Assess RNA integrity (RIN ≥7 recommended); critical for successful library preparation [10]. |
| rRNA Depletion Kits | RiboMinus, Ribo-Zero | Remove abundant ribosomal RNA; essential for total RNA sequencing without poly-A selection [84] [10]. |
| Strand-Specific Library Prep | dUTP Second Strand Marking | Maintain strand orientation information during library prep; crucial for accurate annotation of antisense transcripts [10]. |
| Unique Molecular Identifiers | UMI Adapters | Molecular barcodes added during reverse transcription; enable accurate digital counting by correcting for PCR duplicates [85]. |
Application to Differential TSS Usage Analysis
The integration of 5'-end-specific RNA-seq with TSS peak calling enables powerful analyses of differential promoter usage across biological conditions. Application of this approach to brain-related samples revealed a genome-wide shift in TSS usage between fetal and adult brain, with downstream TSSs preferentially used in adult brain and upstream TSSs utilized in fetal brain and in vitro differentiated neurons [83]. Similarly, studies of bovine embryonic genome activation employed STRT-N RNA-seq to identify transcription factors involved in embryonic development by profiling 5' ends of transcripts in single oocytes and preimplantation embryos [85].
For robust differential TSS analysis:
- Experimental Design: Include a minimum of 3 biological replicates per condition to adequately capture biological variation [86].
- Normalization: Normalize read counts using spike-in controls and/or housekeeping genes with stable TSS usage.
- Statistical Testing: Employ specialized tools for differential TSS usage (e.g., CAGEr or custom scripts) that test for significant changes in TSS peak height between conditions while accounting for overall gene expression changes.
This comprehensive approach to assessing 5'-end specificity and TSS peak calling provides researchers with a validated framework for promoter analysis, enabling insights into gene regulation mechanisms relevant to development, disease pathogenesis, and drug response.
Evaluating the Impact of Trimming and Pre-processing on Final Results
Within the established framework of a paired-end RNA-seq alignment thesis utilizing the STAR protocol, the initial data pre-processing phase is frequently underestimated. Although subsequent alignment and differential expression analysis often receive greater focus, the quality and integrity of the raw sequencing data serve as the foundational layer upon which all subsequent biological interpretations are built. Trimming and pre-processing are not merely optional preliminary steps; they are critical procedures that directly influence the accuracy, reliability, and reproducibility of the final results. This assessment explores the tangible impact of these initial steps on key analytical outcomes, providing detailed protocols and quantitative evaluations to guide researchers in optimizing their RNA-seq pipelines. Evidence suggests that carefully selected pre-processing methods can significantly enhance mapping efficiency and improve the detection power of downstream differential expression analysis [32] [76]. The decisions made at this early stage can either safeguard the integrity of the entire dataset or introduce biases that compromise all subsequent findings.
Quantitative Impact of Trimming on Data Quality and Alignment
The process of read trimming primarily aims to remove adapter sequences, low-quality bases, and other technical artifacts from raw sequencing reads. Systematic comparisons of various trimming tools reveal significant differences in their performance, which in turn has a measurable effect on downstream alignment success.
A comprehensive study evaluating 288 distinct RNA-seq pipelines on fungal data demonstrated that the choice of trimming algorithm directly influences base quality and subsequent mapping rates. The tool fastp, for instance, was shown to enhance the proportion of high-quality Q20 and Q30 bases by 1% to 6% compared to raw data [32]. Furthermore, the specific parameters used during trimming are equally critical. Aggressive trimming can unnecessarily reduce read length and available data, while overly permissive parameters may leave behind adapter sequences and low-quality bases that interfere with alignment [20] [87]. Another large-scale assessment of 192 alternative methodological pipelines on human RNA-seq data confirmed that trimming improves read mapping rates, though it emphasized that this process must be applied non-aggressively to avoid unpredictable changes in gene expression signals [76].
Table 1: Performance Comparison of Trimming Tools
| Tool | Key Characteristics | Impact on Base Quality | Impact on Alignment Rate | Considerations |
|---|---|---|---|---|
| fastp | Rapid operation; integrated quality control [32] | Increases Q20/Q30 bases by 1-6% [32] | Positive effect on mapping rate [32] | Balances speed and effective cleaning |
| Trim Galore | Wrapper integrating Cutadapt and FastQC [32] | Enhances base quality [32] | Positive effect on mapping rate [32] | May cause unbalanced base distribution in read tails [32] |
| Trimmomatic | High configurability; complex parameters [20] [76] | Effectively removes adapters and low-quality bases [20] | Improves mapping rate [76] | Parameter setup is complex; no speed advantage [32] |
| Cutadapt | Effective adapter removal [20] [76] | Effective adapter removal [20] | Improves mapping rate [76] | Often used within other wrappers |
The relationship between trimming and the subsequent STAR alignment is direct. STAR and other aligners require high-quality sequence ends for accurate splicing detection and unique placement of reads across the genome [9]. Residual adapter sequences can prevent a read from mapping entirely or cause it to map to an incorrect location, leading to inaccurate gene counts. Therefore, a well-trimmed dataset directly translates into a higher mapping rate, more accurate quantification, and a more reliable foundation for identifying differentially expressed genes.
Experimental Protocols for Benchmarking Pre-processing Tools
To objectively evaluate the impact of different trimming tools within a paired-end RNA-seq workflow, the following structured experimental protocol is recommended. This benchmarking approach allows researchers to select the optimal pre-processing strategy for their specific data.
Reagent and Software Toolkit
Table 2: Essential Research Reagents and Tools for RNA-seq Pre-processing Benchmarking
| Category | Item | Function/Description | Example Source/Version |
|---|---|---|---|
| Raw Data | Paired-end FASTQ files | The initial input data for the pipeline. | NCBI GEO, ENA, or SRA [12] |
| Reference Files | Reference Genome (FASTA) | Genomic sequence for read alignment. | ENSEMBL, GENCODE [23] [9] |
| Gene Annotation (GTF) | Defines gene models for read assignment. | ENSEMBL, GENCODE [23] [9] | |
| Software Tools | Trimming Tools (e.g., fastp, Trim Galore) | Removes adapters and low-quality bases. [32] | |
| Quality Control (FastQC, MultiQC) | Assesses raw and trimmed read quality. [20] [12] | ||
| Spliced Read Aligner (STAR) | Aligns RNA-seq reads to the reference genome. [12] [9] | ||
| Quantification Tool (featureCounts) | Counts reads overlapping genes. [20] |
Step-by-Step Benchmarking Procedure
Data Preparation and Initial QC:
- Obtain paired-end RNA-seq datasets. For a robust comparison, include datasets from different species (e.g., human, mouse, plant pathogenic fungi) to assess tool performance across varying genomic architectures [32].
- Run FastQC on the raw FASTQ files to establish a baseline quality profile, checking for adapter contamination, per-base sequence quality, and GC content [20] [12].
Parallel Trimming:
- Process the same raw FASTQ files through multiple trimming tools (e.g., fastp, Trim Galore, Trimmomatic). Use default parameters initially, then explore parameter tuning based on the initial FastQC report.
- Critical Parameter: For tools like Trimmomatic and Cutadapt, specify the adapter sequences and set a minimum read length threshold (e.g., 50 bp) to retain after trimming [76].
- Run FastQC again on all trimmed output files to document the changes in quality metrics.
Alignment with STAR:
- Generate a STAR genome index using the reference genome FASTA and annotation GTF file. The
--sjdbOverhangparameter should be set to (read length - 1), e.g., 100 for 101 bp paired-end reads [23] [9]. - Align the trimmed reads from each pipeline to the reference genome using STAR with consistent parameters (e.g.,
--outSAMtype BAM SortedByCoordinate --quantMode GeneCounts) [23]. - Record key alignment metrics from the STAR log files, particularly the mapping rate and the percentage of reads mapped to multiple loci.
- Generate a STAR genome index using the reference genome FASTA and annotation GTF file. The
Read Quantification:
Downstream Analysis Simulation:
- Perform a basic differential expression analysis (e.g., using DESeq2 or edgeR) on the count matrices from the different pipelines [20] [88].
- Compare the number of differentially expressed genes (DEGs) identified and their statistical significance (p-values, false discovery rates) across the different pre-processing methods [76].
Workflow Visualization
The following diagram illustrates the logical workflow for the benchmarking protocol, showing the parallel processing paths and key evaluation metrics.
The Cascading Effects on Downstream Analysis
The impact of trimming and pre-processing extends far beyond alignment statistics, cascading into all subsequent biological interpretations. The most significant downstream effects are observed in gene expression quantification and the identification of differentially expressed genes (DEGs).
The process of converting aligned reads into a gene count matrix is sensitive to data quality. Ambiguously mapped reads or those with poor alignment quality can lead to incorrect assignment, thereby skewing expression estimates [20] [87]. A consistent finding is that improved pre-processing leads to more accurate and precise raw gene expression quantification, which is the direct input for statistical tests of differential expression [76]. Normalization methods used in tools like DESeq2 and edgeR, while designed to correct for technical biases like sequencing depth, rely on the assumption that the majority of genes are not differentially expressed. Systematic biases introduced during library preparation or inadequate trimming can violate this assumption, reducing the efficacy of these normalization procedures [20].
The ultimate test of a pipeline is its ability to correctly identify DEGs. Studies that validate RNA-seq findings with orthogonal methods like qRT-PCR have demonstrated that pipelines incorporating rigorous trimming often yield DEG lists with higher validation rates [76]. Furthermore, the total number of reported DEGs can vary significantly between different pre-processing workflows. This variation underscores the importance of a optimized and reproducible pre-processing stage to ensure that biological conclusions are driven by true signal rather than technical artifacts [32] [76]. The goal is not necessarily to maximize the number of DEGs, but to enhance the accuracy and reliability of the DEGs that are reported.
The evidence clearly demonstrates that trimming and pre-processing are not routine, inconsequential steps but are critical determinants of success in paired-end RNA-seq studies utilizing the STAR aligner. The choice of tools and parameters directly influences data quality, alignment efficiency, quantification accuracy, and the final list of differentially expressed genes. Based on the current analysis, the following best practices are recommended:
- Always Perform Trimming: Do not skip the trimming step. Raw sequencing data almost always contains adapter sequences and low-quality bases that can negatively impact STAR's alignment performance [20] [76].
- Benchmark Tools on Your Data: There is no universally "best" tool. The optimal choice of trimming software (e.g., fastp, Trim Galore) may depend on the specific dataset and sequencing platform. Conduct a small-scale benchmark, as outlined in the protocol above, to select the best performer for your project [32].
- Avoid Over-Triming: Balance is key. While aggressive trimming can clean data effectively, it can also unnecessarily reduce read length and the total amount of data available for analysis, potentially harming the detection of lowly expressed transcripts [20] [87].
- Implement End-to-End QC: Integrate quality control checks at multiple stages—before trimming, after trimming, and after alignment—using tools like FastQC and MultiQC to monitor the effects of pre-processing on data integrity [20] [12].
- Ensure Reproducibility: Document all parameters and software versions used in the pre-processing stage. This is essential for replicating results and for the long-term reliability of your research findings [89].
By adopting these practices and rigorously evaluating the impact of pre-processing, researchers can establish a robust and reliable foundation for their RNA-seq analyses, ensuring that their biological conclusions are built upon the most accurate data possible.
In paired-end RNA-seq analysis, the alignment of reads using STAR is a critical first step, but the biological insights are ultimately derived from downstream quantification and differential expression (DE) analysis. The integration between the alignment output and these downstream tools is therefore a pivotal phase of the workflow. Successful integration ensures that the rich splicing information captured by STAR is accurately translated into a quantitative format, such as a count matrix, which can then be interrogated for differential expression and splicing. This application note details the protocols for bridging the STAR aligner with the featureCounts quantification tool and subsequent differential expression pipelines using established R/Bioconductor packages. Framed within a broader thesis on RNA-seq protocols, this document provides detailed methodologies and benchmarks to guide researchers and drug development professionals.
The Integrated RNA-Seq Analysis Workflow
The following diagram illustrates the complete workflow, from raw sequencing data to differential expression and splicing analysis, highlighting the integration points between STAR, featureCounts, and downstream statistical tools.
Quantitative Performance of Downstream Workflows
The choice of quantification strategy post-STAR alignment significantly impacts the power and accuracy of downstream statistical analyses. The following table benchmarks the performance of the standard exon-based quantification workflow against the enhanced DEJU (Differential Exon-Junction Usage) workflow, which incorporates exon-exon junction reads.
Table 1: Benchmarking Performance of DEJU versus Standard DEU Workflows on Simulated RNA-seq Data
| Analysis Workflow | Splicing Pattern | Sample Size (n) | False Discovery Rate (FDR) | Statistical Power |
|---|---|---|---|---|
| DEJU-edgeR | Exon Skipping (ES) | 3 | 0.022 | 0.977 |
| DEJU-limma | Exon Skipping (ES) | 3 | 0.043 | 0.975 |
| DEJU-edgeR | Mutually Exclusive Exons (MXE) | 5 | 0.040 | 0.993 |
| DEJU-limma | Mutually Exclusive Exons (MXE) | 5 | 0.062 | 0.995 |
| DEJU-edgeR | Alternative Splice Site (ASS) | 10 | 0.038 | 0.977 |
| DEJU-limma | Alternative Splice Site (ASS) | 10 | 0.047 | 0.979 |
| DEJU-edgeR | Intron Retention (IR) | 10 | 0.042 | 0.964 |
| DEJU-limma | Intron Retention (IR) | 10 | 0.050 | 0.968 |
Source: Adapted from [90] Note: The DEJU workflow integrates exon and exon-exon junction counts, leading to enhanced power for detecting differential splicing events while controlling the FDR.
Detailed Experimental Protocols
Protocol 1: Standard Gene-Level Quantification with featureCounts
This protocol details the steps for generating a gene-level count matrix from STAR-aligned BAM files, which is the standard input for most differential gene expression analysis tools.
- Input Preparation: You will need the aligned BAM file(s) from STAR and the corresponding gene annotation file (GTF format) used during the STAR alignment [12].
- featureCounts Execution: Run the
featureCountsfunction from the RSubread package in R. The critical arguments for this standard analysis are:-a <annotation.gtf>: Path to the GTF annotation file.-o <output.counts.txt>: Path for the output count matrix.-t exon -g gene_id: These parameters specify that reads should be counted for exons and then summarized to the gene level.-p: This flag indicates that the data are from a paired-end experiment.-B: Only count read pairs where both ends are mapped.-T <number_of_threads>: Specify the number of threads/CPUs to use for faster processing [91].
- Output: The primary output is a count matrix where rows are genes and columns are samples. This matrix is directly compatible with DE analysis tools like DESeq2 and edgeR [92] [91].
Protocol 2: The DEJU Workflow for Enhanced Differential Splicing
The Differential Exon-Junction Usage (DEJU) workflow is an advanced protocol that resolves the double-counting issue in standard exon-level analysis and improves the detection of differential splicing by jointly analyzing exons and junctions [90].
- STAR Alignment for DEJU: Perform a 2-pass STAR alignment. The first pass identifies novel junctions from all samples, which are then used to re-index the genome for a more sensitive second pass of alignment. Use the
--outFilterType BySJoutoption to filter out spurious junctions [90]. - Junction-Aware Quantification with featureCounts: Run
featureCountswith specific arguments to generate both exon and junction counts simultaneously.useMetaFeatures = FALSE: This argument is set to produce exon-level counts instead of gene-level summaries.nonSplitOnly = TRUE: This allows the quantification of internal exon read counts.juncCounts = TRUE: This is the crucial argument that directsfeatureCountsto return an extra count matrix for exon-exon junctions [90].
- Data Merging and Filtering: Concatenate the internal exon and processed junction count matrices into a single exon-junction count matrix. Filter out lowly expressed exons and junctions using the
filterByExprfunction in edgeR [90]. - Normalization: Perform normalization for composition biases using the Trimmed Mean of M-values (TMM) method with the
normLibSizesfunction in edgeR [90]. - Differential Splicing Analysis: The final exon-junction count matrix can be analyzed using:
- The
diffSpliceDGEfunction in edgeR, which uses an exon-level negative binomial model and combines feature-level results to the gene level using the Simes method or an F-test [90]. - The
diffSplicefunction in limma, which employs a linear model and uses the Simes method to combine p-values at the gene level [90].
- The
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for RNA-seq Analysis Pipelines
| Tool / Reagent | Primary Function | Role in the Protocol |
|---|---|---|
| STAR Aligner | Spliced Transcripts Alignment to a Reference | Precisely maps RNA-seq reads to the genome, identifying exon-exon junctions [12] [90]. |
| RSubread (featureCounts) | Read Quantification | Assigns aligned reads to genomic features (exons, genes) and generates the count matrix [12] [90]. |
| edgeR | Differential Expression Analysis | Statistical analysis of count data to identify differentially expressed genes or exons [90] [92]. |
| limma | Differential Expression Analysis | Uses linear models to identify differential expression and splicing, particularly powerful with voom transformation [90]. |
| DESeq2 | Differential Expression Analysis | An alternative statistical package for DE analysis, often used with featureCounts output [92]. |
| SAMtools | SAM/BAM File Processing | Utilities for manipulating and indexing alignment files, often used before featureCounts [12]. |
The transition from STAR alignment to biological interpretation hinges on robust and well-executed downstream analysis. While the standard gene-level counting workflow with featureCounts into DESeq2 or edgeR remains a cornerstone for differential expression analysis, incorporating exon and junction-level counts unlocks a deeper layer of transcriptomic regulation. The DEJU workflow demonstrates that strategic integration of junction reads significantly enhances the statistical power to detect differential splicing events, providing a more comprehensive view of gene regulation. By adhering to the detailed protocols and leveraging the benchmarks provided herein, researchers can confidently build upon the STAR alignment foundation to generate reliable, biologically meaningful results in both basic research and drug development contexts.
Conclusion
Mastering paired-end RNA-seq alignment with STAR is a foundational skill that empowers researchers to generate robust and biologically meaningful data. This guide synthesizes key takeaways: the importance of proper genome indexing, the flexibility of STAR's parameters for different applications like fusion detection or novel isoform discovery, and the critical need for rigorous quality assessment. As RNA-seq applications expand into single-cell and long-read sequencing, a solid understanding of this core alignment protocol ensures researchers can adapt to new technologies. For drug development, this translates into more reliable transcriptome data for identifying therapeutic targets and biomarkers, ultimately accelerating the pace of biomedical discovery.
References
- 1. Mastering RNA - seq Read Alignment With STAR [https://www.reneshbedre.com/blog/star-aligner.html]
- 2. STAR: ultrafast universal RNA-seq aligner - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3530905/]
- 3. Alignment with STAR | Introduction to RNA-Seq using high ... [https://hbctraining.github.io/Intro-to-rnaseq-hpc-O2/lessons/03_alignment.html]
- 4. - RNA Tutorial- seq , StringTie and DESeq2* - 1 Learning... STAR [https://cyverse.atlassian.net/wiki/spaces/TUT/pages/258736307/RNA-seq+Tutorial-+STAR+StringTie+and+DESeq2]
- 5. of Evaluation and Kallisto on Single Cell STAR - RNA Data... Seq [https://pmc.ncbi.nlm.nih.gov/articles/PMC7202009/]
- 6. (PDF) Evaluation of STAR and Kallisto on Single Cell RNA - Seq Data... [https://www.academia.edu/66790658/Evaluation_of_STAR_and_Kallisto_on_Single_Cell_RNA_Seq_Data_Alignment]
- 7. Optimizing STAR Aligner for High Throughput Computing ... [https://arxiv.org/html/2409.05886v1]
- 8. rnaseq: Output [https://nf-co.re/rnaseq/2.0/docs/output]
- 9. Mapping RNA-seq Reads with STAR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4631051/]
- 10. A survey of best practices for RNA-seq data analysis [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8]
- 11. A real-world multi-center RNA-seq benchmarking study ... [https://www.nature.com/articles/s41467-024-50420-y]
- 12. Transcriptomics / RNA-seq Alignment with STAR / Hands-on [https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/rna-seq-bash-star-align/tutorial.html]
- 13. Massive mining of publicly available RNA-seq data from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5893633/]
- 14. alexdobin/STAR: RNA-seq aligner [https://github.com/alexdobin/STAR]
- 15. Hardware requirements for bowtie2/STAR RNA-seq ... [https://www.biostars.org/p/482847/]
- 16. rnaseq: Usage [https://nf-co.re/rnaseq/latest/docs/usage/]
- 17. Serverless Approach to Running Resource-Intensive STAR ... [https://arxiv.org/html/2504.05078v1]
- 18. Protocol for conducting a single-cell sequencing assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12305206/]
- 19. NGS analysis: how to handle paired-end reads [https://www.biostars.org/p/311896/]
- 20. a practical guide to RNA sequencing data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599993/]
- 21. Commentary: a review of technical considerations for planning ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12094-8]
- 22. Mapping RNA-seq Reads with STAR [https://www.rna-seqblog.com/mapping-rna-seq-reads-with-star/]
- 23. Mapping with STAR [https://biocorecrg.github.io/RNAseq_course_2019/alnpractical.html]
- 24. Is there a manual to understand STAR output files? [https://www.biostars.org/p/9471213/]
- 25. Star running problem with --sjdbOverhang [https://groups.google.com/g/rna-star/c/q4zGzlPgwXY]
- 26. Performance Analysis and Optimization of the STAR ... [https://arxiv.org/html/2506.12611v1]
- 27. Size for sjdbOverhang for multiple read length datasets #931 [https://github.com/alexdobin/STAR/issues/931]
- 28. STAR Aligner Metrics | WARP - Broad Institute [https://broadinstitute.github.io/warp/docs/Pipelines/Optimus_Pipeline/starsolo-metrics]
- 29. STAR: ultrafast universal RNA-seq aligner. [https://www.encodeproject.org/publications/9d43c048-c70c-42ab-895d-77a69fceb42b/]
- 30. — Janis documentation STAR Aligner [https://janis.readthedocs.io/en/latest/tools/bioinformatics/cold%20spring%20harbor%20laboratory/star_genomegenerate.html]
- 31. chipster.csc.fi/manual/ star - paired - end .html [https://chipster.csc.fi/manual/star-paired-end.html]
- 32. A comprehensive workflow for optimizing RNA-seq data analysis [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10414-y]
- 33. STAR 2.7.9a - CQLS Software Update List [https://software.cqls.oregonstate.edu/updates/star-2.7.9a/]
- 34. [Tutorial] Bulk RNA-seq DE analysis [https://informatics.fas.harvard.edu/resources/tutorials/differential-expression-analysis/]
- 35. Running STAR aligner on paired-end reads [https://www.biostars.org/p/9507364/]
- 36. question about mapping multiple samples [https://groups.google.com/g/rna-star/c/CAfqybt-E1I/m/ra51dsoZCAAJ]
- 37. Alignment of RNA Seq data with STAR [https://research.stowers.org/cws/CompGenomics/Projects/star.html]
- 38. STAR manual with usage examples | BioQueue Encyclopedia [https://open.bioqueue.org/home/knowledge/showKnowledge/sig/star-align]
- 39. Workflow for analysis of paired-end RNA sequencing [https://github.com/etc27/RNAseq-workflow]
- 40. RNASeq Data Analysis - CRCD User Manual - Pitt CRC [https://crc-pages.pitt.edu/user-manual/advanced-genomics-support/RNASeq-data-analysis/]
- 41. 04 Visualizing and counting of the alignments - GitHub Pages [https://scienceparkstudygroup.github.io/rna-seq-lesson/04-bioinformatic-workflow/index.html]
- 42. Which samtools command is used to sort a BAM file? [https://samtools.org/which-samtools-command-is-used-to-sort-a-bam-file/]
- 43. Understanding and manipulating SAM/BAM alignment files [http://homer.ucsd.edu/homer/basicTutorial/samfiles.html]
- 44. Benchmarking RNA-Seq quantification tools - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5003039/]
- 45. Visualizing Base Modifications in Long-Read Alignment Files [https://www.goldenhelix.com/blog/visualizing-base-modifications-in-long-read-alignment-files/]
- 46. Two-pass alignment improves novel splice junction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5006238/]
- 47. Two-pass alignment of RNA-seq reads with STAR [https://www.reneshbedre.com/blog/star-aligner-twopass-mode.html]
- 48. About STAR 2-pass alignment [https://groups.google.com/g/rna-star/c/rBQK-ujtSh8]
- 49. rnaseq: Output [https://nf-co.re/rnaseq/1.4.2/docs/output]
- 50. Sjdboverhang Option In Star [https://www.biostars.org/p/93883/]
- 51. Confused about sjdbOverhang [https://groups.google.com/g/rna-star/c/h9oh10UlvhI/m/BfSPGivUHmsJ]
- 52. Clara-parabricks rna-fq2bam: --sjdbOverhang [https://forums.developer.nvidia.com/t/clara-parabricks-rna-fq2bam-sjdboverhang/275572]
- 53. STAR two-pass basic for differential expression analysis [https://github.com/alexdobin/STAR/issues/1616]
- 54. STAR do i need --twopassMode ? [https://www.biostars.org/p/301944/]
- 55. star 2 pass v. star single pass [https://groups.google.com/g/majiq_voila/c/qRd1EoVsBhc]
- 56. RNAseq short variant discovery (SNPs + Indels) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035531192-RNAseq-short-variant-discovery-SNPs-Indels]
- 57. Re: Chimeric alignments [https://groups.google.com/g/rna-star/c/HUxFCaHSX6c]
- 58. chimJunctionOverhangMin vs chimSegmentMin · Issue #744 [https://github.com/alexdobin/STAR/issues/744]
- 59. Bioinformatics Pipeline: mRNA Analysis - GDC Docs [https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/]
- 60. circrna: Parameters [https://nf-co.re/circrna/latest/parameters]
- 61. What are the best set of options to use while aligning ... [https://www.biostars.org/p/384844/]
- 62. Asking for unmapped genes in RNA sequencing via Galaxy [https://www.biostars.org/p/9614497/]
- 63. - Why total rna - seq usually yields low RNA ? seq mapping rate [https://bioinformatics.stackexchange.com/questions/2897/why-total-rna-seq-usually-yields-low-mapping-rate]
- 64. High percentage of unmapped - too short (~40-55%) perhaps... reads [https://github.com/alexdobin/STAR/issues/793]
- 65. high percentage of multi- RNA - usegalaxy.org... STAR mapped reads [https://help.galaxyproject.org/t/rna-star-high-percentage-of-multi-mapped-reads/7423]
- 66. Very low mapping of mRNA rate seq data [https://www.biostars.org/p/339475/]
- 67. STAR: 速度超快的RNA-seq aligner 原创 [https://blog.csdn.net/u013429737/article/details/116708436]
- 68. Introduction to RNA-Seq using high-performance computing [https://hbctraining.github.io/Intro-to-rnaseq-hpc-salmon/lessons/03_QC_STAR_and_Qualimap_run.html]
- 69. 「RNA-seq分æžè½¯ä»¶ã€RNA-seq比对工具STARå¦ä¹ 笔记原创 [https://blog.csdn.net/u012110870/article/details/102804525]
- 70. How to interpret log.final.out in star [https://www.biostars.org/p/301616/]
- 71. Benchmarking RNA-Seq Aligners at Base-Level and Junction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10935055/]
- 72. Comparison of RNA Sequencing Data Alignment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6617288/]
- 73. Comparison of Short-Read Sequence Aligners Indicates ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.657240/full]
- 74. Comparison of samples to find the best parameters for ... [https://www.biostars.org/p/9462609/]
- 75. A systematic benchmark of Nanopore long-read RNA ... [https://www.nature.com/articles/s41592-025-02623-4]
- 76. Systematic comparison and assessment of RNA-seq ... [https://www.nature.com/articles/s41598-020-76881-x]
- 77. Evaluation and comparison of computational tools for RNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-4002-1]
- 78. Do results obtained with RNA-sequencing require ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7823214/]
- 79. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 80. Kallisto vs. STAR: Alignment and Quantification of Bulk ... [https://www.elucidata.io/blog/kallisto-vs-star-alignment-and-quantification-of-bulk-rna-seq-data]
- 81. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 82. RNA-seq validation: software for selection of reference and ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10511-y]
- 83. Comprehensive comparative analysis of 5' end RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6075671/]
- 84. RNA Sequencing and Analysis - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4863231/]
- 85. RNA sequencing of mRNA 5'-ends reveals regulators of ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12110-x]
- 86. Experimental design considerations | Introduction to RNA-seq ... [https://hbctraining.github.io/Intro-to-rnaseq-fasrc-salmon-flipped/lessons/02_experimental_planning_considerations.html]
- 87. From bench to bytes: a practical guide to RNA sequencing ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1697922/full]
- 88. Review of RNA-Seq Data Analysis Tools [https://www.rna-seqblog.com/review-of-rna-seq-data-analysis-tools/]
- 89. Consistently processed RNA sequencing data from 50 ... [https://www.nature.com/articles/s41597-025-05376-z]
- 90. Incorporating exon–exon junction reads enhances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288301/]
- 91. A Guide to Basic RNA Sequencing Data Processing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12067304/]
- 92. RNA-Seq Alignment & Analysis Workflow [https://www.dnastar.com/workflows/rna-seq/]