A Comprehensive Guide to the GATK Somatic Variant Calling Workflow: From Best Practices to Clinical Application
This article provides a complete overview of the GATK somatic variant calling workflow for researchers, scientists, and drug development professionals.
A Comprehensive Guide to the GATK Somatic Variant Calling Workflow: From Best Practices to Clinical Application
Abstract
This article provides a complete overview of the GATK somatic variant calling workflow for researchers, scientists, and drug development professionals. It covers the foundational principles of the Genome Analysis Toolkit (GATK) and its Mutect2 caller, detailing the step-by-step methodology for accurate SNV and indel discovery in tumor samples. The content explores advanced troubleshooting for common issues like low allele fraction variants and offers optimization strategies for challenging data types, including FFPE samples. Furthermore, it presents a comparative analysis of Mutect2's performance against other callers, guidelines for validation, and the integration of regulatory and quality assurance frameworks essential for clinical translation and compliant biomarker development.
Understanding Somatic Variants and the GATK Ecosystem
The Biological and Clinical Significance of Somatic Mutations in Cancer
Somatic mutations represent the cornerstone of cancer evolution, serving as the fundamental genetic alterations that drive the transformation of normal cells into malignant tumors. These acquired changes, distinct from inherited germline variants, accumulate throughout an individual's lifetime due to interactions with environmental mutagens, endogenous processes, and random replication errors. Within the context of cancer genomics research, the accurate identification and interpretation of somatic mutations through robust bioinformatic workflows like the GATK (Genome Analysis Toolkit) somatic variant calling pipeline is paramount for understanding oncogenesis, tumor heterogeneity, and therapeutic vulnerabilities. This in-depth technical review examines the biological mechanisms through which somatic mutations influence cancer development, explores cutting-edge detection methodologies, and discusses the growing clinical implications for precision oncology, with particular emphasis on the critical role of accurate variant calling in advancing cancer research and drug development.
The transformation of a normal cell into a cancer cell occurs through a sequence of discrete genetic events, establishing cancer properly as a genetic disease of somatic cells [1]. The analogy between organismal evolution and cancer evolution is compelling: in both scenarios, mutation drives change, but Darwinian selection enables clones with a growth advantage to expand, providing a larger target for subsequent mutations [1]. Somatic mutations can be triggered by diverse factors including environmental carcinogens, endogenous metabolic processes, or spontaneous stochastic events during DNA replication [1].
The search for molecular lesions in tumors has been revolutionized by powerful technologies such as microarrays for global gene expression analysis and deep sequencing for comprehensive genome analysis [1]. While next-generation sequencing provides unprecedented insights, it also presents the challenge of distinguishing meaningful driver mutations from biologically inert passenger mutations—a computational challenge that somatic variant calling workflows must overcome through sophisticated filtering and annotation approaches [2] [3].
Biological Significance of Somatic Mutations
Driver versus Passenger Mutations
In the genomic landscape of any cancer, somatic mutations can be categorized into two fundamental classes:
- Driver mutations: These genetic alterations confer selective growth advantage and are directly implicated in cancer development. They occur in cancer genes and have been positively selected during tumor evolution [2]. The ratio of non-synonymous to synonymous substitutions in protein-coding sequences can help estimate the extent of selection pressure on these mutations [2].
- Passenger mutations: These do not contribute to cancer development but represent the imprints of mutational mechanisms that have generated them, providing insights into cancer etiology and pathogenesis without being subject to selection pressures [2].
Mutation Order and Cancer Evolution
Emerging evidence demonstrates that the temporal sequence in which mutations are acquired significantly impacts cancer biology, clinical presentation, and therapeutic response [4]. This represents a paradigm shift from viewing cancer as merely the sum of its mutational parts to understanding it as a dynamically evolving system where historical contingency shapes future trajectories.
In chronic myeloproliferative neoplasms (MPNs), the order of acquisition of JAK2 V617F and TET2 mutations dramatically influences stem and progenitor cell behavior, clonal evolution, and clinical phenotypes [4]. Similar order effects have been observed in mouse models of adrenocortical tumors, where expression of oncogenic Ras before loss of p53 produced highly malignant, metastatic tumors, while the reverse order resulted only in benign tumors [4]. This temporal dependency introduces additional complexity into variant interpretation and underscores the limitations of static mutational catalogs.
Clonal Evolution and Intratumor Heterogeneity
Cancers evolve through a process of sequential subclonal evolution, with competition between subclones and branching evolutionary patterns [4]. Instead of representing a homogeneous mass of genetically identical cells, tumors comprise families of clonally diverse cells with common ancestral origins [4]. This genetic diversity has profound clinical implications, as the pool of genetically diverse clones frequently serves as a source of therapy-resistant populations [4].
Table 1: Key Classes of Somatic Mutations in Cancer
| Mutation Type | Functional Impact | Example Genes | Detection Challenge |
|---|---|---|---|
| Oncogenic activation | Gain-of-function promoting proliferation | PDGFRA, KIT, MET, RET | Distinguishing from benign hypermutation |
| Tumor suppressor loss | Loss-of-function removing growth constraints | p53, APC, VHL, CDKN2A | Identifying haploinsufficient effects |
| Genome stability defects | Compromised DNA repair accelerating mutation rate | ATM, BLM, BRCA2, MSH2 | Interpreting in context of mutational signatures |
| Epigenetic regulator alteration | Changed expression landscape without DNA sequence change | EZH2, DNMT3A, TET2 | Functional interpretation without coding change |
Methodological Approaches: From Detection to Interpretation
Evolution of Somatic Mutation Detection Technologies
The sensitivity and accuracy of somatic mutation detection have progressed dramatically, from early methods with limited throughput to contemporary approaches capable of identifying mutations present in microscopic clones:
- Traditional sequencing: Initially limited to PCR-based resequencing of coding exons to identify base substitutions and small indels [2].
- Whole-genome/exome sequencing: Enabled comprehensive cataloguing of somatic mutations across cancer genomes, revealing remarkable insights into mutational processes [2].
- Error-corrected sequencing: Newer methods like NanoSeq achieve error rates below 5 errors per billion base pairs, enabling detection of mutations in single DNA molecules and revealing the extensive clonal landscapes of normal tissues [5].
The application of targeted NanoSeq to 1,042 non-invasive oral epithelium samples and 371 blood samples demonstrated an extremely rich selection landscape, with 46 genes under positive selection in oral epithelium and evidence of negative selection in essential genes [5]. This approach allows multivariate regression models to study how exposures and cancer risk factors alter the acquisition or selection of somatic mutations [5].
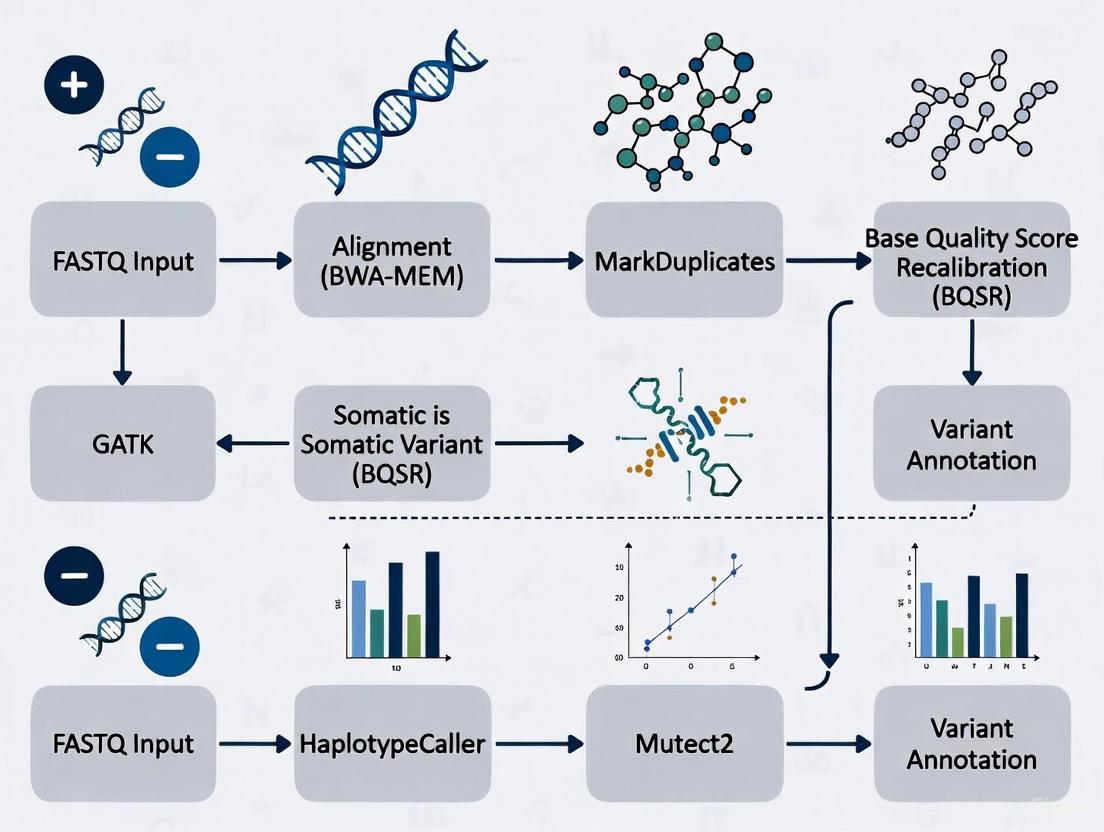
GATK Somatic Variant Calling Workflow
The GATK somatic variant calling pipeline represents a standardized approach for identifying somatic mutations when contrasting tumor and normal samples from the same individual [3]. The workflow encompasses several critical stages:
Diagram 1: GATK somatic variant calling workflow. The pipeline processes tumor and normal samples through alignment, quality control, and specialized somatic calling with Mutect2, incorporating additional resources like Panels of Normals (PoN) and population allele frequencies to improve accuracy.
Key Workflow Components
- Tumor-Normal Analysis: The core approach compares a tumor sample against a matched normal sample from the same individual to identify tumor-specific variants [3].
- Tumor-Only Mode: When matched normal samples are unavailable, the pipeline can operate in tumor-only mode, though this requires additional filtering strategies to address increased false positives [3].
- Panel of Normals (PoN): A critical resource especially important for tumor-only analysis, the PoN contains artifacts and common germline variants identified across multiple normal samples, allowing their systematic removal from tumor results [3].
- External Resources: Integration of population allele frequency data (e.g., gnomAD) and contamination information helps distinguish true somatic variants from germline polymorphisms or technical artifacts [3].
Comparative Analysis of Variant Calling Tools
Table 2: Performance Comparison of Somatic Variant Calling Tools
| Tool | Primary Algorithm | Optimal Use Case | Strengths | Limitations |
|---|---|---|---|---|
| GATK Mutect2 | Bayesian modeling with local assembly | Tumor-normal pairs with matched normal | High accuracy for SNVs/indels, comprehensive filtering | Computational intensity, parameter complexity |
| Strelka2 | Bayesian statistics with local realignment | Sensitive low-frequency somatic detection | Optimized for low VAF, fast operation | Less comprehensive than GATK ecosystem |
| Illumina DRAGEN | Hardware-accelerated graph alignment | Clinical applications requiring speed | Extreme speed, high accuracy | Proprietary hardware dependency |
| VarScan2 | Heuristic/statistical hybrid | Resource-constrained environments | Flexible threshold setting | Higher false positive rate without tuning |
Multiple benchmarking studies have demonstrated that GATK and DRAGEN typically perform best in SNV and indel detection, achieving high F-scores, while Strelka2 shows particular advantage in detecting low allele frequency somatic mutations [6]. The choice of tool depends on specific research requirements, with GATK offering comprehensive functionality and an active community, Strelka2 providing sensitivity for low-frequency variants, and DRAGEN delivering unprecedented speed through hardware acceleration [6].
Research Reagents and Experimental Solutions
Table 3: Essential Research Reagents for Somatic Mutation Analysis
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Panel of Normals (PoN) | Filters common artifacts and germline variants | Critical for tumor-only mode; should be cohort-specific |
| Population AF databases (gnomAD) | Identifies common polymorphisms | Reduces false positives from population variants |
| BWA-MEM aligner | Maps sequencing reads to reference genome | Optimal for short-read Illumina data |
| Control DNA samples | Quality monitoring and cross-experiment standardization | Enables batch effect correction |
| Targeted capture panels | Enrichment for cancer-related genes | Enables deeper sequencing at lower cost |
| Reference genomes (GRCh38) | Standardized genomic coordinate system | Important to use same version across samples |
Clinical Implications and Therapeutic Applications
Mutational Signatures as Exposure Fingerprints
The patterns of somatic mutations in cancer genomes can serve as fingerprints of past mutagen exposures, providing powerful insights into cancer etiology [2]. For example, the comprehensive catalogue of somatic mutations from a malignant melanoma revealed a dominant mutational signature reflective of DNA damage due to ultraviolet light exposure, a known risk factor for this cancer type [2]. Specifically, the majority of somatic base substitutions were C>T/G>A transitions, with a high frequency of CC>TT/GG>AA dinucleotide changes—a pattern characteristic of ultraviolet light exposure [2].
Similar mutational signatures have been identified for tobacco smoke, aristolochic acid, and other environmental carcinogens, creating opportunities for connecting specific exposures to individual cancers [7]. This information is becoming increasingly valuable for understanding lifestyle factors that influence cancer risk, ultimately impacting clinical decisions and public health strategies [7].
Clonal Hematopoiesis and Cancer Predisposition
Recent sensitive sequencing approaches have revealed that as we age, many tissues become colonized by microscopic clones carrying somatic driver mutations [5]. Some of these clones represent a first step toward cancer, while others may contribute to ageing and other diseases [5]. In blood, targeted NanoSeq has identified 14 genes under positive selection, all known clonal hematopoiesis drivers, with approximately 11.9 non-synonymous mutations in these driver genes per donor [5]. This clonal architecture represents a premalignant state that significantly influences cancer risk and may contribute to non-malignant age-associated diseases.
Therapeutic Implications and Resistance Mechanisms
The identification of specific somatic mutations has direct implications for targeted therapy approaches:
- Oncogene-directed therapies: Drugs targeting specific driver mutations in genes like BRAF (V600E) in melanoma or EGFR mutations in lung cancer represent paradigm shifts in cancer treatment [2].
- Mutation order effects: The sequence in which mutations are acquired can influence response to targeted therapies, suggesting that therapeutic strategies may need to account for evolutionary history [4].
- Resistance mechanisms: The pool of genetically diverse subclones within tumors frequently serves as a source of therapy-resistant populations, necessitating combination approaches or evolutionary therapies [4].
Emerging Technologies and Approaches
The field of somatic mutation analysis continues to evolve rapidly, with several promising technological developments:
- Single-molecule sequencing: Methods like NanoSeq with error rates below 5×10^-9 errors per base pair enable accurate mutation detection from single DNA molecules, permitting quantification of mutation rates and signatures in any tissue [5].
- Multivariate regression models: These statistical approaches enable mutational epidemiology studies examining how exposures and cancer risk factors alter the acquisition or selection of somatic mutations [5].
- Integration of epigenetic analyses: Combining mutational data with information on epigenetic changes provides a more comprehensive view of the molecular alterations driving cancer [1].
Somatic mutations represent the fundamental drivers of cancer development, with their identification and interpretation being essential for both biological understanding and clinical application. The GATK somatic variant calling workflow and related bioinformatic pipelines provide robust frameworks for distinguishing true somatic variants from technical artifacts and germline polymorphisms. As detection technologies continue to improve, particularly with the advent of single-molecule sequencing methods, our appreciation of the complex clonal architecture of both tumors and normal tissues has deepened.
The biological and clinical significance of somatic mutations extends from their roles in oncogenesis and tumor evolution to their utility as biomarkers for targeted therapies and indicators of past mutagen exposures. For researchers and drug development professionals, accurate somatic variant calling remains an essential capability, enabling the discovery of new cancer genes, the identification of therapeutic targets, and the development of personalized treatment approaches based on the unique mutational profile of each patient's cancer. As we continue to refine our analytical methods and expand our understanding of mutation order effects, clonal evolution, and mutational signatures, somatic mutation analysis will undoubtedly remain central to cancer research and clinical oncology.
The Genome Analysis Toolkit (GATK) is a structured programming framework developed by the Broad Institute's Data Sciences Platform to enable efficient analysis of high-throughput sequencing data. Established as the industry standard for variant discovery, GATK provides a comprehensive software package with a primary focus on identifying SNPs, indels, and other genetic variants with exceptional accuracy and reliability. Its powerful processing engine and robust architecture make it capable of scaling from small-scale research projects to massive genomic initiatives like the 1000 Genomes Project and The Cancer Genome Atlas. GATK's status as the benchmark in genomic analysis is further solidified by its best practices workflows, which provide battle-tested protocols optimized for computational efficiency and reproducible results across diverse sequencing platforms and organisms.
Technical Architecture and Core Framework
GATK is built on a sophisticated technical foundation that separates data management from analytical algorithms, allowing researchers to focus on biological interpretation rather than computational challenges.
MapReduce Programming Paradigm
GATK implements a functional programming philosophy based on the MapReduce model, which divides computations into discrete, independent pieces that can be efficiently parallelized [8]. This architecture encompasses:
- Traversals: Data access patterns that prepare and present genomic data to analysis modules
- Walkers: Analysis modules that contain the map and reduce methods for processing data
- Data Bundles: Associated genomic information packaged by traversals for consumption by walkers
This separation of data access patterns from analysis algorithms enables the GATK core team to optimize the framework continuously for correctness, stability, and CPU/memory efficiency while providing automatic parallelization capabilities [8].
Supported Platforms and Requirements
GATK is designed to run on Linux and POSIX-compatible platforms, including MacOS X, with no native support for Windows systems [9]. The major system requirement is Java 1.8, with some tools having additional R or Python dependencies. For ease of deployment, the Broad Institute recommends using Docker containers, with an official image available on Dockerhub [9].
The framework has been engineered to support both traditional compute environments and modern cloud infrastructures, leveraging Apache Spark for parallelization and optimized cloud usage [9]. This flexibility allows researchers to scale analyses efficiently across local clusters or cloud platforms like AWS, Google Cloud, and Microsoft Azure [10].
Data Format Support and Compatibility
GATK utilizes the SAM/BAM format as the standard for representing sequencing reads, employing a production-quality SAM library for parsing and querying capabilities [8]. The toolkit has been extensively tested with data from multiple sequencing platforms, including:
- Illumina sequencing technology
- Applied Biosystems SOLiD System
- 454 Life Sciences (Roche)
- Complete Genomics
The framework supports BAM files with alignments from most next-generation sequence aligners and can accommodate various genotyping and variation formats, including VCF, GLF, and the GELI text format [8].
GATK as the Industry Standard
Historical Development and Adoption
GATK emerged to address the computational challenges posed by massive NGS datasets, with the original 2010 publication highlighting its capability to process the "nearly five terabases" of data from the 1000 Genomes Project pilot [8]. The toolkit's structured approach to genomic data analysis filled a critical development gap between sequencing output and biologically interpretable results.
The framework has evolved through multiple generations, with GATK4 representing a significant architectural redesign featuring:
- Apache Spark integration for distributed computing
- Cloud-native execution capabilities
- Expanded variant calling scope beyond germline variants
- Bundled Picard toolkit utilities with harmonized syntax [9]
Industry Partnerships and Ecosystem
GATK's position as the industry standard is reinforced through strategic collaborations with technology leaders, including:
- Intel: Co-development of Cromwell (workflow execution engine) and GenomicsDB (variant storage database) [10]
- Cloudera: Implementation of GATK4 on Apache Spark framework for accelerated genomic research [10]
- Cloud Providers: Partnerships with AWS, Google, IBM, and Microsoft to enable cloud-based GATK access via software-as-a-service models [10]
These collaborations have resulted in performance improvements of two orders of magnitude over previous GATK versions, enabling faster iterative analysis and propelling genomic innovation [10].
Licensing and Accessibility
GATK4 is distributed as open-source software under a BSD 3-clause "New" or "Revised" license [9], which permits:
- Commercial use
- Modification
- Distribution
- Private use
This permissive licensing model has facilitated widespread adoption across academic, clinical, and commercial environments, with over 31,000 registered users benefiting from the toolkit [10].
Somatic Variant Calling Workflow: Methods and Protocols
The GATK Best Practices workflow for somatic short variant discovery (SNVs + Indels) provides a comprehensive, battle-tested protocol for identifying somatic mutations in cancer genomics.
Input Requirements and Data Preparation
The somatic variant discovery workflow requires BAM files for each input tumor and normal sample, which must be pre-processed according to GATK Best Practices for data pre-processing [11]. Key input specifications include:
- Coordinate-sorted BAM files with proper indexing
- Reference genome in FASTA format with sequence dictionary and index
- Optional but recommended: Germline resource (e.g., gnomAD) and panel of normals (PoN)
Step-by-Step Experimental Protocol
Candidate Variant Calling with Mutect2
Purpose: Identify potential somatic SNVs and indels via local de novo assembly of haplotypes.
Methodology:
Technical Details: Mutect2 uses a Bayesian somatic genotyping model that differs from the original MuTect algorithm [12]. The tool performs:
- Local assembly of haplotypes in active regions showing evidence of variation
- Pair-HMM alignment of reads to candidate haplotypes to generate likelihood matrices
- Log odds calculation for alleles being somatic variants versus sequencing errors
Mutect2 can operate in three primary modes:
- Tumor-normal mode: For matched tumor-normal pairs (highest accuracy)
- Tumor-only mode: When only tumor sample is available
- Mitochondrial mode: Optimized for high-depth mitochondrial DNA analysis [12]
Contamination Estimation
Purpose: Estimate cross-sample contamination fraction for each tumor sample.
Tools: GetPileupSummaries, CalculateContamination
Methodology:
Technical Details: CalculateContamination is specifically designed to work without a matched normal, even in samples with significant copy number variation, and makes no assumptions about the number of contaminating samples [11].
Orientation Bias Artifact Modeling
Purpose: Model sequencing artifacts related to read orientation, particularly critical for FFPE tumor samples.
Tool: LearnReadOrientationModel
Methodology:
Technical Details: This step uses the F1R2 counts output from Mutect2 to learn prior probabilities of single-stranded substitution errors for each trinucleotide context prior to sequencing [11].
Variant Filtering
Purpose: Remove false positive calls resulting from correlated errors, alignment artifacts, and other technical artifacts.
Tool: FilterMutectCalls
Methodology:
Technical Details: FilterMutectCalls implements several filtering approaches:
- Hard filters for alignment artifacts
- Probabilistic models for strand and orientation bias artifacts
- Polymerase slippage artifact detection
- Germline variant and contamination filtering
- Bayesian model for overall SNV and indel mutation rate and allele fraction spectrum [11]
The tool automatically sets filtering thresholds to optimize the F score (harmonic mean of sensitivity and precision) [11].
Variant Annotation
Purpose: Add biological and clinical context to filtered somatic variants.
Tool: Funcotator
Methodology:
Technical Details: Funcotator adds comprehensive annotations including:
- Variant classifications (23 distinct types)
- Gene information (affected gene, protein change prediction)
- Database annotations from GENCODE, dbSNP, gnomAD, COSMIC, and other sources [11]
The tool supports both VCF and MAF output formats and features extensible, user-configurable data sources, including cloud-based resources via Google Cloud Storage [11].
Key Research Reagent Solutions
Table: Essential Computational Tools and Resources for GATK Somatic Variant Calling
| Resource Category | Specific Tool/Resource | Function and Purpose |
|---|---|---|
| Variant Caller | Mutect2 [11] [12] | Core somatic variant caller using Bayesian model and local assembly |
| Workflow Management | Cromwell [10] | Portable workflow execution engine for genomic pipelines |
| Variant Storage | GenomicsDB [10] | Optimized database for joint analysis of large variant datasets |
| Data Processing | Picard Toolkit [9] | Data manipulation and QC utilities bundled with GATK4 |
| Germline Resource | gnomAD [11] [12] | Population allele frequencies for germline variant filtering |
| Panel of Normals | CreateSomaticPanelOfNormals [11] | Aggregate normal samples to identify technical artifacts |
| Functional Annotation | Funcotator [11] | Comprehensive variant annotation with clinical and biological context |
| Quality Control | GetPileupSummaries/CalculateContamination [11] | Cross-sample contamination estimation and quality metrics |
Technical Specifications and Performance
GATK Architecture Components
Computational Performance and Scaling
GATK4 represents a significant advancement in computational efficiency through:
- Apache Spark Integration: Enables distributed memory parallelization across compute clusters [10]
- Cloud Optimization: Native support for cloud storage and computing environments [9]
- GenomicsDB Innovation: Provides unprecedented scalability for variant data storage and processing [10]
Benchmarking demonstrates that GATK4 running on Spark with Cloudera Enterprise can perform genomic analysis two orders of magnitude faster than previous GATK versions [10], dramatically reducing iteration time for research and development.
The Genome Analysis Toolkit maintains its position as the industry standard for genomic variant discovery through continuous innovation, comprehensive documentation, and robust community support. Its somatic variant calling workflow, centered on Mutect2 and complementary tools, provides researchers with a rigorously validated methodology for detecting cancer-associated mutations with high confidence. As genomic data continues to grow in scale and complexity, GATK's evolving architecture—with cloud-native execution, Spark-based parallelization, and optimized storage solutions—ensures it remains capable of addressing the computational challenges of modern biomedical research. The toolkit's permissive open-source license, extensive documentation, and integration with commercial and cloud platforms make advanced genomic analysis accessible to researchers across academia, clinical diagnostics, and pharmaceutical development.
The Genome Analysis Toolkit (GATK) Mutect2 is a sophisticated computational tool designed to identify somatic short variants—single nucleotide variants (SNVs) and insertions/deletions (indels)—in cancer sequencing data. Developed by the Broad Institute, Mutect2 represents a significant advancement over its predecessor by incorporating local assembly of haplotypes to detect somatic mutations in one or more tumor samples, with or without a matched normal sample [11] [13]. This capability is crucial for cancer research and drug development, as identifying tumor-specific mutations enables researchers to understand tumor biology, identify therapeutic targets, and guide personalized treatment strategies [14] [15].
In cancer genomics, somatic variant discovery presents unique challenges distinct from germline variant calling. Tumor samples often contain a mixture of cancerous and normal cells, leading to variable mutation allele frequencies. Additionally, artifacts from formalin-fixed, paraffin-embedded (FFPE) samples, tumor heterogeneity, and cross-sample contamination further complicate accurate variant detection [14] [16]. Mutect2 addresses these challenges through a comprehensive probabilistic framework that combines local assembly, powerful filtering, and artifact identification to distinguish true somatic variants from sequencing errors and technical artifacts.
Core Algorithm and Technical Architecture
Local Assembly and Variant Calling
Mutect2 employs an sophisticated approach to variant calling based on local de novo assembly of haplotypes, similar to the GATK HaplotypeCaller but optimized for somatic variant discovery. When Mutect2 encounters a genomic region showing evidence of variation, it performs the following key operations [11]:
- Local Assembly: Discards existing read alignment information and completely reassembles reads in the active region to generate candidate haplotypes. This assembly-based approach is particularly effective for detecting complex variants and indels that may be missed by alignment-based methods.
- Pair-HMM Alignment: Aligns each read to each candidate haplotype using the Pair-Hidden Markov Model (Pair-HMM) algorithm to generate a matrix of alignment likelihoods.
- Bayesian Somatic Likelihoods Model: Applies a specialized Bayesian model to calculate the log odds of alleles being true somatic variants versus sequencing errors. This model forms the statistical foundation for initial variant identification.
Advanced Filtering Framework
Following initial variant calling, Mutect2 employs a multi-tiered filtering approach to remove false positives through the FilterMutectCalls tool. The filtering framework has evolved significantly in recent versions, replacing multiple individual thresholds with a unified approach that filters based on a single quantity: the probability that a variant is a true somatic mutation [17]. Key aspects include:
- Automatic Threshold Optimization: FilterMutectCalls automatically determines the optimal filtering threshold to maximize the F-score (the harmonic mean of sensitivity and precision), eliminating the need for manual threshold tuning [17].
- Somatic Clustering Model: Implements a Dirichlet process binomial mixture model to account for subclonal allele fractions in the tumor. This model clusters variants by their allele fractions, helping to distinguish true somatic variants from artifacts based on their expected frequency distribution [17].
- Artifact-Specific Detection: Incorporates specialized probabilistic models to identify and filter strand bias artifacts, polymerase slippage artifacts, germline variants, and contamination.
Table 1: Key Technical Components of the Mutect2 Algorithm
| Component | Function | Technical Approach |
|---|---|---|
| Variant Calling | Identify candidate somatic variants | Local de novo assembly followed by Pair-HMM alignment and Bayesian likelihood calculation |
| Contamination Estimation | Estimate cross-sample contamination | GetPileupSummaries and CalculateContamination tools using common germline variants |
| Orientation Bias Correction | Correct for strand-specific artifacts | LearnReadOrientationModel using F1R2 counts from Mutect2 |
| Variant Filtering | Remove false positive calls | Unified probabilistic framework optimizing F-score with specialized artifact models |
Complete Mutect2 Workflow
The standard Mutect2 workflow consists of several interconnected steps, each addressing specific challenges in somatic variant discovery.
Figure 1: Complete GATK Mutect2 workflow for somatic variant discovery
Call Candidate Variants
The initial variant calling step uses Mutect2 with appropriate resources to generate raw candidate somatic variants:
Essential resources for this step include [11] [17] [18]:
- Reference genome: The standard reference sequence for alignment (e.g., GRCh38)
- Panel of Normals (PoN): A VCF file containing artifacts identified in normal samples, crucial for filtering systematic technical artifacts
- Germline resource: A population database (e.g., gnomAD) of common germline variants to help distinguish true somatic variants from rare germline polymorphisms
- Intervals: Genomic regions to target for variant calling (essential for exome and targeted sequencing)
Calculate Contamination
Cross-sample contamination can significantly impact variant calling accuracy, particularly for low-frequency variants. Mutect2 uses a two-step process to estimate and account for contamination [11] [18]:
Unlike other contamination tools, CalculateContamination is designed to work effectively even in samples with significant copy number variation and without matched normal samples [11].
Learn Orientation Bias Artifacts
Sequencing artifacts that occur on a single DNA strand before sequencing are common in FFPE samples and certain sequencing platforms. Mutect2 includes a specialized workflow to identify and correct these artifacts [17]:
This step uses the F1R2 counts output from Mutect2 to learn prior probabilities of single-stranded substitution errors for each trinucleotide context, which is particularly important for FFPE tumor samples and Illumina NovaSeq data [17].
Filter Variants
The FilterMutectCalls step integrates all available information to filter false positive variants:
This step accounts for correlated errors, alignment artifacts, strand and orientation bias, polymerase slippage, germline variants, and contamination. It also incorporates the learned somatic clustering model to refine variant probabilities based on the observed allele fraction spectrum in the tumor [11] [17].
Annotate Variants
Functional annotation is the final step in the workflow, adding biological context to the identified variants:
Funcotator adds gene-level information, variant classifications, protein change predictions, and annotations from various datasources including GENCODE, dbSNP, gnomAD, and COSMIC [11]. The output can be generated in either VCF or Mutation Annotation Format (MAF), facilitating integration with downstream analysis tools.
Performance Characteristics and Benchmarking
Understanding the performance characteristics of Mutect2 is essential for proper experimental design and data interpretation. Independent benchmarking studies have evaluated Mutect2 against other somatic variant callers across various sequencing depths and variant allele frequencies.
Table 2: Performance Comparison of Somatic Variant Callers at Different Mutation Frequencies
| Mutation Frequency | Sequencing Depth | Mutect2 F-Score | Strelka2 F-Score | Recommended Use Cases |
|---|---|---|---|---|
| ≥20% (High) | 200X | 0.94-0.95 | 0.95-0.96 | Standard tumor samples with high purity |
| 10-20% (Medium) | 300X | 0.90-0.94 | 0.89-0.93 | Mixed samples with stromal contamination |
| 5-10% (Low) | 500X | 0.65-0.95 | 0.64-0.94 | Low-purity tumors, subclonal mutations |
| 1% (Very Low) | 500-800X | 0.32-0.50 | 0.27-0.37 | Early-stage lesions, liquid biopsies |
Impact of Sequencing Depth and Mutation Frequency
Systematic evaluations reveal that sequencing depth and mutation frequency significantly impact variant calling performance [15]:
- For mutations with ≥20% allele frequency, sequencing depths of 200X are sufficient to detect >95% of variants with high precision (>95%)
- For lower frequency mutations (5-10%), increasing sequencing depth to 500X improves recall, but precision may decrease slightly
- At very low mutation frequencies (1%), even high sequencing depths (800X) yield limited recall (<50%), suggesting that improving experimental methods may be more effective than simply increasing sequencing depth
Comparison with Other Somatic Callers
When compared with other widely used somatic variant callers, Mutect2 demonstrates specific strengths and limitations [14] [15]:
- Mutect2 generally shows higher sensitivity for low-frequency variants compared to VarScan2 and Torrent Variant Caller, particularly in Ion Torrent sequencing data
- Compared to Strelka2, Mutect2 performs slightly better at lower mutation frequencies (<10%), while Strelka2 has a small advantage at higher frequencies (≥20%)
- Strelka2 typically executes 17-22 times faster than Mutect2, making it more suitable for time-sensitive clinical applications
- Mutect2 shows particularly strong performance in challenging samples such as FFPE specimens and those with significant contamination
Implementation Considerations
Computational Requirements and Scaling
The computational intensity of Mutect2 varies significantly based on sample type, sequencing depth, and genome size. For whole genome sequencing data, the workflow can be distributed across multiple computational nodes using scatter-gather approaches [17] [19]:
This parallelization strategy significantly reduces execution time for large datasets, though it requires careful management of intermediate files.
Panel of Normals Creation
A critical component for optimal Mutect2 performance is creating a panel of normals (PoN) specific to your sequencing platform and processing pipeline [17] [18]:
The PoN should ideally include 20-40 normal samples processed using the same experimental and computational methods as your tumor samples. Even an imperfect PoN is significantly better than no PoN, as it effectively captures systematic artifacts specific to your sequencing pipeline [18].
Tumor-Only Analysis
For samples without matched normal tissue, Mutect2 can operate in tumor-only mode with appropriate adjustments to filtering parameters. In this scenario, the germline resource and panel of normals become even more critical for distinguishing somatic variants from rare germline polymorphisms [14]. Additional caution is warranted for variants with allele frequencies around 50%, which likely represent heterozygous germline variants rather than somatic mutations.
Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for Mutect2 Analysis
| Resource Type | Specific Examples | Function in Workflow | Availability |
|---|---|---|---|
| Reference Genome | GRCh38, b37 | Baseline for read alignment and variant calling | GATK Resource Bundle |
| Germline Resource | af-only-gnomad.vcf | Population allele frequencies to filter common germline variants | GATK Resource Bundle |
| Panel of Normals | Laboratory-specific PoN | Identifies systematic technical artifacts | Created in-house from normal samples |
| Common Variants Set | chr17smallexaccommon3_grch38.vcf | Known common variants for contamination estimation | GATK Resource Bundle |
| Functional Annotation Data Sources | GENCODE, dbSNP, COSMIC, gnomAD | Provides biological context for identified variants | Funcotator Data Sources |
GATK Mutect2 provides a comprehensive, robust solution for somatic SNV and indel discovery in cancer genomics research. Its assembly-based approach, combined with sophisticated filtering for various artifact types, enables sensitive and specific detection of somatic variants across a wide range of allele frequencies and sample types. The complete workflow—from raw variant calling through contamination estimation, orientation bias correction, and functional annotation—provides researchers with a standardized method for generating high-quality somatic variant calls.
For optimal performance, researchers should ensure adequate sequencing depth based on expected mutation frequencies, create sequencing platform-specific panels of normals, and carefully interpret variants in the context of sample purity and potential technical artifacts. As cancer research increasingly focuses on subclonal mutations and low-frequency variants, proper implementation of the Mutect2 workflow becomes ever more critical for generating biologically meaningful results that can inform drug development and clinical decision-making.
The Importance of the GATK Best Practices Workflows for Reproducibility
In the field of genomics, reproducibility stands as a fundamental requirement for scientific validity, clinical application, and drug development. The Genome Analysis Toolkit (GATK) Best Practices provide the genomic community with standardized, rigorously validated workflows for variant discovery in high-throughput sequencing data. Developed and maintained by the Broad Institute, these workflows represent evolving methodologies that address the substantial technical challenges inherent in sequencing data analysis. For researchers, scientists, and drug development professionals, adherence to these practices is not merely a convenience but a critical factor in ensuring that results are reliable, comparable, and interpretable across different studies and institutions. This technical guide examines the structural components, performance benchmarks, and methodological frameworks that establish GATK Best Practices as a cornerstone for reproducible genomic research, with particular focus on somatic variant calling.
Understanding the GATK Best Practices Framework
Definition and Scope
The GATK Best Practices are defined as the reads-to-variants workflows used at the Broad Institute, providing step-by-step recommendations for variant discovery analysis [20]. These workflows are specifically tailored to particular applications and experimental designs, primarily focusing on:
- Whole genomes and exomes, with adaptability for gene panels and RNAseq [20]
- Different types of variation, including single nucleotide variants (SNVs), insertions/deletions (indels), and copy number variants (CNVs) [21] [22]
- Various experimental designs, from single samples to large cohorts [23]
A critical aspect of the framework is its acknowledgment of limitations. The Broad Institute explicitly notes that these workflows are developed and validated primarily for human data sequenced with Illumina technology, and adaptation may be required for other organisms or technologies [20].
The Three-Phase Analysis Structure
All GATK Best Practices workflows follow a consistent pattern comprising two or three main analysis phases, which creates a standardized approach that enhances reproducibility [20]:
Data Pre-processing: This initial phase converts raw sequence data (FASTQ or uBAM files) into analysis-ready BAM files through alignment to a reference genome and data cleanup operations to correct technical biases.
Variant Discovery: This core phase identifies genomic variation in one or more individuals and applies appropriate filtering methods. The output is typically in VCF format, though some variant classes like CNVs may use other structured formats [21] [20].
Additional Processing: Depending on the application, this may include filtering and annotation using resources of known variation, truth sets, and other metadata to assess and improve accuracy [20].
Quantitative Benchmarks: Establishing Performance Standards
Reproducibility requires not only standardized methods but also predictable performance. Independent benchmarking studies provide crucial quantitative data on the expected accuracy and efficiency of GATK workflows.
Performance of GATK-gCNV for Rare Variants
The GATK-gCNV algorithm enables sensitive discovery of rare copy number variants from sequencing read-depth information. Benchmarking against orthogonal technologies demonstrates its robust performance in exome sequencing data [21].
Table 1: Performance metrics for GATK-gCNV based on validation with 7,962 exomes from autism quartet families
| Metric | Performance | Validation Method |
|---|---|---|
| Recall of rare coding CNVs | Up to 95% | Matched WGS and microarray data |
| Resolution | >2 exons | 7,962 samples with matched WGS |
| Precision Validation Rate | 97% of de novo CNVs | Five orthogonal technologies |
| Application Scale | 197,306 individuals | UK Biobank exome dataset |
Comparative Benchmarking of Variant Callers
A comprehensive 2022 benchmark of state-of-the-art variant calling pipelines evaluated GATK alongside other tools across 14 "gold standard" genome datasets [24]. The study found that while actively developed tools like DeepVariant, Clair3, and Strelka2 also performed well, GATK maintained competitive performance, with accuracy depending more on the variant caller than the read aligner when using established aligners like BWA-MEM [24].
Computational Performance and Optimization
For reproducible workflows, consistent computational performance is essential for practical implementation. Research into GATK optimization reveals significant improvements across versions [25]:
Table 2: Computational performance comparisons between GATK versions
| Version | Speed Improvement | Recommended Use Case | Cost per Sample (Cloud) |
|---|---|---|---|
| GATK3.8 | 29.3% reduction in execution time | Time-critical situations; parallel processing on multiple nodes | $41.60 (4 c5.18xlarge instances) |
| GATK4 | 16.9% reduction in execution time | Cost-effective routine analyses; large population studies | $2.60 (single c5.18xlarge instance) |
GATK4's architecture makes it particularly suitable for large-scale studies, with the ability to process approximately 1.18 samples per hour on a single cloud instance, enabling population-scale analyses while maintaining reproducibility [25].
Experimental Protocols: Implementing Reproducible Workflows
The Somatic Variant Calling Workflow with Mutect2
For somatic mutation detection in cancer research, GATK's Mutect2 represents the industry standard for identifying tumor-specific variants [22]. The detailed protocol ensures reproducible detection of low-frequency variants present in as few as 5% of reads.
Diagram 1: Somatic variant calling workflow with Mutect2
The Mutect2 workflow employs several critical reference resources to ensure accurate variant detection:
- Panel of Normals (PON): Contains common technical artifacts observed across many normal samples, enabling their subtraction from tumor samples [22]
- Germline Resource: Population allele frequencies from gnomAD help distinguish somatic from germline variants [22]
- Common Variants: Used for contamination estimation between tumor-normal pairs [22]
The matched tumor-normal approach provides maximum specificity by eliminating patient-specific germline variants that would otherwise appear as false positive somatic mutations [22].
Germline Short Variant Calling Workflow
The germline variant calling workflow follows a structured three-stage process that emphasizes data quality and joint calling across samples [23]:
Diagram 2: Germline short variant discovery workflow
The germline workflow incorporates several sophisticated statistical models that enhance reproducibility:
- Base Quality Score Recalibration (BQSR): Corrects systematic errors in base quality scores assigned by sequencers, using covariates encoded in read groups [23]
- Joint Calling: Leverages data from multiple samples to improve genotype inference sensitivity, boost statistical power, and reduce technical artifacts [23]
- Variant Quality Score Recalibration (VQSR): Employs Gaussian mixture models to classify variants based on annotation values clustering using training sets of high-confidence variants [23]
Reproducible implementation of GATK workflows requires specific reference files and computational resources. The following table details these essential components:
Table 3: Essential research reagents and resources for GATK workflows
| Resource Type | Specific Examples | Function in Workflow |
|---|---|---|
| Reference Genome | GRCh37, GRCh38 (hg38) | Standardized coordinate system for read alignment and variant calling [22] [24] |
| Known Sites Resources | dbSNP, gnomAD, 1000 Genomes | Population allele frequencies for distinguishing somatic from germline variants [26] [22] |
| Panel of Normals (PON) | 1000g_pon.hg38.vcf.gz | Database of technical artifacts observed across normal samples [22] |
| Contamination Resources | smallexaccommon_3.hg38.vcf.gz | Common variants for estimating cross-sample contamination [22] |
| High-Confidence Callsets | Genome in a Bottle (GIAB) | Gold-standard truth sets for validation and benchmarking [24] |
Implementation and Validation Strategies
Computational Infrastructure Recommendations
Deploying GATK workflows with reproducible performance requires appropriate computational infrastructure. Based on systematic testing, the following configurations are recommended [27]:
- CPU Architecture: x86_64 with minimum AVX instruction set for PairHMM library acceleration, with AVX2 recommended for Smith-Waterman acceleration [27]
- Memory Allocation: Sensible heap sizes (4-8GB) to balance Java memory management with native library requirements [27]
- Storage Solutions: Fast storage systems to mitigate I/O bottlenecks, with CRAM format recommended for efficient data compression [27]
Validation and Concordance Assessment
The GATK Concordance tool provides quantitative measures of reproducibility by comparing variant calls against truth sets [28]. Key metrics include:
- True Positives (TP): Variants correctly identified in both test and truth datasets
- False Positives (FP): Variants identified in test data but absent in truth set
- False Negatives (FN): Variants present in truth set but missed in test data
- Recall and Precision: Calculated as TP/(TP+FN) and TP/(TP+FP) respectively [28]
Proper interpretation of these metrics requires ensuring that compared variant sets are on the same genome build, as build discrepancies can artificially inflate false positive and false negative counts [28].
The GATK Best Practices workflows represent an essential framework for achieving reproducibility in genomic variant discovery. Through standardized methodologies, comprehensive benchmarking, detailed experimental protocols, and validated computational infrastructure, these workflows provide the scientific community with a reliable foundation for both research and clinical applications. The continued evolution of these practices—including the recent development of tools like GATK-gCNV for copy number variant detection and Mutect2 for somatic analysis—ensures that the framework adapts to new challenges while maintaining the rigorous standards required for reproducible science. For drug development professionals and researchers, adherence to these practices provides the consistency and reliability necessary to translate genomic discoveries into meaningful clinical applications.
The identification of somatic variants using the GATK framework requires a carefully curated set of input files and resources. Unlike germline variant discovery, somatic calling presents unique challenges, including the need to distinguish true somatic events from technical artifacts and germline variants present at low allele fractions. The GATK somatic short variant discovery workflow is specifically designed to identify somatic short variants (SNVs and Indels) in one or more tumor samples from a single individual, with or without a matched normal sample [11]. Success in this endeavor hinges on the quality and appropriateness of core inputs—primarily pre-processed BAM files, germline reference resources, and a Panel of Normals (PoN). These elements form the foundational layer upon which the variant calling process is built, directly impacting the sensitivity and specificity of the final results. Proper understanding and preparation of these prerequisites are essential for researchers, scientists, and drug development professionals aiming to generate reliable data for downstream analysis in cancer genomics.
Core Input Requirements: The Essential Components
Primary Input: Analysis-Ready BAM Files
The primary input for the GATK somatic variant discovery workflow is sequence data in BAM file format. The workflow requires BAM files for each input tumor and, if available, matched normal sample [11]. It is critical that these input BAMs have undergone comprehensive pre-processing as described in the GATK Best Practices for data pre-processing. This pre-processing typically includes steps such as alignment to a reference genome, duplicate marking, base quality score recalibration (BQSR), and indel realignment, which collectively ensure that the data is in an optimal state for variant calling. The matched normal sample, typically derived from healthy tissue (often blood), serves as a crucial control to identify germline variants present in the patient and filter them from the final somatic callset.
Beyond the primary sequence data, the somatic workflow depends on several key resources to function accurately and effectively. The requirements for these resources vary based on the specific use case and availability of a matched normal sample.
Table: Core Resource Requirements for Somatic Variant Calling
| Resource Type | Mandatory/Optional | Description & Purpose | Use Case |
|---|---|---|---|
| Reference Genome | Mandatory | A FASTA file of the reference sequence (e.g., GRCh38, b37/hg19) and its associated index (fai) and dictionary (dict) files. |
Universal; required for all analysis steps. |
| Germline Resource | Highly Recommended | A sites-only VCF containing population allele frequencies (e.g., gnomAD). Used to classify variants as potential germline events. | Essential for tumor-only mode; highly beneficial for tumor-normal pairs. |
| Panel of Normals (PoN) | Highly Recommended | A sites-only VCF of artifacts common across a set of normal samples. Filters recurrent technical artifacts. | Universal; significantly reduces false positives. |
| Matched Normal BAM | Conditional | A BAM file from a normal tissue of the same individual. The most effective resource for removing patient-specific germline variants. | Tumor-Normal Pair mode; not used in Tumor-Only mode. |
The Panel of Normals (PoN): A Cornerstone for Artifact Filtering
Definition and Purpose of a PoN
A Panel of Normals (PoN) is a critical resource in somatic variant analysis whose primary purpose is to capture recurrent technical artifacts present in sequencing data [29]. It is created from a collection of normal samples—derived from healthy tissue believed to be free of somatic alterations—that have been processed using the same experimental and analytical protocols intended for the tumor samples. The PoN works by identifying sites where variants consistently appear across multiple normal samples. Since these normals are from healthy tissue, true somatic variants should not be present. Therefore, variants appearing in the PoN are classified as systematic errors—arising from factors like sequencing chemistry, mapping ambiguities, or other platform-specific biases—and are filtered out from the tumor sample results. This process dramatically increases the specificity of the final somatic callset by removing false positives that would otherwise appear as recurrent artifacts across multiple samples in a cohort.
Construction and Best Practices for PoNs
Creating an effective Panel of Normals is a multi-step process that requires careful sample selection and processing. The established method for short variants involves running Mutect2 on each normal sample individually and then combining the results [29] [17].
Table: Methodology for Creating a Short Variant Panel of Normals
| Step | Tool | Action | Key Parameters |
|---|---|---|---|
| 1. Call Variants on Normals | Mutect2 |
Run in tumor-only mode on each normal BAM. | --max-mnp-distance 0 |
| 2. Consolidate Calls | GenomicsDBImport |
Import the per-normal VCFs into a GenomicsDB datastore. | Interval list is required. |
| 3. Create PoN | CreateSomaticPanelOfNormals |
Generate the final sites-only VCF from the datastore. | Optional: --germline-resource to exclude germline sites. |
Several best practices govern the creation and use of a PoN. The most important selection criterion for normals is technical similarity to the tumor samples; they should come from the same exome or genome preparation methods, sequencing technology, and analysis software versions [29]. Biologically, samples should ideally come from young, healthy subjects to minimize the chance of including an undiagnosed tumor. While there is no absolute minimum, a PoN with at least 40 samples is recommended in practice, with larger panels (hundreds of samples) providing greater power to identify rare artifacts [29]. For researchers without the resources to build a custom PoN, the GATK team provides public panels of normals for common reference genomes (e.g., hg38 and b37) as part of the GATK resource bundle, which can be directly used or serve as a starting point [29].
In tumor-only analyses, where a matched normal sample is unavailable, a germline resource becomes essential. This resource is a sites-only VCF containing population allele frequencies, such as a subset of gnomAD [17] [30]. Mutect2 uses the allele frequencies from this resource to calculate the prior probability that a variant is germline, allowing it to filter out common germline polymorphisms that would otherwise appear as false positive somatic calls [31]. When a matched normal is available, the germline resource still provides valuable power to distinguish somatic from germline variants, especially in regions with low coverage in the normal sample.
The reference genome is a mandatory input for all steps in the workflow, from variant calling with Mutect2 to filtering and annotation [17] [30]. The GATK resource bundle provides reference sequences and a wide array of supporting files for common human genome builds like GRCh38/hg38 and b37/hg19 [30]. These bundles include reference FASTA files, index files, known variant sites from projects like dbSNP and the 1000 Genomes Project, which are used for base quality recalibration in pre-processing. While the bundle is hosted on Google Cloud storage, its files can be accessed via a web browser with a Google account or downloaded for local analysis.
Diagram 1: A simplified workflow of the GATK4 Mutect2 somatic variant calling pipeline, showing the integration points for its key inputs, including BAM files, reference genome, germline resource, and Panel of Normals.
Table: Essential Materials and Resources for GATK Somatic Variant Calling
| Item | Category | Critical Function | Example/Format |
|---|---|---|---|
| Pre-processed BAM Files | Primary Data | Analysis-ready aligned sequences for tumor and (optionally) matched normal samples. | Coordinate-sorted, duplicate-marked, BQSR-applied BAM. |
| Reference Genome | Core Reference | Standard sequence for read alignment and variant comparison. | GRCh38 or b37 FASTA file with .fai and .dict indices. |
| Germline Resource VCF | Filtering Resource | Provides population allele frequencies to flag common germline variants. | gnomAD subset (sites-only VCF). |
| Panel of Normals (PoN) | Filtering Resource | Captures recurrent technical artifacts to remove systematic false positives. | Sites-only VCF generated from normal samples. |
| Known Sites Resources | Pre-processing | Used for Base Quality Score Recalibration (BQSR) during data pre-processing. | dbSNP, Mills indels, 1000G indels (VCF format) [30]. |
The reliability of a somatic variant analysis is fundamentally determined by the quality and appropriateness of its inputs. The journey from raw BAM files to a confident somatic callset is underpinned by several non-negotiable prerequisites: meticulously pre-processed sequence data, a high-quality reference genome, and robust filtering resources like a Panel of Normals and a germline resource. These components work in concert to empower the GATK Mutect2 workflow to distinguish true somatic mutations from the vast background of technical artifacts and inherited germline variation. For researchers in drug development and clinical science, a rigorous approach to gathering these inputs is not merely a technical formality but a critical step in ensuring that subsequent biological insights and therapeutic decisions are based on a foundation of accurate genomic data.
A Step-by-Step Guide to the Mutect2 Somatic Calling Pipeline
The Genome Analysis Toolkit (GATK) somatic variant calling workflow represents the industry standard for identifying somatic single nucleotide variants (SNVs) and insertions/deletions (indels) in cancer genomics research [9]. This end-to-end pipeline transforms raw sequencing data from tumor and normal samples into a high-confidence set of annotated somatic variants suitable for downstream biological interpretation. The GATK Best Practices for somatic short variant discovery provide a battle-tested framework that optimizes the balance between sensitivity (finding true variants) and specificity (excluding false positives) [32] [20]. This technical guide details the core components, experimental protocols, and analytical methodologies comprising a production-grade somatic variant calling workflow, providing researchers and drug development professionals with the foundational knowledge required to implement robust variant detection in cancer studies.
Core Workflow Components and Process
The GATK somatic variant discovery workflow follows a structured, multi-phase approach that systematically processes sequencing data through quality control, alignment, preprocessing, variant calling, and filtration stages [20]. Each phase employs specialized tools and methodologies designed to address specific technical challenges in somatic variant detection.
Table 1: Core Workflow Phases in Somatic Variant Discovery
| Phase | Primary Input | Primary Output | Key Tools | Main Purpose |
|---|---|---|---|---|
| Data Pre-processing | Raw FASTQ files | Analysis-ready BAM files | BWA, Picard, GATK | Convert raw reads into aligned, quality-processed data [33] [34] |
| Variant Discovery | Analysis-ready BAMs | Raw variant calls (VCF) | Mutect2 | Identify potential somatic variants [12] |
| Filtering & Annotation | Raw variant calls | Filtered, annotated VCF | FilterMutectCalls, Funcotator | Remove false positives and add biological context [12] [17] |
The computational foundation of this workflow leverages an industrial-strength infrastructure and engine that handles data access, conversion, and traversal, with high-performance computing features including parallelization using Apache Spark and optimized usage of cloud infrastructure [9]. This engineering enables the processing of projects of any scale, from targeted gene panels to whole genomes.
Figure 1: Comprehensive Somatic Variant Calling Workflow. The end-to-end process transforms raw sequencing data into filtered, annotated variants using a structured, multi-phase approach with integrated quality control and external resources.
Successful implementation of the somatic variant calling workflow requires both computational tools and carefully curated biological resources. These components work in concert to distinguish true somatic mutations from technical artifacts and germline variation.
Table 2: Essential Research Reagents and Computational Resources
| Resource Category | Specific Examples | Purpose & Function | Implementation Notes |
|---|---|---|---|
| Reference Genome | GRCh37, GRCh38 | Standardized coordinate system for read alignment and variant reporting | Must include sequence, dictionary, and index files [34] |
| Germline Resource | gnomAD, 1000 Genomes | Provides population allele frequencies to distinguish somatic from germline variants | VCF format with population AF annotations; critical for tumor-only mode [12] [34] |
| Panel of Normals (PON) | Broad Institute PON, custom-built PON | Identifies systematic technical artifacts present in normal samples | Created from Mutect2 calls on multiple normal samples; sites present in ≥2 samples included [17] [34] |
| Known Variants | dbSNP, COSMIC | Context for variant interpretation and filtering | Helps distinguish common polymorphisms from potential somatic events [34] |
| Intervals/Targets | WGS, WES, panel intervals | Defines genomic regions for analysis in targeted sequencing | BED or interval list format; essential for exome/panel sequencing [34] |
The germline resource and Panel of Normals serve distinct but complementary functions. The germline resource contains population frequency data for common polymorphisms, enabling Mutect2 to distinguish potential somatic mutations from inherited variants [12]. In contrast, the Panel of Normals specifically captures systematic technical artifacts that arise from sequencing methodologies, mapping biases, or sample preparation protocols [34]. When constructing a custom Panel of Normals, the GATK Best Practices workflow recommends running Mutect2 in tumor-only mode on each normal sample, then combining the results using GenomicsDBImport and CreateSomaticPanelOfNormals to generate a sites-only VCF suitable for reuse [17].
Detailed Experimental Protocols and Methodologies
Data Pre-processing and Quality Control
The initial phase transforms raw sequencing data into analysis-ready BAM files through a series of computational steps that improve data quality and mitigate technical artifacts:
- Read Alignment: Process raw FASTQ files using BWA-MEM to align sequencing reads to the reference genome [33] [34]. This tool efficiently handles the large volumes of sequencing data generated by modern instruments while accounting for genetic variation.
- Duplicate Marking: Identify and flag PCR duplicates using Picard MarkDuplicates to prevent artificial inflation of variant supporting reads [33]. PCR duplicates typically represent 5-15% of sequencing reads in a standard exome capture [35].
- Base Quality Score Recalibration (BQSR): Apply GATK's BQSR to empirically adjust base quality scores using an established error model [33]. This step corrects for systematic technical errors in base quality scores, though evaluations suggest improvements may be marginal for modern sequencing technologies [35] [34].
- Quality Control Metrics: Generate comprehensive QC reports using tools such as Picard and VerifyBAMID to confirm sample identity, check for contamination, and verify that sufficient sequencing coverage was achieved [35]. For paired tumor-normal samples, confirm expected genetic relationships using the KING algorithm [35].
Somatic Variant Calling with Mutect2
The Mutect2 algorithm represents the core somatic variant discovery engine in the GATK workflow. Unlike germline callers that assume fixed ploidy, Mutect2 employs a Bayesian somatic genotyping model that accommodates the varying allele fractions characteristic of tumor samples due to factors such as tumor purity, subclonality, and copy number alterations [12] [31].
Fundamental Mutect2 Execution Modes:
Tumor with Matched Normal (Recommended):
This mode provides the highest specificity by leveraging the matched normal to directly identify and filter germline variants [12].
Tumor-Only Mode:
Tumor-only analysis requires both a germline resource and Panel of Normals to compensate for the absence of a matched normal, though it produces higher false positive rates [12].
Mitochondrial Mode:
The
--mitochondriaflag automatically optimizes parameters for calling variants in mitochondrial DNA, including setting appropriate initial LOD thresholds and allele frequency priors [12].
Advanced Filtering Strategies
Post-calling filtration represents a critical step in the somatic workflow, leveraging multiple orthogonal approaches to remove false positives while retaining true biological variants:
Read Orientation Artifact Filtering: For samples susceptible to strand-specific artifacts (e.g., FFPE tumors or NovaSeq data), implement a three-step orientation bias filter [17]:
- Step 1: Generate F1R2 counts during Mutect2 execution using the
--f1r2-tar-gzargument - Step 2: Learn the orientation model using
LearnReadOrientationModel - Step 3: Apply the learned model during filtering with
FilterMutectCalls --ob-priors
- Step 1: Generate F1R2 counts during Mutect2 execution using the
Contamination Estimation: Calculate cross-sample contamination using GetPileupSummaries and CalculateContamination [17]. This is particularly important for large sequencing batches where sample mixing may occur.
Probabilistic Filtering: FilterMutectCalls employs a unified probabilistic framework that calculates the probability each variant is a true somatic mutation [17]. The tool automatically determines the optimal threshold that maximizes the F-score (harmonic mean of sensitivity and precision), though users can adjust the sensitivity-precision balance using the
-f-score-betaparameter.
Technical Considerations and Benchmarking
Computational Requirements and Optimization
Somatic variant calling represents a computationally intensive process, with a typical whole-genome sequencing sample requiring approximately 84.5 hours on a 36-core machine using standard implementation [33]. Several strategies can significantly improve computational efficiency:
- Parallelization Frameworks: Leverage Spark-based implementations such as Halvade Somatic to distribute workload across multiple cores and nodes, reducing runtime by 4.3× on a single node and up to 62.4× across multiple nodes [33].
- Scattering by Genomic Region: Execute Mutect2 concurrently across multiple genomic intervals, then merge results using MergeMutectStats [17].
- Cloud-Native Implementation: Utilize WDL workflows with Cromwell execution engine for portable, scalable analysis across cloud and local compute environments [20].
Performance Validation and Benchmarking
Rigorous validation using established benchmarking resources represents a critical component of clinical-grade variant calling [35]. Key benchmarking resources include:
- Genome in a Bottle (GIAB): Provides consensus "ground truth" variant calls for reference samples, though primarily focused on germline variation [35].
- Synthetic Diploid (Syndip): Offers less biased benchmarking across challenging genomic regions through synthetic datasets with known variant positions [35].
- Tumor-Normal Benchmark Sets: Utilize well-characterized cancer cell lines with orthogonal validation to establish sensitivity and specificity metrics.
When evaluating pipeline performance, employ sophisticated comparison tools that account for subtle differences in variant representation and focus analysis on high-confidence genomic regions where benchmark truth sets are most reliable [35].
The GATK somatic variant calling workflow provides a comprehensive, production-grade framework for detecting cancer-associated mutations from high-throughput sequencing data. Through its structured approach encompassing data pre-processing, variant discovery, and sophisticated filtering, the workflow achieves an optimal balance between sensitivity for true somatic variants and specificity against technical artifacts and germline polymorphisms. The integration of external resources such as germline databases and panels of normals further enhances the biological accuracy of the resulting variant calls. As sequencing technologies continue to evolve and new capabilities such as long-read single-molecule sequencing emerge, the fundamental principles underlying this workflow—rigorous quality control, appropriate use of reference resources, and comprehensive validation—will remain essential components of reliable somatic variant discovery in both research and clinical settings.
In the context of cancer genomics, identifying somatic mutations—acquired variants present in tumor cells but absent from the germline—is fundamental for understanding tumorigenesis, progression, and therapeutic targeting. The Genome Analysis Toolkit's (GATK) Mutect2 is a specialized tool designed to call somatic short mutations (single nucleotide variants and indels) via local assembly of haplotypes [12]. Its operation differs fundamentally from germline variant callers, as it employs a Bayesian somatic genotyping model tailored to the unique characteristics of cancer genomes, such as variable purity, heterogeneous subclones, and aneuploidy [31]. This technical guide details the core algorithmic step of calling candidate variants, which forms the foundation of the GATK somatic short variant discovery workflow [11].
The Core Algorithm: Local Re-assembly of Haplotypes
Mutect2, like the HaplotypeCaller, operates on the principle that accurate variant discovery, especially for indels in repetitive regions, requires moving beyond the initial read alignments. It does this by performing local de-novo assembly of haplotypes within genomic regions that show evidence of variation, known as ActiveRegions [36] [11]. This process allows Mutect2 to reconstruct the true biological sequences present in the sample, leading to superior accuracy compared to methods that rely solely on pre-aligned reads.
The following diagram illustrates the multi-stage workflow of the local assembly and variant evaluation process.
Reference Graph Assembly
The process begins by constructing a reference assembly graph, which is a directed DeBruijn graph [36]. The reference sequence for the ActiveRegion is decomposed into a succession of kmers (small sequence subunits 'k' bases long), where each kmer overlaps the previous one by k-1 bases. This creates a graph where nodes represent unique kmers and edges represent the sequential relationship between them. By default, Mutect2 builds two separate graphs using kmer sizes of 10 and 25, then selects the most effective one [36]. A hash table of unique kmers is also built to track node positions.
Threading Reads and Graph Refinement
Once the initial graph is built, each read is "threaded" through it [36]. The algorithm compares each read's kmers against the hash table. Matching kmers are mapped to their existing nodes, while new, unique kmers (indicating potential variation) are added as new nodes. Edges are created or reinforced based on read support; the weight of an edge is increased by one for every read that supports that connection. After all reads are processed, the graph undergoes pruning to remove sections supported by very few reads (default: fewer than 2 reads), which are likely stochastic sequencing errors [36]. This step is crucial for reducing noise and maintaining computational efficiency.
Haplotype Determination and Site Identification
After pruning, the program identifies the most plausible haplotypes by traversing all possible paths in the cleaned graph [36]. Each path's likelihood is scored based on the transition probabilities of its edges, which are derived from the supporting read counts. To limit computational load, Mutect2 selects a maximum number of the best-scoring haplotypes (default: 128 per kmer size) [36]. Finally, each candidate haplotype is aligned to the original reference sequence using the Smith-Waterman algorithm to reconstruct a CIGAR string and identify the precise locations of potential variants (SNVs and indels) relative to the reference [36]. This yields a super-set of candidate variant sites for the final step.
Evaluating Evidence for Candidate Variants
The list of candidate haplotypes and sites must now be rigorously evaluated against the read data. This is done using the PairHMM (Pair Hidden Markov Model) algorithm [37].
Mutect2 aligns each individual read to every candidate haplotype (including the reference) using the PairHMM, which produces a likelihood score expressing the probability of observing the read given a specific haplotype, while accounting for base quality scores [37]. This generates a matrix of per-read likelihoods for each haplotype. These haplotype-level likelihoods are then marginalized over individual alleles to produce per-read likelihoods for each candidate allele at a given site [37]. For each read and each candidate allele, the algorithm selects the highest likelihood among all haplotypes supporting that allele. The total evidence for an allele is the product of all per-read likelihoods supporting it. Sites with sufficient evidence for at least one non-reference allele are passed on as candidate somatic variants [37].
Key Parameters and Research Reagents
The following table summarizes the function of key resources and parameters used in a standard Mutect2 experiment, which are essential for achieving high-specificity somatic calls [12] [17].
Table 1: Essential research reagents and parameters for Mutect2 analysis
| Item | Type | Function / Purpose |
|---|---|---|
| Reference Genome | Reagent | A reference sequence in FASTA format and its index, used for read alignment and variant calling [12]. |
| BAM Files | Input Data | Processed sequence data from the tumor and (if available) matched normal sample(s). Input BAMs should be pre-processed as per GATK Best Practices [11]. |
| Germline Resource | Reagent | A VCF of common germline variants (e.g., gnomAD) used to distinguish common germline polymorphisms from true somatic mutations [12] [31]. |
| Panel of Normals (PoN) | Reagent | A VCF of artifacts and common germline variants found in a cohort of normal samples, used to filter systematic technical artifacts [12] [17]. |
--af-of-alleles-not-in-resource |
Parameter | The prior allele frequency imputed for alleles absent from the germline resource. Dynamically adjusted based on calling mode (e.g., tumor-only defaults to 5e-8) [12]. |
--minPruning |
Parameter | Minimum number of supporting reads to retain a graph path during pruning (default: 2). Increasing improves speed/specificity but decreases sensitivity [36]. |
-maxNumHaplotypesInPopulation |
Parameter | Maximum number of haplotypes to consider per graph (default: 128). Used to limit computational complexity [36]. |
Advanced Considerations and Methodological Variations
Modes of Operation
Mutect2 supports several operational modes, each requiring specific inputs and parameter adjustments [12]:
- Tumor-Normal Mode: The standard and recommended approach. A matched normal sample from the same individual is provided, allowing Mutect2 to perform an early filter of obvious germline events, saving computational resources and improving specificity [12].
- Tumor-Only Mode: Runs on a single sample. While possible, it produces a higher rate of false positives from undiscovered germline variants and requires a Panel of Normals and germline resource for basic filtering [12] [38]. It is also used for creating the PoN.
- Mitochondrial Mode: Activated with the
--mitochondriaflag, this mode automatically adjusts key parameters (e.g., sets initial LOD thresholds to 0) for the high-depth, unique genetics of mitochondrial DNA [12]. - Force-Calling Mode: Instructs Mutect2 to call all alleles specified in a provided VCF in addition to any it discovers de novo, useful for targeted analyses [12].
Distinction from Germline Calling
It is critical to understand that Mutect2 is not simply a modified germline caller. While it shares the assembly and haplotype determination engine with HaplotypeCaller, its core genotyping model is fundamentally different [31]. HaplotypeCaller uses a ploidy-based model to calculate genotype likelihoods, making it ideal for germline variants. In contrast, Mutect2 uses a Bayesian model to calculate the likelihood that a variant is somatic, which does not assume fixed ploidy and is designed to handle the variable allele fractions common in tumors due to sample impurity and subclonality [31]. Using the wrong tool for a given use case will lead to suboptimal results.
Integration with the Broader Somatic Workflow
Calling candidate variants with Mutect2 is only the first main step in the somatic discovery workflow [11]. The raw VCF output from Mutect2 requires further filtering to remove residual artifacts. The subsequent step, FilterMutectCalls, employs probabilistic models to account for correlated errors, strand bias, germline events, and contamination [17] [11]. Furthermore, additional preprocessing steps like LearnReadOrientationModel (for FFPE and other orientation bias artifacts) and CalculateContamination are often essential components of a robust somatic variant discovery pipeline [17] [11]. For a complete experimental protocol, these downstream steps are indispensable for generating a high-confidence somatic callset.
In somatic variant calling, the accurate detection of true positive mutations is fundamentally compromised by two major technical challenges: sequencing artifacts and sample cross-contamination. Artifacts, such as those introduced by formalin-fixed paraffin-embedded (FFPE) sample processing or specific sequencing platforms, generate false variant signals that mimic true somatic mutations [17] [39]. Simultaneously, cross-sample contamination, where DNA from multiple samples mixes during processing, can obscure true variants and introduce false positives [40]. Without robust computational correction, these factors severely compromise the integrity of variant calls, leading to biologically misleading results. Within the GATK Mutect2 best practices workflow, three specialized tools—LearnReadOrientationModel, GetPileupSummaries, and CalculateContamination—form an essential pipeline for quantifying and filtering these technical noises [41] [17]. This guide provides an in-depth technical examination of their operation, integration, and critical parameters, enabling researchers to achieve the high-precision variant calling required for reliable cancer genomics and therapeutic development.
Tool Theoretical Foundations and Methodologies
LearnReadOrientationModel: Quantifying Strand Bias Artifacts
LearnReadOrientationModel addresses a specific category of sequencing artifacts characterized by substitution errors that occur on a single DNA strand before sequencing. These artifacts are particularly prevalent in FFPE-treated tumor samples and data from Illumina NovaSeq platforms [17] [39]. The tool operates by analyzing raw data generated during the Mutect2 calling step to build a probabilistic model of these strand-specific errors.
The methodology begins during variant calling, where Mutect2 is executed with the --f1r2-tar-gz parameter to produce a file containing raw count data for read orientations [17] [39]. This file encapsulates the distribution of forward and reverse reads supporting reference and alternate alleles across different sequence contexts. LearnReadOrientationModel then processes this data using an expectation-maximization (EM) algorithm to estimate the prior probabilities of observing strand-specific artifacts [42]. The number of EM iterations can be controlled via the --num-em-iterations parameter to ensure model convergence [42]. The output is a model file that FilterMutectCalls subsequently uses to identify and filter variants likely explained by these orientation biases, thereby preventing false positives arising from technical artifacts rather than biological mutations.
GetPileupSummaries & CalculateContamination: Estimating Cross-Sample Contamination
The GetPileupSummaries and CalculateContamination tools work in tandem to estimate the fraction of reads originating from cross-sample contamination, a critical quality metric in somatic calling [40].
GetPileupSummaries functions by tabulating pileup metrics at a predefined set of known variant sites, typically from a resource like a small common ExAC subset [17] [42]. It processes a BAM file and a VCF of known sites, summarizing read support (reference and alternate counts) across these positions. This summary provides the foundational data for contamination inference.
CalculateContamination employs a methodology borrowed from ContEst but with significant enhancements [40]. It estimates contamination by analyzing the presence of reference reads at homozygous alternate sites. The key innovation lies in relaxing the assumptions of a diploid genotype with no copy number variation and independent contaminating reads, making it robust in the presence of CNVs and an arbitrary number of contaminating samples [40]. The tool can operate in both tumor-only and matched-normal modes, with the latter providing improved accuracy by incorporating a matched normal BAM file [40]. The output is a contamination table that quantifies the contamination fraction, which FilterMutectCalls uses to adjust genotype likelihoods and filter suspect variants.
Table 1: Core Methodological Principles of Artifact and Contamination Tools
| Tool Name | Primary Input | Core Algorithm/Method | Primary Output |
|---|---|---|---|
| LearnReadOrientationModel | F1R2 count tar.gz file from Mutect2 | Expectation-Maximization (EM) for learning priors | Tar.gz file containing read orientation bias model |
| GetPileupSummaries | BAM file + VCF of known common sites | Pileup summarization at known polymorphic sites | Table of pileup statistics |
| CalculateContamination | Pileup summary table | Estimation from ref reads at hom-alt sites; robust to CNVs | Table containing contamination fraction |
Integrated Workflow and Experimental Protocol
The three tools function not in isolation but as sequentially dependent components within the broader Mutect2 somatic variant calling workflow. The following diagram and protocol detail their exact integration and execution.
Step-by-Step Execution Protocol
Generate Raw Data for Artifact Analysis with Mutect2: Execute Mutect2 with the
--f1r2-tar-gzargument to embed the collection of read orientation counts within the variant calling process.Note: When scattering Mutect2 across multiple compute nodes, you must combine the resulting F1R2 outputs from all scatters before proceeding, using multiple
-Iarguments for the next step [17].Learn the Read Orientation Model: Pass the F1R2 data to LearnReadOrientationModel to generate the artifact prior model.
Summarize Pileup Metrics for Contamination Estimation: Run GetPileupSummaries on the tumor BAM file, targeting a set of known common variant sites.
Calculate Contamination Fraction: Use the pileup summary table as input for CalculateContamination.
Apply Filters in FilterMutectCalls: Integrate the outputs from both artifact and contamination pipelines into the final filtering step.
Technical Specifications and Parameters
Optimizing the performance of these tools requires a detailed understanding of their key arguments. The default parameters are robust for most scenarios, but specific cases (e.g., targeted panels or high-contamination samples) may require adjustment.
Table 2: Critical Tool Arguments and Configuration Parameters
| Tool | Argument | Default Value | Description & Use Case |
|---|---|---|---|
| LearnReadOrientationModel | --num-em-iterations |
Not Specified | Increases iterations for model convergence, useful for complex artifact patterns [42]. |
| GetPileupSummaries | -V / -L |
None | Required. VCF and intervals of common sites (e.g., small ExAC). Use same file for both [17] [42]. |
| CalculateContamination | --matched-normal |
None | Input pileup table from matched normal BAM for significantly improved contamination estimation [40]. |
--tumor-segmentation |
None | Outputs a table segmenting the tumor by minor allele fraction [40]. | |
-low-coverage-ratio-threshold |
0.5 | Filters out sites with coverage below this ratio relative to the median [40]. | |
-high-coverage-ratio-threshold |
3.0 | Filters out sites with coverage above this ratio relative to the mean [40]. | |
| FilterMutectCalls | --ob-priors |
None | Required. Input for the read orientation model from LearnReadOrientationModel [17]. |
--contamination-table |
None | Required. Input for the contamination table from CalculateContamination [43] [42]. |
The following reagents, reference files, and software tools are indispensable for the successful implementation of this workflow.
Table 3: Essential Resources for the Artifact and Contamination Pipeline
| Resource Name | Type | Critical Function in Workflow |
|---|---|---|
| Reference Genome (FASTA) | Reference Data | Same reference used for read alignment and all GATK tools (Mutect2, GetPileupSummaries, FilterMutectCalls) [17]. |
| af-only-gnomAD VCF | Germline Resource | Used by Mutect2 for germline filtering; also used in creating a Panel of Normals [17] [39]. |
| Panel of Normals (PoN) | Somatic Resource | A VCF of artifacts found in normal samples, used by Mutect2 to filter recurrent technical artifacts [43]. |
| Common Variants VCF (e.g., small ExAC) | Known Sites | A VCF of known common polymorphic sites, used by GetPileupSummaries for contamination estimation [17] [42]. |
| Java & GATK Jar | Software | GATK tools are Java-based and require a compatible Java version and the GATK software package. |
| Interval Lists | Genomic Coordinates | Specifies target regions (e.g., exome capture kit intervals) for focused analysis in Mutect2 and GetPileupSummaries [17]. |
Impact on Analytical Results: A Quantitative Perspective
The implementation of this workflow has a measurable and profound impact on the quality of somatic variant calls. Benchmarking studies demonstrate that failure to account for cross-sample contamination can inflate the number of putative somatic mutations by orders of magnitude. One analysis of Patient-Derived Xenograft (PDX) models, a context with inherent mouse contamination, found that the number of exonic mutations detected without contamination removal was about 30 times higher than after proper bioinformatic processing (mean of 1,127 vs. 38) [44]. Furthermore, these false-positive mutations, or "artifacts," typically exhibit a significantly lower variant allele fraction (VAF) compared to bona fide somatic mutations, a key characteristic that contamination-aware filtering exploits [44].
For FFPE and other samples prone to strand-biased artifacts, the LearnReadOrientationModel filter is equally critical. While the exact quantitative improvement varies by sample type and sequencing platform, its integration is considered a best practice for all somatic analyses, as it removes a specific class of false positives with minimal computational overhead and no impact on sensitivity [17] [39]. The combined use of these tools ensures that the final variant catalog is highly enriched for biological signal, providing a reliable foundation for downstream biological interpretation and clinical decision-making.
FilterMutectCalls is a critical component of the GATK4 somatic short variant discovery workflow, responsible for distinguishing true somatic variants from the numerous technical artifacts and false positives generated by the initial variant calling step performed by Mutect2 [11]. This tool applies a sophisticated series of filters to the raw output of Mutect2, transforming a comprehensive but noisy callset into a highly confident somatic variant dataset suitable for downstream biological analysis[ citation:1] [45]. The filtering process accounts for correlated errors that Mutect2's independent error model cannot address, including alignment artifacts, strand and orientation bias, polymerase slippage, germline variants, and cross-sample contamination[ citation:8].
The tool forms an essential part of the GATK Best Practices workflow for somatic short variant discovery, which has become the industry standard for identifying SNPs and indels in cancer genomics research[ citation:4] [11]. When deployed within a comprehensive thesis on somatic variant calling, understanding FilterMutectCalls's mechanisms and parameters becomes crucial for producing publication-quality variant calls and accurately interpreting results in translational research and drug development contexts.
Core Filtering Mechanisms and Algorithms
Probabilistic Filtering Framework
FilterMutectCalls employs a Bayesian statistical framework to refine the initial likelihoods calculated by Mutect2 [11]. The tool learns the mutation rate and allele fraction spectrum specific to the tumor sample, then uses this information to calculate posterior probabilities for each variant being a true somatic mutation versus various artifact types[ citation:8]. This approach allows for automated, data-driven threshold setting rather than relying solely on fixed, predetermined cutoffs.
The tool offers multiple thresholding strategies through the --threshold-strategy parameter, with OPTIMAL_F_SCORE serving as the default[ citation:1]. This strategy automatically determines the filtering threshold that optimizes the harmonic mean of precision and recall (F-score), with the relative weight of recall to precision adjustable via the --f-score-beta argument[ citation:1]. Alternatively, researchers can employ a FALSE_DISCOVERY_RATE strategy, which controls the maximum false discovery rate at a user-defined level (default 0.05)[ citation:1].
Contamination-Based Filtering
A critical filtering mechanism addresses cross-sample contamination, which can introduce false positive calls. When provided with a contamination table (generated by CalculateContamination), FilterMutectCalls filters variants with a high posterior probability of being due to contamination[ citation:1] [45]. The --max-contamination-probability parameter controls this filter's stringency, with a default value of 0.1 [45]. This integration with CalculateContamination is particularly valuable as the latter tool is specifically designed to work effectively even in samples with significant copy number variation and without matched normals[ citation:8].
Technical Artifact Detection
FilterMutectCalls implements multiple specialized filters to address common technical artifacts in sequencing data:
- Read Orientation Artifacts: Using prior probabilities generated by
LearnReadOrientationModel, the tool filters variants that exhibit significant strand bias patterns characteristic of FFPE-derived DNA and other sequencing artifacts [46] [11]. This is implemented through the--orientation-bias-artifact-priorsargument. - Polymerase Slippage Artifacts: The tool identifies and filters variants likely resulting from PCR slippage events in short tandem repeat (STR) regions, with sensitivity adjustable via the
--min-slippage-lengthand--pcr-slippage-rateparameters [46]. - Mapping Artifacts: Multiple filters target variants supported by poor-quality alignments, including thresholds for median base quality (
--min-median-base-quality), mapping quality (--min-median-mapping-quality), and read position (--min-median-read-position) of alternative allele reads [46] [45].
Quantitative Filtering Parameters and Thresholds
Table 1: Key Filtering Parameters and Default Thresholds in FilterMutectCalls
| Filter Category | Parameter | Default Value | Description |
|---|---|---|---|
| Statistical Thresholds | --normal-artifact-lod |
0.0 [45] | LOD threshold for filtering variants as artifacts in the normal sample |
--tumor-lod |
5.3 [45] | LOD threshold for calling tumor variants (Tumor_LOD) | |
--false-discovery-rate |
0.05 [46] | Maximum FDR when using FDR threshold strategy | |
--max-germline-posterior |
0.1 [45] | Maximum posterior probability for germline classification | |
| Base & Mapping Quality | --min-median-base-quality |
20 [46] [45] | Minimum median base quality of alt reads |
--min-median-mapping-quality |
30 [46] [45] | Minimum median mapping quality of alt reads | |
--min-median-read-position |
1 [46] (5 [45]) | Minimum median distance of variants from read ends | |
| Strand & Orientation | --max-strand-artifact-probability |
0.99 [45] | Maximum probability for strand artifact filter |
--min-strand-artifact-allele-fraction |
0.01 [45] | Minimum allele fraction for strand artifact application | |
--min-reads-per-strand |
0 [46] | Minimum alt reads on both forward and reverse strands | |
| Contamination | --max-contamination-probability |
0.1 [45] | Maximum posterior probability of contamination |
| Slippage & INDELs | --min-slippage-length |
8 [46] | Minimum reference bases in STR to suspect slippage |
--log10-somatic-snv-prior |
-6.0 [46] | Log10 prior probability of a somatic SNV | |
--log10-somatic-indel-prior |
-7.0 [46] | Log10 prior probability of a somatic indel |
Table 2: Specialized Parameters for Mitochondrial and Advanced Use Cases
| Parameter | Default Value | Application Context |
|---|---|---|
--mitochondria-mode |
false [46] | Activates mitochondrial-specific filtering defaults |
--max-numt-fraction |
0.85 [46] | Maximum fraction of alt reads from nuclear mitochondrial segments (NuMTs) |
--unique-alt-read-count |
0 [46] [45] | Minimum unique (deduplicated) reads supporting alternate allele |
--max-events-in-region |
2 [46] [45] | Filter all variants if assembly region exceeds event count |
--max-median-fragment-length-difference |
10000 [46] [45] | Maximum difference in median fragment length between alt and ref reads |
Integrated Workflow for Somatic Variant Filtering
Diagram 1: FilterMutectCalls workflow integration and data dependencies
The filtering process requires specific inputs generated by preceding tools in the GATK somatic workflow. The contamination table from CalculateContamination and artifact priors from LearnReadOrientationModel provide critical information for specialized filters [46] [11]. For samples with unique characteristics such as FFPE-derived DNA, an optional additional filtering step using FilterByOrientationBias may be employed to further address sequencing artifacts [45].
Table 3: Essential Inputs and Resources for FilterMutectCalls
| Resource Type | Specific Tool/File | Role in Filtering Process |
|---|---|---|
| Required Inputs | Raw Mutect2 VCF [46] | Primary input containing unfiltered somatic calls |
| Reference genome (FASTA) [46] | Genomic context for variant positioning and annotation | |
| Optional Inputs | Contamination table [46] [45] | Enables contamination-based filtering |
| Tumor segmentation files [46] | Output from CalculateContamination for allelic copy number | |
| Orientation bias priors [46] | Enables read orientation artifact filtering | |
| Mutect2 stats file [46] | Provides additional statistical context for filtering | |
| Supporting Tools | CalculateContamination [11] | Generates contamination estimates for tumor samples |
| LearnReadOrientationModel [11] | Generates priors for orientation bias artifacts | |
| GetPileupSummaries [11] | Supports contamination calculation |
Experimental Protocol and Implementation
Basic Filtering Command
The fundamental command structure for FilterMutectCalls requires minimal parameters while leveraging default filtering thresholds:
This implementation produces a filtered VCF file with low-confidence variants either removed or flagged in the FILTER field [46] [45]. The tool automatically generates a comprehensive set of filtering statistics when the --filtering-stats argument is provided [45].
Comprehensive Filtering Protocol
For enhanced filtering sensitivity, particularly with challenging sample types like FFPE-derived DNA, researchers should implement the complete integrated protocol:
This protocol significantly enhances artifact removal, particularly for FFPE samples where orientation bias artifacts are prevalent [11]. The approach has been validated in comparative studies, demonstrating robust performance across sample types [47].
Validation and Performance Assessment
In a comparative analysis of somatic variant callers, Mutect2 (with FilterMutectCalls) was evaluated alongside Streka2, VarScan2, and Shimmer on matched fresh-frozen and FFPE samples [47]. The study found that while individual callers showed variable performance, ensemble approaches incorporating multiple callers increased both precision and sensitivity [47]. Specifically, the research demonstrated that with appropriate filtering strategies, the majority of clonal single nucleotide variants can be recovered from FFPE samples with high precision and sensitivity, enabling their use in cohort studies [47].
For researchers implementing FilterMutectCalls in production environments, the tool is designed to run on Linux and other POSIX-compatible platforms, including MacOS X, with Java 1.8 as the major system requirement [9]. The GATK Docker container provides a simplified deployment environment with all dependencies pre-configured [9].
Functional annotation is the critical final step in the somatic variant calling workflow that transforms raw variant calls into biologically interpretable data. The Functional Annotator (Funcotator) tool within the Genome Analysis Toolkit (GATK) provides a standardized framework for adding functional context to discovered variants, enabling researchers to distinguish driver mutations from passenger mutations and prioritize candidates for further investigation. As a functional annotation tool, Funcotator analyzes variants for their potential functional impact by retrieving information from a comprehensive set of biological databases and data sources, then produces the analysis in a specified output file format [48] [49].
Funcotator was designed to handle both somatic and germline use cases, serving as the recommended annotation method following variant filtering in the GATK Best Practices Somatic Pipeline [11] [48]. Its integration into the broader variant discovery workflow ensures consistent annotation across projects and provides researchers with standardized outputs that include gene-level information, variant classifications, and associations to established datasources such as GENCODE, dbSNP, gnomAD, and COSMIC [11]. The tool's extensible architecture allows laboratories to incorporate custom datasources, making it adaptable to specialized research needs while maintaining compatibility with community-standard resources.
Funcotator's Role in the GATK Somatic Workflow
Within the comprehensive GATK somatic short variant discovery workflow for SNVs and Indels, Funcotator serves as the final annotation step that enables biological interpretation [11]. The complete workflow begins with data preprocessing and proceeds through variant calling with Mutect2, contamination assessment, orientation bias modeling, and variant filtering before reaching the functional annotation stage. The diagram below illustrates Funcotator's position in this multi-step process:
Figure 1: Funcotator's position in the GATK somatic variant calling workflow
As shown in Figure 1, Funcotator operates on the filtered variant call format (VCF) file produced by FilterMutectCalls [11]. This sequential positioning is crucial because it ensures that only high-confidence somatic variants undergo comprehensive functional annotation, conserving computational resources and focusing analytical efforts on biologically relevant mutations. The input to Funcotator is a VCF file containing somatic short variants (SNVs and Indels) identified in one or more tumor samples from a single individual, with or without a matched normal sample [11].
Funcotator's annotation process adds critical biological context to these variants, labeling each with one of twenty-three distinct variant classifications and providing gene information such as the affected gene and predicted variant amino acid sequence [11]. This transformation from a raw variant list to an annotated dataset enables researchers to answer fundamental questions about the potential functional consequences of somatic mutations in their experimental data.
Funcotator Technical Architecture and Annotation Mechanism
Funcotator employs a modular datasource-driven architecture that enables flexible and comprehensive variant annotation. The core annotation mechanism involves matching input variants against a curated set of biological databases through configurable matching criteria. The diagram below illustrates Funcotator's internal workflow and annotation mechanism:
Figure 2: Funcotator's technical architecture and annotation mechanism
Data Source Organization and Management
Funcotator's annotation capabilities are powered by its datasource architecture, which follows a specific directory structure for organization [48] [49]. Each datasource resides in its own sub-directory within a main datasources folder, with further sub-directories for specific genome builds (e.g., hg19, hg38). The reference-specific datasource directory contains a configuration file with metadata about the datasource and instructions for matching variants to the data [48]. A typical datasource directory structure is organized as follows:
Table 1: Funcotator datasource directory structure
To simplify deployment, GATK provides pre-packaged datasources optimized for specific use cases [48]. These include:
- Somatic datasources: Curated for somatic variant annotation, including cancer-relevant resources
- Germline datasources: Optimized for germline variant annotation
These pre-packaged datasources are available as versioned gzip archives that can be downloaded using the FuncotatorDataSourceDownloader tool [49]. The download commands are:
Supported Datasource Types
Funcotator supports multiple datasource formats, each with specialized matching capabilities [49]:
- simpleXSV: Arbitrarily separated value tables (e.g., CSV, TSV) keyed by gene name or transcript ID
- locatableXSV: Separated value tables keyed by genomic position using contig, start, and end columns
- GENCODE: Custom datasource class for GENCODE annotations with protein change prediction
- COSMIC: Custom datasource class for COSMIC cancer mutation database
- VCF: Custom datasource class for Variant Call Format files
This flexible architecture enables Funcotator to integrate heterogeneous biological data while maintaining efficient variant matching performance. The tool can handle both local datasources and cloud-based resources, with support for querying data directly from Google Cloud Storage buckets [49].
Implementation Guide: Running Funcotator
Input Requirements
Funcotator requires several specific inputs to execute successfully [49]:
- A reference genome sequence in FASTA format
- The version of the reference genome being used (e.g., hg19, hg38)
- An input VCF file of variant calls to annotate
- A path to a folder of datasources formatted for Funcotator
- The desired output format (VCF or MAF)
Basic Command Structure
The fundamental command structure for running Funcotator is as follows [49]:
In this command, the --variant argument specifies the input VCF file, --reference provides the reference genome, --ref-version declares the genome build, --data-sources-path points to the datasources directory, and --output with --output-file-format define the annotation output.
Practical Implementation Example
In a practical research setting, Funcotator is typically executed after completing the Mutect2 variant calling and filtering steps [50]. A real-world example from a tumor epidemiology study demonstrates this implementation:
This example shows the annotation of a somatic variant callset from a kidney cancer study, where the output is written in VCF format with annotations added to the INFO field [50] [51].
Configuration for Different Use Cases
Funcotator provides several optional parameters to customize annotation for specific research needs [48]:
--ignore-filtered-variants: Annotate only variants that pass all filters--transcript-selection-mode: Control how transcripts are selected for annotation--transcript-list: Provide a custom list of transcripts for annotation--annotation-default: Set default values for specific annotations--annotation-override: Override specific annotation values
These parameters enable researchers to tailor the annotation process to their specific experimental questions and analytical requirements.
Output Formats and Annotation Content
Output Format Options
Funcotator supports two primary output formats, each serving different analytical needs [48]:
- VCF Format: Adds annotations to the INFO field of the input VCF file, maintaining all original variant information while adding functional annotations as key-value pairs
- MAF Format: Generates a Mutation Annotation Format file, a standardized format widely used in cancer genomics that represents variants in a tabular format optimized for downstream analysis
The selection between output formats depends on the intended downstream applications. VCF output is preferable when maintaining the original file structure with added annotations, while MAF format is better suited for integration with cancer genomics pipelines and tools like MAFtools [50].
Funcotator's pre-packaged datasources provide comprehensive annotations for somatic variants. Key annotation categories include:
Table 2: Key annotation categories provided by Funcotator
| Category | Annotation Elements | Biological Significance |
|---|---|---|
| Variant Classification | 23 distinct classifications including MissenseMutation, NonsenseMutation, FrameShiftIns | Categorizes functional impact of variants based on their effect on protein coding sequences |
| Gene Information | Affected gene, transcript ID, protein change prediction, amino acid changes | Identifies specific genes and transcripts affected by variants and predicted molecular consequences |
| Population Frequency | gnomAD allele frequencies across populations and subpopulations | Provides context on variant prevalence in general populations, helping filter common polymorphisms |
| Clinical Significance | COSMIC annotations, dbSNP identifiers, known cancer associations | Links variants to known clinical and cancer-related information from established databases |
gnomAD Integration
The pre-packaged datasources include a subset of gnomAD (Genome Aggregation Database), a large database of known variants [48]. This integration provides allele frequency data across different populations, which is crucial for distinguishing rare somatic mutations from common germline polymorphisms. Due to the size of gnomAD, it is disabled by default but can be enabled by extracting the gnomAD tar.gz files in the datasources directory [48]:
The gnomAD subset includes numerous population-specific allele frequency fields, such as AFafr (African ancestry), AFamr (Latino ancestry), AFeas (East Asian ancestry), AFnfe (non-Finnish European ancestry), and AF_sas (South Asian ancestry), among others [48]. This comprehensive population data enables researchers to assess the rarity of variants across global populations.
The Scientist's Toolkit: Research Reagent Solutions
Implementing Funcotator in a research environment requires specific computational resources and biological data assets. The following table details the essential components:
Table 3: Essential research reagents and resources for Funcotator implementation
| Resource | Function | Example/Format |
|---|---|---|
| Reference Genome | Provides reference sequence for variant context and coordinate mapping | FASTA file (e.g., Homosapiensassembly38.fasta) with corresponding index and dictionary files |
| Pre-processed BAM Files | Input alignment data containing sequence reads mapped to reference genome | Binary Alignment/Map files with sorted read alignments, generated through GATK Best Practices preprocessing |
| Funcotator Datasources | Curated biological databases for variant annotation | Pre-packaged somatic or germline datasources in organized directory structure with configuration files |
| gnomAD Resource | Population frequency data for filtering common polymorphisms | VCF format with allele frequencies across populations, available through Broad Institute resources |
| COSMIC Database | Catalog of somatic mutations in cancer with clinical annotations | Curated cancer mutation database, integrated into Funcotator datasources |
| GENCODE Annotations | Comprehensive gene annotation set for variant consequence prediction | GTF format with gene models, transcripts, and coding sequences for accurate protein effect prediction |
Troubleshooting and Common Technical Considerations
Reference Genome Compatibility
A common issue when running Funcotator is reference genome compatibility between the input VCF and the datasources [51]. Funcotator performs contig matching between the input variant file and the configured datasources, and will produce warnings if there are discrepancies. To resolve these issues:
- Ensure the
--ref-versionparameter matches the genome build of your input VCF and datasources - Verify that all datasources are available for your specific genome build (hg19, hg38, etc.)
- Use consistent reference genome versions throughout the entire variant calling pipeline
Performance Considerations
Funcotator performance can be impacted by several factors [48]:
- Internet connectivity: When using gnomAD without localization, Funcotator queries remote data sources, which can slow processing
- Datasource localization: For improved performance, localize all datasources to the machine running Funcotator
- Memory allocation: Large variant sets may require increased memory allocation to the Java virtual machine
For large-scale analyses, consider using the --ignore-filtered-variants option to annotate only variants that pass all filters, reducing the computational burden by focusing on high-confidence calls [48].
Funcotator represents an essential component of the GATK somatic variant calling workflow, transforming raw variant calls into biologically interpretable data through comprehensive functional annotation. Its integration with established biological databases, support for multiple output formats, and flexible datasource architecture make it a powerful tool for cancer researchers seeking to understand the functional significance of somatic mutations in tumor samples.
By following the implementation guidelines outlined in this technical guide, researchers can effectively incorporate Funcotator into their analysis pipelines, enabling standardized annotation of somatic variants and facilitating the identification of biologically relevant mutations in cancer genomics studies. The tool's position at the culmination of the variant discovery process underscores its importance in translating computational findings into biologically actionable insights.
Within the comprehensive framework of GATK somatic variant calling workflow research, understanding the distinct operational modes of its core tool, Mutect2, is paramount. The Genome Analysis Toolkit (GATK) Mutect2 serves as the industry standard for identifying somatic short variants (SNVs and Indels) [9]. This technical guide delineates the application, configuration, and methodological considerations for its three primary scenarios: tumor-normal matched analysis, tumor-only analysis, and mitochondrial variant discovery. Each mode addresses specific experimental constraints and biological questions, requiring tailored inputs, parameters, and filtering strategies to optimize the accuracy and sensitivity of somatic mutation detection [52] [11].
Mutect2 call somatic short mutations via local assembly of haplotypes, combining a Bayesian somatic genotyping model with assembly-based machinery [52] [53]. Its application, however, diverges significantly across different experimental designs. The following table summarizes the core characteristics and command-line specifications for its three main operational modes.
Table 1: Comparative Overview of Mutect2 Operational Modes
| Aspect | Tumor-Normal Mode | Tumor-Only Mode | Mitochondrial Mode |
|---|---|---|---|
| Primary Use Case | Standard somatic variant discovery with a matched normal control from the same individual [52] | Situations with no matched normal available; also used for creating Panels of Normals (PoN) [52] | Sensitive calling at high depths in mitochondrial DNA [52] |
| Key Inputs | Tumor BAM, Normal BAM, Germline Resource (recommended), PoN (recommended) [52] | Tumor BAM, Germline Resource (recommended), PoN (recommended) [52] | Mitochondrial BAM, --mitochondria flag [52] |
| Sample Specification | -I tumor.bam -I normal.bam -normal normal_sample_name [52] |
-I tumor.bam [52] |
-I mitochondria.bam --mitochondria [52] |
| Critical Parameters | --germline-resource, --panel-of-normals [52] |
--germline-resource, --panel-of-normals [52] |
Automatically sets --initial-tumor-lod 0, --tumor-lod-to-emit 0, --af-of-alleles-not-in-resource 4e-3 [52] |
| Output Nature | Somatic variants not present in the matched normal [31] | Raw somatic calls requiring stringent filtering [52] | Somatic variants in mitochondrial DNA [52] |
| Typical Filtering | FilterMutectCalls [11] | FilterMutectCalls, often with additional functional filtering (e.g., Funcotator) [52] | FilterMutectCalls [52] |
Tumor-Normal Mode: The Standard for Somatic Discovery
Methodology and Experimental Protocol
The tumor-normal mode is the most robust and widely recommended approach for somatic variant discovery. It is designed to call somatic variants by contrasting evidence between a tumor sample and a matched normal (germline) sample from the same individual [31]. The core protocol involves the following steps:
- Data Pre-processing: Input BAM files for both tumor and normal samples must be pre-processed according to GATK Best Practices, including steps like duplicate marking, base quality score recalibration, and indel realignment [11].
- Variant Calling with Mutect2: Execute Mutect2 with the required and recommended inputs. The following command represents a standard execution for a tumor-normal pair, which can be extended to multiple tumors and normals from the same individual [52]:
The
--germline-resource(e.g., gnomAD) provides population allele frequencies, allowing Mutect2 to distinguish common germline polymorphisms from true somatic events. The--panel-of-normals(PoN) contains sites of technical artifacts found in a set of normal samples, enabling their systematic removal [52] [31]. - Contamination Estimation (Optional but Recommended): This step is crucial for accuracy.
- Use
GetPileupSummariesto summarize read support at known variant sites. - Use
CalculateContaminationwith the output to estimate the fraction of cross-sample contamination in the tumor sample [11].
- Use
- Variant Filtering: The raw Mutect2 output must be filtered using
FilterMutectCalls. As of GATK 4.1.1.0, this step requires the same reference file and automatically uses the stats file generated by Mutect2 [17] [52].
Workflow Diagram
The following diagram illustrates the logical sequence and data flow for a comprehensive tumor-normal somatic variant discovery workflow.
Figure 1: Comprehensive Tumor-Normal Somatic Variant Discovery Workflow.
Tumor-Only Mode: Navigating the Challenges
Methodology and Experimental Protocol
Tumor-only analysis is applied when a matched normal sample is unavailable. This mode is inherently more susceptible to false positives from undiscovered germline variants and technical artifacts, making the use of a germline resource and a Panel of Normals (PoN) critical [52]. The protocol involves:
- Variant Calling with Mutect2: The command is similar to tumor-normal mode but omits the normal sample input.
- Creating a Panel of Normals (PoN): A PoN is essential for tumor-only analysis and is created from a collection of normal samples [17] [52].
- Step 1: Run Mutect2 in tumor-only mode on each normal sample.
- Step 2: Import the normal VCFs into a GenomicsDB.
- Step 3: Create the PoN using
CreateSomaticPanelOfNormals.
- Filtering and Annotation: After calling variants on the tumor sample, proceed with
FilterMutectCalls. Given the higher false positive rate, additional functional annotation with a tool likeFuncotatoris highly recommended to prioritize potentially actionable mutations [52] [11].
Workflow Diagram
The process for tumor-only analysis and the parallel creation of a Panel of Normals is depicted below.
Figure 2: Tumor-Only Analysis and Panel of Normals Creation Workflow.
Mitochondrial Mode: Calling Variants at High Depth
Methodology and Experimental Protocol
Mitochondrial DNA (mtDNA) analysis presents unique challenges, including high copy number and potential heteroplasmy (mixtures of mutant and wild-type molecules). Mutect2's mitochondrial mode is specifically configured for this context, optimizing sensitivity for detecting low allele fraction variants in high-depth sequencing data [52]. The protocol is streamlined:
- Variant Calling with Mitochondrial Flag: The key is to use the
--mitochondriaflag, which automatically sets appropriate parameters for mtDNA analysis. The mode accepts only a single sample (which can be provided as multiple BAM files) [52]. This flag automatically configures critical parameters: it sets--initial-tumor-lodand--tumor-lod-to-emitto 0 for increased sensitivity, adjusts--af-of-alleles-not-in-resourceto 4e-3, and sets an advanced--pruning-lod-thresholdto -4 [52]. - Filtering: The resulting VCF is filtered using
FilterMutectCallsas in other modes, leveraging the learned somatic clustering model to distinguish true variants from errors [17].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of the GATK somatic variant calling workflows relies on a set of key reagents and data resources. The following table details these essential components and their functions.
Table 2: Key Research Reagent Solutions for GATK Somatic Variant Calling
| Item Name | Function / Purpose | Specifications & Notes |
|---|---|---|
Reference Genome (-R) |
The standard sequence against which reads are aligned and variants are called [52]. | Must be the same build used for read alignment (e.g., GRCh38). A dedicated mitochondrial reference is often used for mtDNA analysis [52]. |
Germline Resource (--germline-resource) |
A VCF containing population allele frequencies used to identify and filter common germline polymorphisms [52] [31]. | Typically an "af-only-gnomad" VCF. Alleles not found in this resource are assigned a low, configurable prior probability (--af-of-alleles-not-in-resource) [52]. |
Panel of Normals (PoN) (--panel-of-normals) |
A VCF of technical artifacts discovered in a collection of normal samples, enabling their systematic removal from tumor samples [17] [31]. | Created using CreateSomaticPanelOfNormals. Sites listed in the PoN are filtered out during the Mutect2 call [17]. |
Read Orientation Model (--ob-priors) |
A model file providing prior probabilities for strand-specific sequencing artifacts (e.g., common in FFPE samples) [17]. | Generated by LearnReadOrientationModel from Mutect2's --f1r2-tar-gz output. Passed to FilterMutectCalls to filter orientation bias artifacts [17]. |
The GATK Mutect2 toolkit provides a versatile and powerful framework for somatic variant discovery across diverse research scenarios. The choice between tumor-normal, tumor-only, and mitochondrial modes dictates the specific workflow, required resources, and potential limitations. The tumor-normal mode remains the gold standard for its ability to control for germline variation and technical artifacts. When a matched normal is unavailable, the tumor-only mode offers a viable alternative, provided a robust Panel of Normals and germline resource are employed. Finally, the mitochondrial mode addresses the unique challenges of calling variants in high-depth, non-nuclear genomes. A thorough understanding of the methodologies, parameters, and reagents underlying each mode is fundamental for researchers and drug development professionals to generate high-confidence somatic variant calls that can drive scientific discovery and clinical insights.
Solving Common Mutect2 Issues and Enhancing Detection Sensitivity
Within the structured framework of the GATK somatic variant calling workflow, the accurate detection of somatic mutations, particularly low-allele-fraction variants, is paramount for oncology research and therapeutic development. Researchers often encounter the critical problem of missing variants in their final call sets, which can skew biological interpretations and compromise the validity of downstream analyses. Two parameters in the Mutect2 caller, --initial-tumor-lod and --max-reads-per-alignment-start, are frequently central to this issue, as they directly govern the sensitivity and computational feasibility of variant discovery [54] [53]. This guide provides an in-depth examination of these parameters, offering evidence-based troubleshooting methodologies to optimize their settings within the broader context of the GATK somatic analysis Best Practices workflow [11].
The following table summarizes the key characteristics, mechanisms, and optimization strategies for the two parameters under investigation.
Table 1: Key Parameters for Troubleshooting Missing Variants
| Parameter | Default Value | Primary Function | Impact on Variant Calling | Recommended Adjustment for Missing Variants |
|---|---|---|---|---|
--initial-tumor-lod |
Not explicitly stated in sources | Threshold for the initial Log Odds (LOD) that a site is somatic versus artifact/error. Controls which sites are considered for full analysis. | A threshold that is too high may pre-filter legitimate low-allele-fraction somatic variants before genotyping. | Consider lowering the value to allow more potential variant sites into the assembly and genotyping steps. |
--max-reads-per-alignment-start |
50 [55] [56] | Downsamples reads that share the exact same start position to control memory usage and computational load. | In deep sequencing, can artificially limit coverage at genuine variant sites, leading to missed calls. | Set to 0 to disable downsampling in deep sequencing projects (>1000X) where uniform, high coverage is available [57]. |
The GATK Somatic Variant Calling Workflow: A Framework for Troubleshooting
Understanding the role of --initial-tumor-lod and --max-reads-per-alignment-start requires placing them within the comprehensive GATK somatic short variant discovery workflow [11]. The diagram below illustrates the key steps, highlighting where these parameters exert their influence.
Diagram 1: Key Parameters in the GATK Somatic Variant Calling Workflow. The parameter --max-reads-per-alignment-start affects the early "Define Active Regions" step, while --initial-tumor-lod influences the "Calculate Somatic LOD" step.
Workflow Step Details and Experimental Context
The GATK Best Practices workflow for somatic short variant discovery involves two main steps: calling candidate variants with Mutect2 and then filtering them [11]. Mutect2, the core variant discovery tool, operates by first identifying active regions of the genome that show evidence of variation. It then performs local de-novo assembly of haplotypes in these regions, discarding existing read alignments to overcome mapping artifacts [54] [11]. Finally, it applies a Bayesian somatic genotyping model to calculate the likelihood of a site being a true somatic variant versus a sequencing error [11].
The Role of
--max-reads-per-alignment-start: This parameter functions during the initial processing of reads, before active region determination. It is designed as a computational safeguard to prevent the software from spending excessive time and memory on genomic "hotspots" with extreme, often artifactual, coverage (e.g., 10,000x in a 100x exome) caused by mapping errors [57]. By downsampling reads that start at the exact same base position, it ensures computational efficiency but risks discarding valid data in uniformly deep sequencing.The Role of
--initial-tumor-lod: This parameter acts as a sensitivity threshold during the statistical evaluation of assembled haplotypes. The LOD score represents the confidence that observed evidence supports a somatic variant. Sites with an initial LOD below this threshold are not subjected to the full, computationally intensive genotyping process [54] [53]. Consequently, a threshold that is too stringent can cause low-allele-fraction variants, which are common in heterogeneous tumor samples or subclonal populations, to be prematurely discarded.
Successful somatic variant analysis relies on a suite of bioinformatic reagents and resources. The following table details the essential components used in the GATK Best Practices workflow.
Table 2: Key Research Reagent Solutions for Somatic Variant Analysis
| Item | Function in the Workflow | Implementation Example |
|---|---|---|
| Germline Resource | A population VCF (e.g., gnomAD) used to pre-filter sites and impute allele frequencies for alleles not found in the resource, helping to distinguish somatic from rare germline variants. | --germline-resource af-only-gnomad.vcf.gz [54] [53] |
| Panel of Normals (PoN) | A VCF created from normal samples used to filter out common technical artifacts that are not present in the matched normal. | --panel-of-normals pon.vcf.gz [54] [11] |
| Filtering Tools Suite | Tools applied post-Mutect2 to account for correlated errors and artifacts, producing a high-confidence callset. | GetPileupSummaries, CalculateContamination, LearnReadOrientationModel, FilterMutectCalls [11] |
| Functional Annotation Tool | Adds biological and clinical context to filtered variants, such as gene effect and protein change prediction. | Funcotator for generating VCF or MAF files [11] |
Advanced Scenarios and Parameter Interaction Logic
The decision to adjust --initial-tumor-lod and --max-reads-per-alignment-start is highly dependent on the experimental design and data characteristics. The following diagram outlines the logical decision process for optimizing these parameters.
Diagram 2: Decision Logic for Parameter Adjustment. This flowchart guides the optimization of parameters based on experimental goals and data properties.
Detailed Experimental Protocols for Parameter Optimization
Protocol for Investigating
--max-reads-per-alignment-start:- Baseline Analysis: Run Mutect2 on your deep sequencing BAM file (e.g., >1000X coverage) using the default setting of
--max-reads-per-alignment-start 50. - Optimized Analysis: Re-run the analysis on the same data with
--max-reads-per-alignment-start 0to disable downsampling. - Comparative Evaluation: Use tools like
vcftoolsorbcftoolsto compare the two output VCFs. Focus on variants with low allele frequencies (e.g., <10%) and check their depth (DP) in the original BAM. Variants that appear only in the second run and have high original depth were likely missed due to the default downsampling. - Validation: Verify the newly discovered, low-AF variants using an orthogonal method, such as digital PCR or deep amplicon sequencing, to confirm they are not false positives.
- Baseline Analysis: Run Mutect2 on your deep sequencing BAM file (e.g., >1000X coverage) using the default setting of
Protocol for Investigating
--initial-tumor-lod:- Gather Evidence: First, ensure that missing variants are not due to other factors, such as filtering in the
FilterMutectCallsstep. Examine the raw VCF output from Mutect2 before filtering. - Parameter Adjustment: If a variant is absent from the raw VCF but visible in the BAM file with supporting reads, lower the
--initial-tumor-lodthreshold incrementally (e.g., from its default to 2.0, then 1.0) and re-run Mutect2. - Monitor Output: As you lower the threshold, monitor the increase in the number of candidate variants in the raw VCF. Be aware that this will increase computational runtime and the burden on the subsequent
FilterMutectCallsstep to remove false positives. - Benchmarking: If a validated truth set is available for your data, use it to quantify the trade-off between sensitivity (recall) and precision as you adjust this parameter, aiming to maximize the F-score [11].
- Gather Evidence: First, ensure that missing variants are not due to other factors, such as filtering in the
Within the sophisticated framework of the GATK somatic variant calling workflow, the parameters --initial-tumor-lod and --max-reads-per-alignment-start serve as critical control points for balancing sensitivity and specificity. Troubleshooting missing variants necessitates a systematic approach that begins with a clear understanding of one's experimental data—specifically its depth and the target allele fraction—and proceeds through logical, iterative parameter optimization as outlined in this guide. By integrating these tuning strategies with the mandatory use of complementary resources like a germline resource and panel of normals, researchers and drug developers can significantly enhance the fidelity of their somatic variant datasets, thereby ensuring the biological insights and clinical conclusions drawn from them are both robust and reliable.
Optimizing for Low Allele Fraction Variants and Subclonal Populations
The accurate identification of somatic variants with low allele fractions is a paramount challenge in cancer genomics, with direct implications for understanding tumor heterogeneity, monitoring minimal residual disease, and developing targeted therapies. Subclonal populations, which represent genetically distinct groups of cells within a tumor, often harbor mutations at low variant allele frequencies (VAFs), typically below 10% [15]. These low-frequency variants present significant technical challenges for detection using next-generation sequencing (NGS) due to the inherent difficulty in distinguishing true biological signals from sequencing artifacts and alignment errors [58]. In the context of the broader GATK somatic variant calling workflow, specialized approaches are required to optimize sensitivity for these subclonal events while maintaining high specificity to prevent false positive calls that could mislead clinical interpretation and therapeutic decisions.
The fundamental challenge stems from several biological and technical factors. Intratumor heterogeneity leads to extensive genetic diversity among different tumor cells within the same tumor, resulting in mutations present in only a subset of cancer cells [58]. Additionally, tumor samples are commonly contaminated with normal cells, further diluting the VAF of true somatic variants [31]. Sequencing artifacts, such as errors during library preparation, sequencing, or data processing, can create false signals that resemble low-frequency variants, while cross-contamination between tumor and normal tissues can introduce additional spurious results [58]. The GATK Mutect2 pipeline, specifically designed for somatic variant discovery, incorporates multiple advanced features to address these challenges through sophisticated statistical modeling and filtering strategies [17] [31].
Computational Workflow for Low-Frequency Variant Detection
Enhanced Mutect2 Configuration for Sensitivity
The standard GATK Mutect2 workflow requires specific modifications to optimize detection of low-frequency variants. The standard command for variant calling includes:
For low-frequency variants, additional parameters must be considered to enhance sensitivity. While earlier versions of Mutect2 utilized a --tumor-lod threshold for variant emission, version 4.1.1.0 and higher replaced this with a more sophisticated probabilistic approach that automatically determines the optimal threshold balancing sensitivity and precision [17]. However, users can influence this balance using the -f-score-beta parameter, where values greater than the default of 1 bias results toward greater sensitivity, which may be desirable when prioritizing detection of low-frequency variants [17].
The --af-of-alleles-not-in-resource parameter defines the germline variant prior used in likelihood calculations, which is particularly important for distinguishing rare germline variants from true somatic events in low-VAF ranges [31]. Additionally, the --log-somatic-prior parameter influences the somatic variant prior in Mutect2's likelihood calculations [31]. For subclonal variant detection, the initial assembly phase benefits from increased sensitivity settings, though specific parameters must be tailored to the expected VAF distribution and tumor purity.
Advanced Filtering Strategies with FilterMutectCalls
Following variant calling, the FilterMutectCalls step applies sophisticated filtering to remove false positives while retaining true low-frequency variants. In GATK 4.1.1.0 and higher, this tool implements a unified filtering strategy based on a single quantity: the probability that a variant is a true somatic mutation [17]. The command structure is:
A critical advancement in recent versions is the incorporation of a somatic clustering model that leverages a Dirichlet process binomial mixture model to account for an unknown number of subclones [17]. This model helps distinguish true somatic variants from errors by learning the spectrum of subclonal allele fractions present in the tumor sample. If the tumor contains subclones with distinct allele fractions (e.g., variants occurring in 40% of cells), the model learns to reject calls with allele fractions significantly different from the observed clusters while retaining those that fit the pattern [17].
The FilterMutectCalls step also automatically determines the probability threshold that optimizes the F-score (the harmonic mean of sensitivity and precision), eliminating the need for manual threshold tuning for parameters such as -tumor-lod and -normal-artifact-lod that were required in earlier versions [17]. This automated optimization generally provides a better balance between sensitivity and precision than manually tuned thresholds.
Read Orientation Artifact Detection
Sequencing artifacts that occur on a single strand before sequencing represent a significant source of false positives in low-frequency variant detection. This is particularly problematic for FFPE tumor samples and samples sequenced on Illumina Novaseq machines [17]. The updated Mutect2 workflow includes a dedicated three-step process to address these artifacts:
Step 1: Generate F1R2 counts during Mutect2 execution
Step 2: Learn read orientation model
Step 3: Apply orientation bias filtering
This workflow has been optimized in GATK 4.1.1.0 to minimize computational cost while maintaining effectiveness, making it practical to run even when orientation artifacts are not initially suspected [17].
Contamination Estimation and Panel of Normals
For low-frequency variants, accurate estimation and correction of sample contamination is crucial. The GetPileupSummaries and CalculateContamination tools provide this functionality:
The resulting contamination table can then be provided to FilterMutectCalls using the --contamination-table argument [17].
The panel of normals (PoN) remains essential for filtering systematic artifacts across all variant frequency ranges. The updated CreateSomaticPanelOfNormals workflow uses GenomicsDB for improved scalability and now includes additional INFO fields (FRACTION and BETA) that characterize artifacts across normal samples [17]. While not currently used by FilterMutectCalls, these enhancements provide a foundation for future improvements in artifact filtering.
Table 1: Performance Comparison of Somatic Variant Callers at Different Mutation Frequencies
| Variant Caller | Mutation Frequency 1% | Mutation Frequency 5-10% | Mutation Frequency ≥20% | Computational Efficiency |
|---|---|---|---|---|
| Mutect2 | F-score: 0.05-0.50 (depth-dependent) | F-score: 0.65-0.95 | F-score: 0.94-0.96 | 17-22 times slower than Strelka2 |
| Strelka2 | F-score: 0.06-0.37 (depth-dependent) | F-score: 0.64-0.94 | F-score: 0.94-0.965 | Reference for speed comparison |
| Consensus Approach | Highest precision at low frequencies | Balanced sensitivity and precision | Excellent performance | Variable based on callers used |
Note: Performance metrics derived from systematic comparisons using downsampled datasets across multiple sequencing depths (100X-800X). F-score represents the harmonic mean of precision and recall [15].
Experimental Design and Sequencing Considerations
Optimal Sequencing Depth for Low-Frequency Variants
Sequencing depth requirements become increasingly important as the target variant allele frequency decreases. Systematic performance comparisons reveal that for higher mutation frequencies (≥20%), a sequencing depth of ≥200X is sufficient to call 95% of mutations [15]. However, for low-frequency variants (≤10%), the relationship between sequencing depth and detection performance becomes more complex.
Research has demonstrated that increasing sequencing depth from 100X to 200-800X can improve recall rates by 0.6-44% for low-frequency variants [15]. While deeper sequencing generally improves sensitivity, there are diminishing returns and potential trade-offs. Studies have observed a slight decrease in precision (less than 0.7%) when sequencing depth increases beyond 200X, though the overall F-score typically improves [15]. At the lowest mutation frequency (1%), even with high sequencing depths (800X), recall rates remain limited to 2.7-34.5% across different tools, indicating fundamental limitations in detecting very rare variants [15].
For variants in the 5-10% VAF range, sequencing depths of 300-500X generally provide the best balance of sensitivity and precision, with recall rates of 48-96% and precision rates of 68.9-100% across different callers [15]. Notably, for the most challenging low-frequency variants (≤10%), the study authors recommend improving experimental methods rather than indefinitely increasing sequencing depth, suggesting that techniques such as unique molecular identifiers (UMIs) or enhanced library preparation methods may provide better returns on investment [15].
Integration with Complementary Tools
While Mutect2 provides excellent performance for low-frequency variant detection, recent evidence suggests that ensemble approaches combining multiple callers can further improve accuracy. The Musta pipeline, which integrates six variant callers including Mutect2, demonstrated the lowest root mean square error (RMSE) compared to individual callers when benchmarking against validated variant sets [58]. In one hepatocellular carcinoma dataset, nearly 99% of somatic mutations identified by Musta were detected by at least four out of the six variant callers, indicating high confidence in the consensus calls [58].
For tumor-only samples (without matched normal), the challenges of low-frequency variant detection are amplified. In these scenarios, newer approaches such as ClairS-TO, which uses an ensemble of two disparate neural networks trained for opposite tasks, have shown promising results [59]. ClairS-TO outperformed Mutect2 in tumor-only benchmarks, particularly for variants with low VAFs, though Mutect2 remains the gold standard for paired tumor-normal analyses [59].
Table 2: Sequencing Depth Recommendations for Different Variant Frequency Ranges
| Variant Frequency Range | Minimum Recommended Depth | Optimal Depth Range | Expected Recall Rate | Expected Precision Rate |
|---|---|---|---|---|
| ≥20% | 200X | 200-300X | ≥95% | ≥95% |
| 10-20% | 300X | 300-500X | 92-97% | ≥95% |
| 5-10% | 500X | 500-800X | 48-96% | 95.5-95.9% (Mutect2) |
| 1-5% | 800X | 800X+ | 2.7-34.5% | 68.9-100% |
Note: Performance metrics based on systematic comparisons using mixed samples with known variant frequencies. Precision and recall ranges represent performance across multiple callers and replicate experiments [15].
Technical Implementation and Workflow Integration
Scalable Execution for Large-Scale Studies
For large-scale studies or clinical applications, the computational efficiency of the variant calling pipeline becomes increasingly important. The GATK team provides reference implementations of the Best Practices workflows in WDL (Workflow Description Language) format, which can be executed using the Cromwell execution engine [20]. These workflows are optimized for scalability and can be deployed on local computing clusters or cloud-based platforms.
When scattering Mutect2 execution across multiple genomic intervals (e.g., by chromosome), special attention must be paid to file handling:
This scattered approach significantly reduces runtime for whole-genome sequencing data while ensuring that the statistical models have sufficient data across the entire genome for accurate filtering [17].
Validation and Quality Control
Robust quality control measures are essential when prioritizing sensitivity for low-frequency variants. The filtering statistics output from FilterMutectCalls (.filtering_stats.tsv) provides valuable information about the learned parameters of somatic clustering, the probability threshold chosen to optimize the F-score, and the expected numbers of false positives and false negatives for each filter [17].
Additional quality metrics should include:
- Cross-sample contamination estimates: Particularly important for low-frequency variants where minor contamination can generate false positives
- Tumor segmentation data: Helps identify copy number variations that affect expected VAF distributions
- Coverage uniformity: Ensures adequate coverage across target regions, as coverage drops can dramatically impact low-frequency variant detection
- Duplicate rates: High duplication rates can indicate limited sequencing complexity, which affects diversity of observation for low-frequency variants
For clinical applications, benchmarking against reference materials such as the Genome in a Bottle (GIAB) or synthetic datasets like Syndip provides objective performance measures [60]. These resources enable quantitative assessment of pipeline sensitivity and specificity across different VAF ranges.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Resources for Optimized Somatic Variant Calling
| Resource Category | Specific Tools/Resources | Function in Workflow | Key Considerations |
|---|---|---|---|
| Reference Sequences | GRCh38, GRCh37 | Genome alignment and variant calling | Use consistent version throughout pipeline |
| Germline Resources | gnomAD, 1000 Genomes | Filtering common germline variants | Population-specific frequencies improve filtering |
| Panel of Normals | Custom-built from normal samples | Filtering systematic technical artifacts | Should be sequencing protocol-matched |
| Variant Annotation | Funcotator, VEP | Functional consequence prediction | Funcotator requires more resources but provides comprehensive annotation |
| Benchmarking Resources | GIAB, Platinum Genomes, Syndip | Pipeline validation and optimization | Essential for establishing sensitivity limits |
| Workflow Management | WDL, Nextflow, Snakemake | Reproducible pipeline execution | Must handle scattered execution and file merging |
Workflow Visualization
Low-Frequency Variant Detection Workflow
Optimizing the GATK Mutect2 workflow for low allele fraction variants and subclonal populations requires a multifaceted approach combining appropriate sequencing depth, specialized computational methods, and rigorous validation. The key considerations include: (1) implementing the complete orientation bias workflow to eliminate strand-specific artifacts; (2) leveraging the updated somatic clustering model in FilterMutectCalls that automatically learns subclonal allele fraction patterns; (3) selecting appropriate sequencing depths based on target variant allele frequencies; and (4) implementing robust validation strategies using benchmark materials. While Mutect2 provides excellent performance across the VAF spectrum, ensemble approaches combining multiple callers may offer additional improvements for the most challenging low-frequency variants. As sequencing technologies continue to evolve, with emerging approaches such as long-read sequencing and advanced error correction methods, the capabilities for detecting and characterizing subclonal populations will continue to improve, enabling new insights into tumor evolution and heterogeneity.
Formalin-Fixed Paraffin-Embedded (FFPE) specimens represent an invaluable resource in clinical oncology, with millions of samples archived worldwide alongside detailed clinical records [61]. However, the chemical processes involved in formalin fixation introduce extensive DNA damage that manifests as sequencing artifacts, profoundly impacting variant discovery in cancer genomics [62] [61]. The core challenge lies in the formalin-induced chemical modifications that systematically alter DNA, leading to false positive variant calls that can obscure true somatic mutations and compromise biomarker detection [61].
The molecular pathology of FFPE-derived DNA damage encompasses multiple mechanistic pathways. Formaldehyde engages in addition reactions with nucleophilic groups on DNA bases, creating modified bases with altered pairing properties [61]. These modified bases can further form methylene bridges, establishing covalent cross-links with nearby nucleophilic groups that potentially block polymerase progression during sequencing library amplification [61]. Perhaps most significantly, formalin fixation accelerates glycosidic bond cleavage, generating apurinic/apyrimidinic (AP) sites that are highly susceptible to DNA backbone fragmentation and mis-incorporation during amplification [61]. Additionally, spontaneous cytosine deamination to uracil occurs with high frequency in FFPE samples due to inactivation of cellular repair enzymes, leading to C>T/G>A transitions that mimic true somatic variants [62] [61]. These chemical alterations collectively create a complex landscape of sequencing artifacts that must be addressed through specialized computational methods within somatic variant calling workflows.
Table 1: Primary Chemical Alterations in FFPE-DNA and Their Sequencing Consequences
| Alteration Mechanism | Chemical Effect | Primary Sequencing Artifact | Impact on Variant Calling |
|---|---|---|---|
| Cytosine Deamination | Converts cytosine to uracil | C>T / G>A transitions | False positive SNVs, inflated TMB |
| Formaldehyde Addition | Adds hydroxymethyl group to bases | Altered base pairing | Base substitution errors |
| DNA Cross-linking | Covalent bonds between DNA molecules | PCR amplification failure | Reduced sensitivity in affected regions |
| Glycosidic Bond Cleavage | Creates apurinic/apyrimidinic sites | DNA fragmentation & mis-incorporation | False indels and base substitutions |
| Polydeoxyribose Fragmentation | DNA backbone cleavage | Shortened fragment lengths | Reduced mapping quality, coverage dropouts |
The Read Orientation Model: Computational Methodology
Within the GATK somatic variant calling workflow, the Read Orientation Model constitutes a specialized probabilistic framework designed to identify and filter artifacts arising from orientation bias in FFPE-derived sequencing data [63] [11]. This model specifically addresses the statistical overrepresentation of artifactual variants supported predominantly by reads aligning to a single DNA strand, a common phenomenon in FFPE samples due to localized DNA damage and subsequent biased amplification [64].
The model operates through an expectation-maximization (EM) algorithm that iteratively learns the prior probabilities of artifact occurrence across different trinucleotide contexts [63]. The mathematical foundation of this approach involves calculating maximum likelihood estimates of artifact prior probabilities within a orientation bias mixture model, which then informs the filtering decisions in downstream processing [63]. The algorithm continues this iterative refinement until either the parameter distance between iterations falls below a defined convergence threshold (default: 1.0E-4) or until reaching the maximum number of EM iterations (default: 20) [63]. This learned model of orientation bias artifacts is subsequently applied within the FilterMutectCalls step to distinguish true somatic variants from strand-biased artifacts with high precision [11].
Table 2: Key Parameters of the LearnReadOrientationModel Tool
| Parameter | Default Value | Function | Impact on Model Performance |
|---|---|---|---|
--convergence-threshold |
1.0E-4 | Stopping criterion for EM algorithm | Lower values increase precision but extend compute time |
--num-em-iterations |
20 | Maximum iterations for EM algorithm | Prevents infinite loops; may need adjustment for complex datasets |
--max-depth |
200 | Sites with higher depth are grouped | Maintains statistical robustness for high-coverage regions |
--input (-I) |
Required | F1R2 count tables from CollectF1R2Counts | Multiple inputs allowed for scattered execution |
--output (-O) |
Required | Output artifact prior tables | Contains learned priors for FilterMutectCalls |
Experimental Protocols and Implementation
Integrated Workflow for FFPE Somatic Variant Discovery
The implementation of the Read Orientation Model requires careful execution within the broader GATK somatic variant calling framework [11]. The initial stage involves generating candidate somatic variants using Mutect2, which performs local de novo assembly of haplotypes and applies a Bayesian somatic likelihoods model to identify potential SNVs and indels [11]. A critical parallel step involves running CollectF1R2Counts (either as part of Mutect2 or as a separate tool) to quantify forward and reverse read support for each potential variant, generating the required count data for orientation bias modeling [63].
The core orientation modeling is performed using LearnReadOrientationModel, which processes the F1R2 count tables to learn artifact prior probabilities [63]. The tool accepts multiple inputs, allowing researchers to process data from multiple samples or scattered executions simultaneously [63]. The resulting artifact prior model is then passed to FilterMutectCalls via the --ob-priors argument, where it contributes to the comprehensive filtering that accounts for correlated errors, strand bias, contamination, and other artifactual sources [11]. This integrated approach is particularly crucial for FFPE samples, where orientation bias artifacts can otherwise dominate the variant call set [64].
Quantitative Impact on Variant Calling Precision
Empirical studies demonstrate the critical importance of proper orientation artifact filtering for FFPE samples. Research analyzing 56 matched FF-FFPE sample pairs revealed that FFPE processing results in a median 20-fold enrichment in artifactual calls across mutation classes, severely impairing detection of clinically relevant biomarkers such as homologous recombination deficiency (HRD) [62]. Without specialized filtering, consensus calling approaches alone show limited efficacy for small variants, with the median proportion of FFPE-specific mutations remaining unacceptably high at 62% for SNVs and 73% for indels [62].
The practical consequences of inadequate artifact filtering are starkly evident in real-world implementations. In one reported case, enabling the orientation bias filter in FilterMutectCalls reduced PASS calls from 39,718 to just 197 in a replication-repair deficient sample, highlighting both the pervasive nature of orientation artifacts and the potential for over-filtering if model parameters are not properly calibrated [64]. This underscores the necessity of method optimization, particularly for samples with inherently high mutation loads where the balance between sensitivity and specificity becomes crucial.
Table 3: Performance Metrics of FFPE Variant Calling With and Without Specialized Filtering
| Variant Class | Precision Without Orientation Filter | Precision With Orientation Filter | Fold-Reduction in Artifacts | Impact on Clinical Biomarkers |
|---|---|---|---|---|
| SNVs | 50% | >90% (estimated) | 20-152x | Enables accurate TMB calculation |
| Indels | 62% | >90% (estimated) | 2.4-42x | Improves driver mutation detection |
| Structural Variants | 80% | Minimal additional benefit | 0.76x (median) | Maintains SV calling precision |
| Coding TMB | Unaffected | Unaffected | Not significant | Consistent with panel assays |
| Genome-wide TMB | 7x inflated | Normalized to FF levels | 7x reduction | Enables non-coding biomarker analysis |
Validation and Research Applications
The Scientist's Toolkit: Essential Research Reagents and Tools
Table 4: Key Research Reagent Solutions for FFPE Sequencing Analysis
| Reagent/Tool | Function | Application in FFPE Workflow |
|---|---|---|
| GATK LearnReadOrientationModel | Learns artifact prior probabilities | Identifies strand-biased artifacts from F1R2 counts |
| GATK CollectF1R2Counts | Generates forward/reverse read counts | Provides input data for orientation bias modeling |
| GATK FilterMutectCalls | Applies comprehensive artifact filtering | Integrates orientation priors for final variant filtering |
| DNA Repair Enzymes (e.g., UDG) | Repairs cytosine deamination damage | Reduces C>T artifacts prior to library preparation |
| Target Enrichment Panels | Selects genomic regions of interest | Mitigates coverage dropouts in fragmented DNA |
| FFPE DNA Extraction Kits | Optimized for cross-linked DNA | Maximizes yield and fragment length recovery |
Analytical Framework for Method Validation
The validation of read orientation model implementation requires a multifaceted approach comparing variant calls with and without the orientation bias filter. Researchers should first establish baseline variant counts from Mutect2 output, then systematically apply the orientation filter while monitoring the distribution of filtering annotations [64]. Successful implementation typically shows a significant reduction in variants flagged with orientation bias filters while preserving known true positives, particularly in clinically relevant genes [64].
For orthogonal validation, comparison with matched fresh frozen samples represents the gold standard, enabling direct quantification of precision and sensitivity [62]. In cases where matched FF samples are unavailable, comparison with targeted panel sequencing results provides clinical ground truth for validation [62]. Additionally, monitoring the spectrum of mutation signatures offers important quality metrics; successful artifact filtering should reduce the contribution of FFPE-associated signatures like SBS37 while preserving true biological signatures [62]. The distribution of variant allele frequencies (VAFs) also provides crucial validation - effective filtering should preferentially remove variants with intermediate VAFs (5-20%) that are characteristic of artifacts while preserving clonal variants with higher VAFs [61].
The integration of the Read Orientation Model within the GATK somatic variant calling workflow represents a critical advancement for unlocking the potential of FFPE specimens in cancer genomics. By systematically addressing the strand-biased artifacts that plague FFPE-derived sequencing data, this computational approach enables researchers to leverage vast archival sample collections while maintaining the precision required for clinical and research applications. As WGS continues to expand beyond fresh frozen samples to encompass the rich diversity of FFPE biospecimens, sophisticated artifact modeling frameworks will remain essential for distinguishing true biological signals from technical artifacts, ultimately advancing precision oncology through more comprehensive genomic analysis.
Parameter Tuning for Extreme Depth or Challenging Genomic Regions
Within somatic variant calling research, a core challenge is maintaining high sensitivity and specificity when analyzing data from extreme sequencing depths or challenging genomic regions. These conditions, common in cancer genomics and specialized assays, push standard analysis parameters beyond their limits, necessitating tailored configurations. This guide details specialized parameter tuning for the GATK Mutect2 tool to optimize performance under these demanding conditions, providing a crucial methodological foundation for reproducible somatic variant discovery in drug development and clinical research.
Mutect2 Parameter Adjustments for Extreme Depth
Specialized Operational Modes
Mutect2 provides dedicated operational modes pre-configured for specific high-depth scenarios. The most notable is mitochondrial mode, designed for the extreme depths typical in mitochondrial DNA sequencing [12].
Mitochondrial Mode Activation:
Using the --mitochondria flag automatically sets four critical parameters appropriate for mitochondrial calling [12]:
--initial-tumor-lod: Set to 0--tumor-lod-to-emit: Set to 0--af-of-alleles-not-in-resource: Set to 4e-3--pruning-lod-threshold(advanced): Set to -4
For non-mitochondrial extreme depth data (e.g., 20,000X), Mutect2 versions v4.1.0.0 onwards inherently accommodate these depths without additional configuration, representing a significant advancement over previous versions [12].
Critical Parameters for Tuning
Table 1: Key Mutect2 Parameters for Extreme Depth and Challenging Regions
| Parameter | Standard Default | Extreme Depth Setting | Functional Impact |
|---|---|---|---|
--af-of-alleles-not-in-resource |
0.001 (tumor-normal) | 4e-3 (mitochondrial) | Population allele frequency for alleles absent from germline resource |
--initial-tumor-lod |
Not specified | 0 (mitochondrial) | Initial tumor LOD threshold |
--tumor-lod-to-emit |
Not specified | 0 (mitochondrial) | Tumor LOD threshold for emitting variants |
--pruning-lod-threshold |
Not specified | -4 (mitochondrial) | Advanced parameter for assembly graph pruning |
--f-score-beta (FilterMutectCalls) |
1 | >1 for increased sensitivity | Controls sensitivity/specificity trade-off [65] |
The --af-of-alleles-not-in-resource parameter is particularly crucial, as it provides the imputed allele frequency for variants not found in the germline resource, with Mutect2 dynamically adjusting defaults based on analysis mode [12]:
- Tumor-only calling: Default 5e-8
- Tumor-normal calling: Default 1e-6
- Mitochondrial mode: Default 4e-3
For organisms other than human, this parameter should be adjusted to 1/(ploidy × number of samples in resource) [12].
Experimental Design and Benchmarking
Performance Optimization Strategies
Optimizing variant calling workflows requires strategic resource management and parallelization. GATK4 is largely single-threaded except for the PairHMM portion of HaplotypeCaller, making workflow-level parallelization essential for timely results [66].
Scatter-Gather Methodology: The recommended approach for speeding up Mutect2 analysis involves scattering execution across genomic intervals [65] [67]:
Computational Resource Recommendations:
- RAM: ~4GB sufficient for most exome samples [65]
- CPUs: Mutect2 is largely single-threaded; use scattering rather than multithreading [65]
- Parallelization: Implement via genomic interval scattering in WDL/Terra or custom scripts [65] [67]
Table 2: Benchmarking Results for Variant Callers on GIAB Gold Standard Datasets
| Software | SNV Precision (%) | SNV Recall (%) | Indel Precision (%) | Indel Recall (%) | Runtime (minutes) |
|---|---|---|---|---|---|
| DRAGEN Enrichment | >99 | >99 | >96 | >96 | 29-36 |
| GATK Mutect2 | Benchmark data not specified in search results | Not specified | Not specified | Not specified | Not specified |
| Partek Flow (GATK) | Not specified | Not specified | Not specified | Not specified | 216-1782 |
| CLC Genomics | Not specified | Not specified | Not specified | Not specified | 6-25 |
Data derived from benchmarking on GIAB samples HG001, HG002, HG003 [68]
Addressing False Positives in High-Depth Data
High-depth sequencing often increases false positive rates, with FilterMutectCalls typically filtering out 90% or more of initial Mutect2 calls [69]. This aggressive filtering is by design, as Mutect2 employs a permissive calling strategy, leaving rigorous filtering to FilterMutectCalls [69].
Strategies to Reduce False Positives:
- Employ Panel of Normals (PoN): Use PoN to filter common artifacts [69]
- Utilize Germline Resources: Implement population frequency filters (e.g., gnomAD) [69]
- Orientation Bias Correction: Collect F1R2 metrics with
--f1r2-tar-gzand use LearnReadOrientationModel for FFPE samples [12] - Tumor Purity Adjustment: Adjust minimum allele fraction filters based on estimated tumor purity [69]
Visualization of Parameter Tuning Strategy
The following diagram illustrates the decision workflow for parameter tuning based on data characteristics:
Figure 1: Decision workflow for parameter tuning strategy based on sequencing depth and genomic region characteristics. The path highlighted in red indicates the specialized mitochondrial mode configuration.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Resources
| Resource | Type | Function in Analysis | Source/Example |
|---|---|---|---|
| Panel of Normals (PoN) | Computational Resource | Filters common sequencing artifacts | Broad Institute PoN [65] |
| Germline Resource | Population Data | Provides population allele frequencies | gnomAD (af-only-gnomad.vcf.gz) [12] |
| High-Confidence Truth Sets | Benchmarking Data | Validation of variant calls | GIAB Gold Standard Datasets [68] |
| BWA-MEM | Alignment Software | Maps sequencing reads to reference | Burrows-Wheeler Aligner [67] |
| FilterMutectCalls | Filtering Tool | Applies sophisticated filtering models | GATK Suite [12] |
| LearnReadOrientationModel | Artifact Correction | Models and corrects orientation bias | GATK Suite [12] |
Optimizing Mutect2 for extreme depth and challenging genomic regions requires a multifaceted approach combining specialized operational modes, careful parameter adjustment, and appropriate filtering strategies. The mitochondrial mode demonstrates how domain-specific configurations can address unique variant calling challenges, while strategic parameter tuning enables researchers to maintain sensitivity without sacrificing specificity. As sequencing technologies continue to evolve, producing ever-deeper datasets from increasingly complex genomic regions, these tuning methodologies provide a critical foundation for robust somatic variant discovery in both research and clinical applications.
System Requirements and Computational Optimization for Scalable Analysis
The Genome Analysis Toolkit (GATK) has established itself as the industry standard for identifying SNPs and indels in germline DNA and RNAseq data, with its scope expanding to include somatic short variant calling, copy number (CNV) and structural variation (SV) [9]. The computational workload for somatic variant discovery is substantial, involving processing high-throughput sequencing data through multiple analytical steps. Efficient execution requires understanding both the fundamental system requirements and advanced optimization strategies to balance computational cost against processing time, which is particularly crucial in clinical and large-scale research settings where timely results and resource efficiency are paramount [25].
System Requirements and Specifications
Core Platform Requirements
GATK is designed to run on Linux and other POSIX-compatible platforms, which includes MacOS X. Windows systems are not supported. The major system requirement is Java 1.8, while some tools have additional R or Python dependencies [9]. For ease of deployment, the use of Docker containers is recommended, with an official container available on Dockerhub [9].
The toolkit's powerful processing engine and high-performance computing features make it capable of taking on projects of any size. In addition to supporting traditional compute environments such as local clusters, GATK4 has been engineered to play well with cloud environments and leverage Spark architectures wherever possible [9].
Hardware Configuration Recommendations
Optimal hardware configuration depends on the specific GATK tools being used and the chosen parallelization strategy. Different tools within the GATK ecosystem have varying computational profiles and resource requirements:
Table 1: Recommended Hardware Configurations for GATK Tools
| Tool | Available Parallelization Modes | Recommended CPU Threads | Recommended Memory | Cluster Nodes |
|---|---|---|---|---|
| BaseRecalibrator | NCT, SG | 8 | 4 GB | 4 |
| HaplotypeCaller | NCT, SG | 4 | 16 GB | 4 |
| PrintReads | NCT | 4-8 | 4 GB | 1 |
| UnifiedGenotyper | NT, NCT, SG | 3-24 | 4-32 GB | 4 |
Source: Adapted from GATK documentation [70]
For cost-effective routine analyses or large population studies, GATK4 running multiple samples on one node is recommended, with total walltime of approximately 34.1 hours on 40 samples using a c5.18xlarge instance on Amazon Cloud [25]. For time-sensitive situations, GATK3.8 with sample splitting across multiple nodes can achieve walltime under 4 hours for a 20X whole human genome at higher computational cost [25].
Parallelism Strategies for GATK Optimization
Fundamental Concepts of Parallel Computing
Parallelism is a method to make a program finish faster by performing several operations concurrently rather than sequentially. The principle can be illustrated by a simple analogy: if you need to cook rice for sixty-four people but your rice cooker only handles four people at a time, using eight rice cookers in parallel reduces the time nearly eightfold compared to sequential cooking [71].
In computational terms, this involves transforming a linear set of instructions into several subsets that can be executed simultaneously. Most GATK runs consist of many small independent operations that can be parallelized, though with important tradeoffs. Parallelized jobs require management overhead, memory allocation, file access regulation, and result collection, making it crucial to balance costs against benefits and avoid dividing work into excessively small jobs [71].
Multi-threading Implementation
Multi-threading operates at the core and machine levels, where a thread of execution is a set of instructions issued to the processor. In single-threading mode, a program sends one thread at a time to the processor, while multi-threading mode sends several threads simultaneously [71].
In GATK4, multi-threading is implemented using Apache Spark, an open-source industry-standard software library, replacing the home-grown code used in earlier versions [71]. The two primary arguments for multi-threading in GATK are:
-nt / --num_threads: Controls the number of data threads sent to the processor-nct / --num_cpu_threads_per_data_thread: Controls the number of CPU threads allocated to each data thread [70]
Memory management is crucial when implementing multi-threading. Each data thread needs the full memory allocation of a single-threaded run, so memory requirements scale linearly with the number of data threads. However, CPU threads share memory allocated to their "mother" data thread [70].
Figure 1: GATK Parallelization Decision Workflow
Scatter-Gather Methodology
Scatter-gather operates at the cluster or cloud level and involves separate GATK program copies running independently under a workflow execution system. The preferred pipelining solution for scatter-gather GATK analysis uses Cromwell as the workflow engine, executing scripts written in WDL [71].
In this approach, the execution engine creates separate GATK commands based on workflow script instructions, with each command running on different data intervals in parallel. For example, in somatic variant calling with Mutect2, the genome can be scattered across chromosomes:
The results are subsequently gathered and merged for downstream analysis [17]. This method is particularly effective for tools like HaplotypeCaller in GATK4, where internal multithreading options have been largely abandoned in favor of scatter-gather parallelization [67].
Performance Comparison of Parallelization Methods
Table 2: Performance Characteristics of GATK Parallelization Methods
| Parallelization Method | Implementation Level | Optimal Use Case | Speedup Factor | Key Considerations |
|---|---|---|---|---|
| Multi-threading (-nct) | CPU/core level | Single machine with multiple cores | 5x with 16 threads [25] | Suboptimal scalability beyond 16 threads |
| Data-level Parallelization (-nt) | Machine level | Single machine with abundant memory | Varies by tool | Memory requirements scale linearly with threads |
| Scatter-Gather | Cluster/cloud level | Large datasets and compute clusters | 4x with 4 intervals [67] | Requires workflow management system |
| Spark-based | Distributed computing | Spark-enabled environments | Up to 4.85x for WGS [72] | Still in development for some tools |
Experimental Protocols for Performance Benchmarking
Benchmarking Methodology for GATK Performance
To systematically evaluate GATK performance, researchers should establish controlled benchmarking environments and methodologies. The following protocol outlines a comprehensive approach:
Hardware Standardization: Conduct tests on consistent hardware configurations, ideally using Skylake Xeon Gold 6148 processors with 40 cores and 192 GB RAM, or equivalent cloud instances like Amazon c5.18xlarge [25].
Data Selection: Use standardized datasets such as NA12878 WGS data sequenced to ~20X depth, with aligned BAM files as starting point. For preliminary tests, chromosome 21 data alone can be used to reduce computation time during multiple parameter tests [25].
Metric Collection: Monitor walltime, CPU utilization, memory consumption, and I/O operations for each tool and parameter combination. The Linux top command can track cores engaged and threads spawned [25].
Java Environment Optimization: Configure Java garbage collection parameters to reduce overhead. For GATK4, parallel garbage collection should be enabled with appropriate heap size settings, which can effectively cut overall analysis time in half [67].
Resource Optimization Experiments
Memory and Heap Size Optimization: Experimental trials demonstrate that optimized Java garbage collection and heap size settings for GATK applications including SortSam, MarkDuplicates, HaplotypeCaller, and GatherVcfs can reduce overall analysis time by approximately 50% [67]. The optimal settings vary by specific tool and data size, requiring empirical determination for each workflow.
Native PairHMM Thread Configuration:
For HaplotypeCaller, the native pair Hidden Markov Model (HMM) implementation thread count (controlled by --native-pair-hmm-threads) significantly impacts performance. Testing reveals that four native pairHMM threads provides the best runtime for individual HaplotypeCaller processes, while using a single thread enables more parallel processes in scatter-gather approaches [67].
Tool-Specific Thread Scalability: Performance benchmarking shows different GATK tools have distinct scaling characteristics. BaseRecalibrator and HaplotypeCaller achieve approximately 5-fold speedup with 16 threads compared to single-threaded execution, but do not scale effectively beyond this point. PrintReads shows optimal performance with only 3 threads, with degraded performance at higher thread counts [25].
Table 3: Essential Research Reagents and Computational Resources for GATK Analysis
| Resource Category | Specific Tool/Resource | Function in Analysis Workflow |
|---|---|---|
| Variant Callers | MuTect2 [17] | Primary somatic variant detection using Bayesian statistical methods |
| Germline Filters | Germline resource (e.g., gnomAD) [17] | Filters out common germline variants from somatic calls |
| Artifact Detection | Panel of Normals (PON) [17] | Identifies technical artifacts present in normal samples |
| Workflow Management | Cromwell with WDL scripts [71] | Enables scatter-gather parallelization across distributed systems |
| Data Processing | Apache Spark [71] [72] | Provides distributed computing framework for data-intensive operations |
| In-Memory Analytics | Apache Arrow [72] | Enables efficient columnar in-memory data representation to minimize I/O |
| Containerization | Docker/Singularity [9] [67] | Ensures reproducibility and simplifies software deployment |
| Reference Resources | Reference genome (e.g., hg19, hg38) [17] | Provides standardized coordinate system for variant calling |
Advanced Optimization Techniques
In-Memory Data Frameworks
Traditional GATK workflows experience significant disk I/O bottlenecks due to repeated file access. Integration with Apache Arrow, a cross-language development platform providing columnar in-memory data format, can dramatically improve performance [72].
Implementation of Apache Arrow's in-memory SAM format and shared memory objects in genomics data processing applications enables:
- Avoidance of (de)-serialization and copy overheads between processes
- Better cache locality through columnar data representation
- Parallel processing capabilities through shared memory objects
Experimental results demonstrate this approach achieves speedup of 4.85x for WGS and 4.76x for WES data in overall variant calling workflow execution time [72].
Workflow-Specific Optimizations
Somatic Variant Calling with Mutect2: For scattered execution of Mutect2 across cluster nodes, additional steps are required for proper results integration. Stats files from each scatter must be merged using MergeMutectStats before filtering with FilterMutectCalls [17]:
Read Orientation Artifact Handling: The Mutect2 read orientation filter requires a three-step process:
- Generate F1R2 counts during Mutect2 execution using
--f1r2-tar-gz - Learn orientation bias model with LearnReadOrientationModel
- Apply the model during filtering with FilterMutectCalls using
--ob-priors[17]
Figure 2: Optimized Somatic Variant Calling Workflow with Parallelization
Optimizing GATK for scalable analysis requires a multifaceted approach addressing system configuration, parallelization strategies, and workflow-specific tuning. The fundamental requirements include Linux-based systems with adequate Java support, while performance optimization leverages both multi-threading for single-machine deployments and scatter-gather for distributed computing environments [9] [71].
Emerging technologies like Apache Arrow for in-memory analytics [72] and continued improvements in GATK's Spark implementation [9] promise further performance enhancements. However, researchers must balance speed against computational costs, selecting the appropriate optimization strategy based on their specific use case, dataset size, and available infrastructure [25].
For somatic variant calling in time-sensitive situations such as clinical applications, GATK3.8 with scatter-gather across multiple nodes provides the fastest processing times, while for cost-effective large-scale population studies, GATK4 processing multiple samples on a single node offers better resource utilization [25]. By implementing the system requirements and optimization strategies outlined in this guide, researchers can achieve efficient, scalable genomic analysis pipelines suited to their specific research objectives.
Benchmarking Performance and Ensuring Clinically Actionable Results
The accurate detection of somatic variants is a critical component of cancer genome analysis, with direct implications for understanding tumorigenesis, developing targeted therapies, and advancing precision oncology [73]. Within the genomic analysis ecosystem, the Genome Analysis Toolkit (GATK) from the Broad Institute provides a comprehensive framework for variant discovery, with its Best Practices workflows serving as industry standards for many applications [9] [32]. Among its toolkit, Mutect2 has emerged as a widely adopted solution for somatic short variant discovery, functioning as the core engine in GATK's somatic variant calling workflow [11] [12].
The performance characteristics of somatic variant callers vary significantly based on their underlying algorithms, filtering strategies, and the specific biological and technical contexts in which they are deployed [74]. While Mutect2 represents a powerful and versatile option, its performance must be evaluated against other established callers such as Strelka2, FreeBayes, and emerging deep-learning approaches to provide meaningful guidance for researchers and clinicians. This technical assessment provides a comprehensive benchmarking analysis of Mutect2 against contemporary somatic variant callers, focusing on empirical performance metrics across diverse experimental conditions and offering evidence-based recommendations for implementation within genomic research and drug development pipelines.
Methodological Approaches for Comparative Benchmarking
Experimental Designs for Variant Caller Evaluation
Rigorous benchmarking of somatic variant callers requires carefully designed methodologies that balance controlled simulations with real-world biological data. The studies reviewed herein employed multiple complementary approaches to assess caller performance. Synthetic datasets with known ground truth variants provide the most controlled environment for calculating precise performance metrics. One comprehensive evaluation introduced 4,709 single-nucleotide variants (SNVs) from the COSMIC database into a variant-free BAM file using BAMSurgeon (v1.4.1), creating artificial tumor and germline samples with mean variant allele frequency (VAF) of 50% and mean sequencing depth of 99× at variant positions [74] [75]. This approach enabled direct measurement of recall (sensitivity) and precision (specificity) for each variant caller.
Alternative methodologies have employed mixed cell line samples to create reference standards with biologically authentic variant profiles. One large-scale benchmark utilized 39 mixtures of six pre-genotyped normal cell lines to generate 354,258 control positive mosaic SNVs and indels alongside 33,111,725 control negatives, creating a robust ground truth for evaluating variant detection accuracy across a wide VAF spectrum (0.5-56%) [76]. This design specifically mimicked mutation acquisition processes during cellular differentiation and enabled testing of both shared and sample-specific mosaic variants.
For real-world performance validation, researchers have turned to established cancer cell lines with extensively characterized variant profiles. Studies utilizing COLO829 (metastatic melanoma) and HCC1395 (breast cancer) cell lines leveraged their well-defined truth sets—42,993 SNVs and 985 indels for COLO829, and 39,447 SNVs and 1,602 indels for HCC1395—to evaluate caller performance under biologically authentic conditions [73]. These approaches typically implement stringent criteria for variant inclusion in benchmarking, requiring minimum coverage (≥4×), alternative allele support (≥3 reads), and VAF thresholds (≥0.05) to ensure reliable performance assessment.
Analysis Workflows and Performance Metrics
Consistent bioinformatic processing is essential for meaningful variant caller comparisons. The evaluated studies typically employed standardized pipelines such as nf-core/sarek (v3.5.0) for data preprocessing, including read alignment to reference genomes (GRCh38) using BWA-MEM and duplicate marking [74] [75]. Variant callers were run with default parameters using their recommended filtering strategies: Mutect2 and Strelka2 outputs were filtered to retain only "PASS" variants, while FreeBayes required additional post-processing with quality and strand bias filters (QUAL ≥1, SAF>0, SAR>0, RPL>1, RPR>1) [74].
Performance assessment incorporated multiple complementary metrics. Recall (TP/[TP+FN]) and precision (TP/[TP+FP]) were calculated for synthetic datasets where ground truth was known. For real samples without complete ground truth, concordance analysis and variant characteristics (VAF, read depth) provided insights into caller behavior. More comprehensive evaluations employed area under the precision-recall curve (AUPRC) and F-scores (harmonic mean of precision and recall) to evaluate performance across different VAF ranges and sequencing depths [73] [15] [76]. Ensemble approaches such as SomaticSeq were also implemented to assess potential performance gains from integrating multiple callers [74].
Performance Benchmarking Results
Comparative Performance Across Variant Callers
Multiple studies have consistently demonstrated that somatic variant callers exhibit distinct performance characteristics influenced by their underlying algorithms and filtering approaches. A systematic evaluation using synthetic WES data with 4,709 known SNVs revealed that while all three major callers (Mutect2, Strelka2, and FreeBayes) achieved high precision (~99.9%), their recall rates varied substantially: Mutect2 achieved the highest recall (63.1%), followed by Strelka2 (46.3%) and FreeBayes (45.2%) [74]. This pattern of high specificity with variable sensitivity was echoed in real ovarian cancer WES samples, where FreeBayes detected the largest number of variants overall, but only 5.1% of SNVs were consistently identified across all three callers, highlighting substantial differences in variant detection preferences [74] [75].
Table 1: Performance Comparison of Somatic Variant Callers in Synthetic WES Data
| Variant Caller | Recall (%) | Precision (%) | Key Characteristics |
|---|---|---|---|
| Mutect2 | 63.1 | ~99.9 | Highest recall; Bayesian model with local assembly; best for VAF >10% |
| Strelka2 | 46.3 | ~99.9 | Position-wise probabilistic model; efficient for high-confidence calls; detects VAF down to ~5% |
| FreeBayes | 45.2 | ~99.9 | Originally for germline; flexible; reports VAF as low as 0.01-0.05 with higher false positive risk |
Analysis of caller-exclusive variants revealed systematic differences in the types of variants detected. Mutect2-specific calls typically exhibited higher VAFs and coverages, aligning with its reported strength in detecting variants above ~10% VAF [74]. In contrast, Strelka2 demonstrated enhanced capability for detecting lower-frequency variants, with performance advantages at VAFs down to ~5% [74] [15]. FreeBayes, while detecting the largest total number of variants, exhibited a more permissive profile with increased potential for false positives, particularly in tumor-only analysis contexts [74].
Performance Across Variant Allele Frequencies and Sequencing Depths
Variant caller performance is strongly influenced by technical parameters such as variant allele frequency and sequencing depth. A comprehensive evaluation across 30 combinations of sequencing depth and mutation frequency revealed important patterns in caller behavior [15]. For variants with higher mutation frequency (≥20%), sequencing depths ≥200× were sufficient to call 95% of mutations with both Mutect2 and Strelka2 performing comparably well, though Strelka2 showed a slight advantage in these conditions [15].
At lower VAF ranges (≤10%), Mutect2 demonstrated superior performance compared to Strelka2, particularly as sequencing depth increased [15]. This advantage became more pronounced at the lowest VAF (1%), where Mutect2's F-score surpassed Strelka2 when sequencing depth reached 500× [15]. These findings highlight the context-dependent strengths of each caller and suggest that optimal caller selection depends on the specific variant characteristics expected in a given study.
Table 2: Performance at Different Sequencing Depths and VAF Ranges
| Condition | Mutect2 Performance | Strelka2 Performance | Recommendations |
|---|---|---|---|
| High VAF (≥20%) | Strong performance | Slightly better performance | Sequencing depth ≥200× sufficient for 95% detection |
| Low VAF (5-10%) | Better recall, slightly lower precision | Higher precision, lower recall | Mutect2 preferred for sensitivity; Strelka2 for specificity |
| Very Low VAF (1%) | F-score surpasses Strelka2 at 500× | Better at lower depths (100×-300×) | Mutect2 benefits more from increased sequencing depth |
| Tumor-Only Analysis | Moderate precision, high sensitivity | N/A | Additional filtering essential; emerging tools like ClairS-TO show promise |
Emerging Callers and Specialized Applications
Recent advances in somatic variant calling have introduced new approaches, particularly deep-learning methods that show promise in specific applications. ClairS-TO, a deep-learning-based method designed for long-read tumor-only somatic variant calling, employs an ensemble of two disparate neural networks trained for complementary tasks—determining how likely a candidate is a somatic variant (affirmative network) and how likely it is not a somatic variant (negational network) [73]. Benchmarking using ONT and PacBio long-read data demonstrated that ClairS-TO outperformed DeepSomatic, and in Illumina short-read data at 50-fold coverage, it surpassed Mutect2, Octopus, Pisces, and DeepSomatic [73].
In the specialized context of mosaic variant detection, comprehensive benchmarking of 11 detection approaches revealed distinct performance patterns across variant types and VAF ranges [76]. For mosaic SNVs, Mutect2 in tumor-only mode (MT2-to) and MosaicForecast showed the best performance in low to medium VAF ranges (4-25%), with MT2-to exhibiting higher sensitivity and lower precision than MosaicForecast [76]. The performance gain for both tools was most pronounced at low VAFs (<10%), though with elevated false positive rates (0.69 and 0.71 false positives per 1 Mbp for MT2-to and MF, respectively) [76].
Implementation Guidelines and Best Practices
Recommendations for Caller Selection
Based on the comprehensive benchmarking evidence, caller selection should be guided by specific research objectives, sample characteristics, and technical parameters. For standard tumor-normal paired analysis where sensitivity is prioritized, particularly for variants with VAF >10%, Mutect2 represents an optimal choice based on its superior recall rates while maintaining high precision [74]. When analyzing samples with expected low-frequency variants (5-10% VAF), Mutect2 provides better sensitivity, though Strelka2 may be preferred when prioritizing specificity or computational efficiency [15].
For tumor-only analysis contexts, where matched normal samples are unavailable, additional precautions are necessary. While Mutect2 can be run in tumor-only mode, this approach generates substantially more false positives without careful filtering [12] [77]. In these scenarios, implementation of robust panel-of-normal (PoN) resources, germline frequency filters, and additional annotation-based filtering is essential [73] [12]. Emerging approaches like ClairS-TO show promise for tumor-only contexts but require further validation in diverse sample types [73].
In resource-constrained environments or high-throughput settings where computational efficiency is paramount, Strelka2 offers significant advantages, with reported execution speeds 17-22 times faster than Mutect2 on average [15]. However, for projects where comprehensive variant detection is the primary objective, particularly those involving deeper sequencing (≥500×), Mutect2's enhanced sensitivity at lower VAFs justifies its additional computational requirements [15].
Ensemble Approaches and Enhanced Workflows
Given the substantial differences in variants detected by individual callers, ensemble approaches represent a powerful strategy for maximizing detection accuracy. Evidence demonstrates that integrating calls from multiple callers using tools like SomaticSeq in consensus mode (combining Mutect2 and Strelka2) retains variants with stronger allelic signals—typically exhibiting higher VAFs and coverages—while filtering caller-specific false positives [74]. This approach leverages the complementary strengths of different callers, potentially overcoming limitations inherent in any single algorithm.
The GATK Best Practices workflow for somatic short variant discovery provides a comprehensive framework for Mutect2 implementation, incorporating critical preprocessing steps, contamination estimation (GetPileupSummaries, CalculateContamination), orientation bias modeling (LearnReadOrientationModel), and specialized filtering (FilterMutectCalls) [11] [12]. For challenging sample types such as FFPE-derived DNA, additional processing to address formalin-induced artifacts is essential, including the generation of F1R2 metrics for read orientation analysis [12].
The creation and application of panels of normals (PoNs) represents another crucial enhancement for somatic variant detection, particularly in tumor-only contexts. Mutect2's PoN functionality allows systematic filtering of technical artifacts and common germline variants not adequately represented in population frequency databases [12]. For specialized applications, such as mitochondrial variant calling or detection of somatic mosaicism, Mutect2 offers dedicated modes with optimized parameters for these specific contexts [12] [76].
Table 3: Key Experimental Resources for Somatic Variant Detection Studies
| Resource/Reagent | Function/Application | Implementation Notes |
|---|---|---|
| BAMSurgeon | Spiking known variants into BAM files for synthetic dataset generation | Parameters: --mindepth 50 --maxdepth 500 --minmutreads 5; enables precision/recall calculation [74] |
| nf-core/sarek | Standardized pipeline for variant calling analysis | Provides consistent processing baseline; version 3.5.0 used in benchmarking [74] |
| Panel of Normals (PoN) | Filtering technical artifacts and common germline variants | Created from normal samples using Mutect2 in tumor-only mode + CreateSomaticPanelOfNormals [12] |
| germline-resource (e.g., gnomAD) | Allele frequency database for germline filtering | Critical for tumor-only analysis; provides population allele frequencies [12] |
| SomaticSeq | Ensemble approach for integrating multiple callers | Consensus mode combines Mutect2 + Strelka2; improves variant confidence [74] |
| Cell Line Mixtures (COLO829, HCC1395) | Benchmarking with known truth sets | COLO829: 42,993 SNVs, 985 indels; HCC1395: 39,447 SNVs, 1,602 indels [73] |
Comprehensive benchmarking of Mutect2 against contemporary somatic variant callers reveals a complex performance landscape where optimal tool selection depends on specific research contexts and objectives. Mutect2 demonstrates particular strengths in overall sensitivity, especially for variants with VAF >10% and in low-VAF contexts with sufficient sequencing depth. Strelka2 offers advantages in computational efficiency and high-VAF precision, while emerging deep-learning approaches like ClairS-TO show promise for tumor-only and specialized applications.
The substantial discordance in variants detected across different callers—with as little as 5.1% overlap reported in some studies—strongly supports the adoption of ensemble approaches that leverage the complementary strengths of multiple callers. Implementation of the complete GATK Best Practices workflow for somatic variant discovery, including proper preprocessing, contamination estimation, and specialized filtering, is essential for maximizing Mutect2's performance in both research and clinical contexts.
As somatic variant calling continues to evolve with new technologies like long-read sequencing and deep-learning methodologies, ongoing benchmarking and validation will remain crucial for ensuring accurate variant detection in cancer genomics. The evidence-based guidelines presented here provide a foundation for informed tool selection and implementation, ultimately supporting more reliable genomic analysis in both basic research and drug development applications.
The Impact of Sequencing Depth and Mutation Frequency on Caller Accuracy
Within the framework of research on the GATK somatic variant calling workflow, the accurate detection of somatic mutations is a cornerstone of cancer genomics, directly influencing the understanding of tumorigenesis, heterogeneity, and therapeutic target identification [15] [78]. The precision of this detection is not absolute but is governed by critical experimental parameters, principally sequencing depth and mutation frequency [15]. Sequencing depth, or coverage, determines the number of times a genomic base is sequenced, influencing the power to distinguish true variants from stochastic sequencing errors. Mutation frequency, often reflecting tumor purity or subclonality, presents a significant challenge, particularly for low-frequency variants that may drive resistance or early progression [15]. The complex interplay between these parameters and the choice of variant calling algorithm, such as the widely adopted GATK Mutect2, defines the sensitivity and specificity of the entire analytical pipeline [11] [15] [79]. This whitepaper provides an in-depth technical analysis of how sequencing depth and mutation frequency impact caller accuracy, synthesizing recent benchmarking studies to offer a quantitative guide for researchers and drug development professionals optimizing somatic variant discovery workflows.
Core Concepts and Workflow
The Somatic Variant Calling Workflow with GATK Mutect2
The GATK (Genome Analysis Toolkit) is an industry-standard suite for variant discovery, with its Mutect2 tool specifically engineered for somatic short variant discovery (SNVs and Indels) [9] [11]. The Best Practices workflow for somatic variants is a multi-step process designed to maximize accuracy and minimize false positives.
caption: Simplified GATK Somatic Variant Discovery Workflow
The workflow begins with pre-processed BAM files for tumor and normal samples [11]. The core variant calling is performed by Mutect2, which uses local de-novo assembly of haplotypes in active regions showing signs of variation to call SNVs and indels simultaneously [11]. Subsequent steps are critical for filtering artifacts: GetPileupSummaries and CalculateContamination estimate cross-sample contamination; LearnReadOrientationModel identifies and models orientation biases, which is especially important for FFPE samples; and FilterMutectCalls applies probabilistic models to filter alignment artifacts, germline variants, and other false positives [11]. The final step often involves annotation using tools like Funcotator to add clinical and biological context to the variants [11].
Key Definitions: Sequencing Depth and Mutation Frequency
- Sequencing Depth (Coverage): The average number of sequencing reads that align to a specific genomic base. A depth of 100X means each base is covered by 100 reads on average. Higher depth increases the probability of detecting low-frequency variants present in only a fraction of cells.
- Mutation Frequency (Variant Allele Frequency - VAF): The proportion of sequencing reads at a genomic locus that contain a specific alternative allele compared to the total number of reads covering that locus. In cancer genomics, VAF is influenced by tumor purity, ploidy, and the clonal structure of the tumor [15].
Quantitative Impact of Depth and Frequency on Accuracy
A systematic study investigated the performance of Mutect2 and Strelka2 across 30 combinations of sequencing depth (100X, 200X, 300X, 500X, and 800X) and mutation frequency (1%, 5%, 10%, 20%, 30%, and 40%) using down-sampled whole-exome sequencing data from standard DNA samples [15]. The results provide a quantitative framework for decision-making.
Table 1: Impact of Sequencing Depth and Mutation Frequency on Mutect2 F-Score [15]
| Mutation Frequency | 100X | 200X | 300X | 500X | 800X |
|---|---|---|---|---|---|
| 1% | 0.05 - 0.19 | 0.12 - 0.26 | 0.17 - 0.30 | 0.32 - 0.41 | 0.44 - 0.50 |
| 5-10% | 0.65 - 0.82 | 0.83 - 0.91 | 0.88 - 0.93 | 0.91 - 0.94 | 0.92 - 0.95 |
| ≥20% | 0.94 - 0.95 | 0.95 - 0.96 | 0.95 - 0.96 | 0.95 - 0.96 | 0.95 - 0.96 |
F-Score is the harmonic mean of precision and recall, where 1.0 is perfect accuracy.
The data reveals two critical thresholds. First, for mutation frequencies ≥20%, a sequencing depth of 200X is sufficient to achieve excellent accuracy (F-score >0.95), with diminishing returns from further increases in depth [15]. Second, for low-frequency mutations (≤10%), accuracy becomes highly dependent on both parameters. While increasing depth from 100X to 800X can improve the F-score for 5-10% mutations from ~0.65 to ~0.95, the performance at a 1% mutation frequency remains poor (F-score ≤0.50 even at 800X) [15]. This suggests that for very low-frequency variants, improving the experimental method (e.g., increasing tumor purity) is more effective than simply sequencing deeper.
Comparative Performance of Variant Callers
The choice of variant caller is another major factor affecting accuracy. The same systematic study directly compared GATK Mutect2 and Strelka2.
Table 2: Mutect2 vs. Strelka2 Performance Across Mutation Frequencies [15]
| Metric | Condition | Strelka2 Performance | Mutect2 Performance |
|---|---|---|---|
| F-Score | Mutation Frequency ≥20% | Slightly higher (Difference <1%) | Slightly lower (Difference <1%) |
| F-Score | Mutation Frequency 5-10% | Lower (0.64 - 0.94) | Higher (0.65 - 0.95) |
| F-Score | Mutation Frequency 1% (at 500X-800X) | Lower (0.27 - 0.37) | Higher (0.32 - 0.50) |
| Precision | Mutation Frequency 5-10% | Higher (96.2 - 96.5%) | Lower (95.5 - 95.9%) |
| Recall | Mutation Frequency 5-10% | Lower (48 - 93%) | Higher (50 - 96%) |
| Runtime | Across all tests | 17 to 22 times faster | Slower |
The analysis shows that both callers perform excellently for high-frequency mutations (≥20%) [15]. However, Mutect2 demonstrates a significant advantage in sensitivity (recall) for low-frequency mutations (≤10%), achieving a higher F-score in this critical range [15]. A separate, recent benchmarking study using tumor WES data from the FDA-led Sequencing Quality Control Phase 2 project confirmed Mutect2's robust performance, identifying the combinations of BWA with Mutect2 and HISAT2 with Mutect2 as top-performing open-source solutions for SNV and indel detection, respectively [79].
Table 3: Key Research Reagents and Computational Resources for Somatic Variant Discovery
| Item | Function & Application | Example Sources / Tools |
|---|---|---|
| Reference Genome | Baseline for read alignment and variant calling. | GRCh37, GRCh38 [34] |
| BWA-MEM Aligner | Aligns sequencing reads to the reference genome; considered a gold standard. | BWA [79] [24] |
| Panel of Normals (PON) | A VCF of artifacts found in normal samples; used to filter common technical artifacts in tumor-only mode. | Created with Mutect2 [11] [34] |
| Germline Resource | Database of population allele frequencies to help filter common germline variants. | gnomAD [80] [34] |
| Funcotator Datasources | Provide biological and clinical context for called variants (e.g., gene effect, COSMIC annotation). | GENCODE, dbSNP, COSMIC, ClinVar [11] |
| Cloud & HPC Platforms | Provide scalable computational resources for running intensive variant calling workflows. | DNAnexus, Terra, Illumina BaseSpace [34] |
Best Practices and Implementation
Experimental Design and Workflow Recommendations
- For High Purity Tumors (Mutation Frequency ≥20%): Design sequencing experiments with a minimum depth of 200X. This provides an optimal balance of cost and accuracy, as higher depths yield minimal improvement [15].
- For Low Purity/Subclonal Tumors (Mutation Frequency 5-10%): Prioritize sequencing depth of ≥500X and use Mutect2 to maximize sensitivity. Be aware that precision may slightly decrease compared to higher frequency mutations [15].
- For Very Low-Frequency Mutations (~1%): Current WES technologies struggle with reliable detection. Consider error-corrected sequencing techniques or deep targeted sequencing (>1000X) if such variants are of primary interest [15].
- Leverage a Panel of Normals (PON): Always use a PON when available. It is a critical resource for filtering out recurring technical artifacts that otherwise manifest as false positive calls [11] [34].
A Simplified Workflow for Clinical and Research Settings
caption: Decision Framework for Sequencing and Analysis
The accuracy of somatic variant calling is a direct function of deliberate experimental and bioinformatic design. The quantitative data demonstrates that a one-size-fits-all approach is inadequate. For high-frequency mutations, a 200X depth is sufficient, but for the low-frequency variants that are increasingly of clinical interest, a combination of higher sequencing depth (≥500X) and a sensitive caller like GATK Mutect2 is imperative. Researchers must balance the pursuit of sensitivity with the reality of rising costs and computational resources. By aligning their experimental parameters with the established performance characteristics of the GATK somatic variant calling workflow, scientists and drug developers can ensure the reliable detection of mutations necessary to advance cancer research and precision medicine.
The accurate identification and interpretation of somatic variants are fundamental to precision oncology, directly influencing diagnosis, prognosis, and treatment decisions. To standardize this complex process, professional organizations have established guidelines that provide a structured framework for variant classification and reporting. Chief among these are the Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer, developed through a joint consensus recommendation by the Association for Molecular Pathology (AMP), American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP) [81]. These guidelines establish a tiered system for classifying somatic variants based on their clinical significance, which has been widely adopted in clinical care since their publication in 2017 [82].
Within bioinformatics pipelines, such as the GATK (Genome Analysis Toolkit) somatic variant calling workflow, these guidelines provide the critical interpretive layer that transforms raw variant calls into clinically actionable information. The GATK Best Practices offer step-by-step recommendations for variant discovery in high-throughput sequencing data, encompassing data pre-processing, variant discovery, and filtering phases [20]. However, the final determination of a variant's clinical significance relies heavily on the structured evidence-based framework provided by the AMP/ASCO/CAP guidelines. This integration ensures that the technical accuracy of the variant calling process is matched by the clinical validity and utility of the reported results, creating a complete workflow from raw sequencing reads to clinically reportable findings [78].
The AMP/ASCO/CAP Guideline Framework: Core Principles and Updates
Original 2017 Tiered Classification System
The original 2017 AMP/ASCO/CAP guidelines established a four-tiered system for categorizing somatic variants based on their documented or potential clinical significance in diagnostics, prognostics, and therapeutics [81] [83]. This framework was designed to create consistency in how laboratories interpret and report cancer-related sequence variants. The tiers are defined as follows:
- Tier I: Variants with strong clinical significance, subdivided into:
- Tier IA: Variants linked to FDA-approved therapies or recognized by professional guidelines
- Tier IB: Variants with strong clinical significance based on well-powered, consensus-driven studies
- Tier II: Variants with potential clinical significance, including:
- Tier IIC: Variants associated with investigational therapies or approved therapies for different tumor types
- Tier IID: Variants supported by evidence from small published studies or case reports
- Tier III: Variants of unknown clinical significance (VUS)
- Tier IV: Benign or likely benign variants [83]
This structured approach helped standardize variant reporting across laboratories, but implementation revealed challenges in classification consistency, particularly for variants with confirmed oncogenic properties but unclear clinical implications [84].
2025 Guideline Updates: Addressing Classification Gaps
A proposed update to the guidelines in 2025 introduces Tier IIE to resolve a critical ambiguity in the original framework. This new category specifically accommodates variants classified as "oncogenic" or "likely oncogenic" based on biological evidence but lacking clear evidence for diagnostic, prognostic, or therapeutic significance in the specific tumor type tested [83].
This addition addresses a fundamental challenge faced by laboratories: how to classify molecularly confirmed cancer-driving mutations that lack established clinical utility. Before this update, laboratories had to either classify these variants as Tier III (VUS), creating confusion by grouping cancer-driving variants with truly unknown variants, or stretch evidence to fit them into Tier II categories [83]. The Tier IIE designation provides a more nuanced classification that distinguishes the variant's established oncogenic role from its current clinical applicability, thereby preserving the integrity of both the VUS category and the clinical significance tiers [83].
Table 1: AMP/ASCO/CAP Somatic Variant Classification Tiers
| Tier | Category | Clinical Significance | Evidence Level |
|---|---|---|---|
| Tier I | Strong Clinical Significance | FDA-approved therapies or professional guidelines | Level A (IA) / Consensus-driven (IB) |
| Tier II | Potential Clinical Significance | Varying levels of evidence | Investigational therapies (IIC) to case studies (IID) |
| Tier IIE | Oncogenic but Lacking Clinical Evidence | Oncogenic/likely oncogenic but no clear clinical utility | Biological oncogenicity established |
| Tier III | Unknown Significance (VUS) | Uncertain role in cancer | Limited or conflicting evidence |
| Tier IV | Benign/Likely Benign | Not associated with cancer | Population frequency and functional evidence |
Implementing Guidelines in Somatic Variant Analysis Workflows
Integration with GATK Somatic Variant Calling
The GATK Best Practices workflow for somatic short variant discovery provides a standardized computational approach for identifying somatic mutations from next-generation sequencing data [20] [9]. This workflow typically follows three main phases: data pre-processing, variant discovery, and post-processing filtration [20]. The AMP/ASCO/CAP guidelines interface with this pipeline primarily at the variant interpretation stage, after technical variant calling is complete.
The GATK somatic SNVs and indels workflow involves alignment to a reference genome, base quality score recalibration, duplicate marking, and variant calling using tools such as Mutect2 [85] [9]. The resulting variant calls must then be filtered and annotated before the critical step of clinical interpretation according to the AMP/ASCO/CAP framework [20]. This integration ensures that the technical variant identification achieves clinical relevance through standardized interpretation.
Table 2: Key Tools and Resources for Somatic Variant Analysis
| Tool Category | Examples | Primary Function | Guideline Relevance |
|---|---|---|---|
| Variant Callers | GATK Mutect2, Strelka2 | Identify somatic SNVs/indels | Generates raw variant data for interpretation |
| Annotation Tools | ANNOVAR, Ensembl VEP, SnpEff | Functional variant annotation | Provides evidence for classification |
| Clinical Databases | CIViC, COSMIC, ClinVar | Curated clinical evidence | Supports therapeutic, prognostic evidence |
| Classification Tools | QCI Interpret, omnomicsNGS | Semi-automated classification | Applies AMP/ASCO/CAP rules |
| Quality Control | omnomicsQ, Picard | QC metrics monitoring | Ensures analytical validity |
Practical Implementation Framework
Implementing the AMP/ASCO/CAP guidelines requires systematic approaches to address common challenges. Research by Parikh et al. (2020) demonstrated that laboratories can successfully map their internally developed classification systems to the AMP/ASCO/CAP framework, with most "actionable" variants and variants of uncertain significance mapping consistently to Tiers I-III [84]. However, they noted particular variability in classifying certain variants, such as TP53 mutations, highlighting the need for gene-specific expertise in the interpretation process [84].
A successful implementation framework includes:
- Evidence Collection: Systematic gathering of clinical and functional evidence from authoritative databases such as CIViC (Clinical Interpretation of Variants in Cancer), COSMIC (Catalogue of Somatic Mutations in Cancer), and ClinVar [78].
- Multidisciplinary Review: Incorporating perspectives from molecular pathologists, clinical oncologists, and geneticists to evaluate the strength of evidence for each variant.
- Tier Assignment: Applying the AMP/ASCO/CAP criteria consistently across all variants, with special attention to borderline cases that may require discussion at a molecular tumor board.
- Documentation and Reporting: Clearly communicating the tier assignment and supporting evidence in clinical reports, including the specific criteria used for classification [84].
The following diagram illustrates the complete integrated workflow from raw sequencing data to clinically classified variants:
Validation and Quality Assurance in Guideline Implementation
Analytical Validation Frameworks
Ensuring the accuracy and reproducibility of somatic variant classification requires robust validation frameworks that address both analytical and interpretive components. Analytical validation begins with stringent quality control measures throughout the sequencing workflow. Tools such as omnomicsQ provide real-time monitoring of sequencing quality, automatically flagging samples that fall below predefined thresholds, thereby preventing the downstream analysis of low-quality samples that could lead to erroneous variant calls [78].
For the variant interpretation process itself, validation involves verifying that classification decisions align with the AMP/ASCO/CAP guidelines and are consistently applied across all cases. This can be achieved through:
- Internal proficiency testing: Regularly testing a set of well-characterized variants to ensure consistent classification.
- Blinded reclassification: Having different personnel independently classify the same variants to measure inter-reviewer concordance.
- External quality assessment (EQA): Participating in programs such as those offered by EMQN and GenQA, which facilitate cross-laboratory benchmarking and identification of discrepancies [78].
Regulatory Compliance and Standardization
For laboratories operating in clinical diagnostics, adherence to regulatory standards is essential for ensuring patient safety and result reliability. Key regulatory frameworks include:
- ISO 13485:2016: Defines quality management system requirements for medical devices and in vitro diagnostic products, ensuring documented design processes, risk management, and traceability [78].
- IVDR (In Vitro Diagnostic Regulation): Stricter European Union requirements for clinical evidence, performance evaluation, and post-market surveillance of diagnostic tools [78].
- HIPAA (US) and GDPR (EU): Mandate strict protection of patient data and genomic information throughout the testing process [78].
These regulatory frameworks complement the professional guidelines by ensuring that the entire testing process—from wet lab procedures to bioinformatics analysis and final interpretation—meets rigorous quality standards. Laboratories should implement solutions specifically designed to align with these frameworks, supporting regulatory-compliant validation, traceability, and data security [78].
Comparative Analysis of Somatic Variant Classification Systems
Emerging Classification Guidelines
While the AMP/ASCO/CAP guidelines have gained widespread adoption, other classification systems have been developed, including the guidelines published by the Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC) [86]. Understanding the similarities and differences between these frameworks is essential for comprehensive somatic variant interpretation.
A 2025 study compared classifications using the ClinGen/CGC/VICC oncogenicity guidelines with those generated by QIAGEN Clinical Insight (QCI) Interpret One software, which uses a version of the 2015 ACMG/AMP guidelines customized for somatic assessment [86]. The study analyzed 309 variants observed in cancer and found that for variants classified as "oncogenic" and "likely oncogenic" using the ClinGen/CGC/VICC guidelines, the QCI system showed 97.2% concordance [86]. However, the ClinGen/CGC/VICC standards generally led to more conservative classifications, with a larger proportion of variants assigned to the "variant of unknown significance" and "likely benign" categories compared to the software-based approach [86].
Table 3: Comparison of Somatic Variant Classification Systems
| System Attribute | AMP/ASCO/CAP | ClinGen/CGC/VICC | ACMG/AMP (Somatic-modified) |
|---|---|---|---|
| Primary Focus | Clinical actionability in oncology | Oncogenicity assessment | Pathogenicity assessment |
| Classification Structure | Four-tier system (plus IIE update) | Oncogenic/Benign categories | Pathogenic/Benign categories |
| Evidence Types | Therapeutic, prognostic, diagnostic | Functional, genetic, computational | Population, functional, computational |
| Clinical Orientation | Strong emphasis on clinical utility | Focus on biological oncogenicity | Adapted from germline interpretation |
| Reported Concordance | Reference standard | ~80% with QCI system | 97.2% for Oncogenic/Likely Oncogenic |
Resolving Classification Discrepancies
The concurrent use of multiple classification systems can lead to discrepancies that require careful resolution. The study comparing ClinGen/CGC/VICC guidelines and QCI Interpret found that while overall concordance was approximately 80% prior to manual review, certain patterns emerged in the discrepancies [86]. Specifically, the QCI classifications tended more toward "likely pathogenic" over "VUS" and "VUS" over "likely benign" compared to the more conservative ClinGen/CGC/VICC approach [86].
To resolve such discrepancies, laboratories should:
- Establish clear protocols for reviewing discordant classifications, including escalation to senior staff or molecular tumor board discussion.
- Document the rationale for final classification decisions, particularly when overriding automated software classifications.
- Implement ongoing training programs to ensure staff remain current with guideline updates and interpretation standards.
- Utilize clinical decision support software as an aid rather than a replacement for expert review, with supervision by cancer variant interpretation experts recommended when applying variant classifications [86].
The following diagram illustrates the decision pathway for somatic variant classification, highlighting key criteria and evidence sources:
Table 4: Key Research Reagent Solutions for Somatic Variant Analysis
| Resource Category | Specific Tools/Platforms | Primary Application | Guideline Integration |
|---|---|---|---|
| Variant Callers | GATK Mutect2, Strelka2, VarDict | Somatic SNV/indel detection | Generates primary variant data for interpretation |
| Annotation Platforms | ANNOVAR, Ensembl VEP, SnpEff | Functional impact prediction | Provides evidence for oncogenicity assessment |
| Clinical Databases | CIViC, COSMIC, OncoKB, ClinVar | Clinical evidence aggregation | Supports therapeutic, prognostic evidence for tiering |
| Quality Control | omnomicsQ, FastQC, Picard | QC metrics monitoring | Ensures analytical validity pre-classification |
| Classification Software | QCI Interpret, omnomicsNGS | Semi-automated classification | Applies AMP/ASCO/CAP rules consistently |
| Workflow Management | Cromwell, WDL, Snakemake | Pipeline orchestration | Ensures reproducible analysis |
| Visualization Tools | IGV, GenomeBrowse | Visual variant validation | Supports manual review of complex variants |
The field of somatic variant interpretation continues to evolve rapidly, with the AMP/ASCO/CAP guidelines undergoing regular updates to reflect technological advancements and accumulated clinical experience. The proposed 2025 update introducing Tier IIE represents a significant refinement that addresses a key implementation challenge experienced by laboratories [83]. Future developments are likely to focus on standardizing the interpretation of complex biomarkers such as mutational signatures, copy number variations, and structural variants, as well as providing more specific guidance for hematologic malignancies [82].
The integration of machine learning approaches presents another promising direction for enhancing somatic variant analysis. Methods such as VarRNA, which utilizes XGBoost models to classify variants from RNA-Seq data as artifact, germline, or somatic, demonstrate the potential for computational tools to augment but not replace expert interpretation [87]. As these tools mature, guidelines will need to incorporate standards for validating and implementing computational classifications within clinical decision-making frameworks.
In conclusion, adherence to AMP/ASCO/CAP and related professional guidelines provides the essential framework for standardizing somatic variant interpretation in cancer genomics. When integrated with robust bioinformatics pipelines such as the GATK Best Practices, these guidelines ensure that variant classification reflects both analytical accuracy and clinical relevance. As precision oncology continues to advance, maintaining this synergy between technical workflows and interpretive standards will be crucial for delivering clinically actionable results that ultimately improve patient care.
The Role of AI-Based Callers like DeepVariant in the Evolving Landscape
The accurate detection of genetic variants from next-generation sequencing (NGS) data represents a fundamental challenge in genomics, with profound implications for understanding inherited diseases, cancer progression, and therapeutic development. For years, the Genome Analysis Toolkit (GATK) has established itself as the industry standard, utilizing sophisticated statistical models and heuristic rules for variant calling. However, the emergence of artificial intelligence (AI) has catalyzed a transformative shift in computational genomics. AI-based callers like DeepVariant leverage deep learning architectures to achieve unprecedented accuracy by reimagining variant calling as an image classification problem. This technical guide examines the core mechanisms, performance advantages, and practical implementation of AI-based variant callers within the context of somatic variant detection workflows, providing researchers and drug development professionals with a comprehensive framework for evaluating and deploying these cutting-edge tools.
Technical Foundations: From Statistical Models to Deep Learning
GATK's Traditional Approach
The GATK workflow employs a series of statistical algorithms optimized through years of community development. Its HaplotypeCaller engine functions through a multi-step process: first, it identifies active regions based on signs of variation; second, it assembles reads into haplotypes using the DeBruijn graph approach; third, it realigns reads to the most likely haplotypes; and finally, it assigns genotype likelihoods using a pair-hidden Markov model (PairHMM) that calculates the probability of reads given possible haplotypes. This sophisticated but rules-based methodology requires extensive post-calling filtration to remove artifacts, with filters tailored to specific variant types and sequencing technologies. For somatic variant calling, MuTect2 (part of GATK) implements a Bayesian classifier that analyzes tumor and normal samples jointly to distinguish somatic mutations from germline variants and sequencing errors.
DeepVariant's AI Architecture
DeepVariant introduces a fundamentally different paradigm by reformulating variant calling as an image classification task. Rather than relying on hand-crafted statistical models, it employs a deep convolutional neural network (CNN) that learns to identify variants directly from data. The core innovation lies in its transformation of aligned sequencing data into multi-channel "pileup images" where each channel encodes different types of information:
- Reference bases: The reference sequence against which reads are aligned
- Read bases: The actual sequenced nucleotides from the sample
- Base qualities: Confidence scores for each base call from the sequencer
- Mapping qualities: Confidence scores for read alignment positions
- Strand information: Which DNA strand the read originated from
- Read haplotypes: Grouping of reads that support the same haplotype
- Supporting features: Additional sequencing context [88]
These tensor representations are processed through an Inception-style CNN architecture that learns hierarchical features directly from the training data, without relying on manually engineered features or thresholds. The network outputs genotype probabilities for each candidate site, effectively distinguishing true variants from sequencing artifacts through pattern recognition rather than statistical inference.
Evolution to Somatic Variant Calling: DeepSomatic
Building on DeepVariant's success in germline variant calling, Google Research developed DeepSomatic specifically for cancer genomics applications. This extension modifies the pileup image generation to incorporate both tumor and normal reads in a unified tensor representation, with normal sample reads positioned above tumor reads in the image stack. This architectural adaptation enables the model to simultaneously compare variation patterns between matched samples, allowing it to learn the distinguishing characteristics of somatic mutations directly from the data [89] [90]. The system identifies candidate positions that show evidence of variation in the tumor but not the normal sample, then classifies them using a CNN trained on verified somatic variants.
Table 1: Core Architectural Comparison Between GATK and AI-Based Approaches
| Feature | GATK/MuTect2 | DeepVariant/DeepSomatic |
|---|---|---|
| Core Methodology | Statistical models (Bayesian classification, PairHMM) | Deep convolutional neural networks |
| Data Representation | Processed alignment metrics and likelihoods | Multi-channel pileup images/tensors |
| Variant Filtering | Post-hoc filtering based on heuristic thresholds | Integrated classification with minimal filtering |
| Technology Adaptation | Manual parameter tuning per technology | Retraining with technology-specific data |
| Error Model | Explicit error modeling based on predefined assumptions | Implicit error learning from training data |
| Training Dependency | Limited dependency on training data | Heavy dependency on diverse training datasets |
Performance Benchmarking: Quantitative Comparisons
Germline Variant Calling Accuracy
Multiple studies have systematically compared the performance of DeepVariant and GATK across various metrics. A 2022 study published in Scientific Reports analyzed 80 clinical trios (240 exomes) and found that DeepVariant demonstrated significantly lower Mendelian error rates (3.09% ± 0.83%) compared to GATK (5.25% ± 0.91%), indicating more accurate inheritance patterns [91]. Additionally, DeepVariant achieved a higher transition-to-transversion (Ti/Tv) ratio (2.38 ± 0.02 versus 2.04 ± 0.07 for GATK), which is a key quality metric that reflects the proportion of expected versus unexpected nucleotide changes [91]. The concordance rate between the two pipelines was approximately 88.73%, with DeepVariant exhibiting better performance in low-coverage regions and more accurate quality score calibration [91].
In a separate study comparing variant callers in sporadic epilepsy and autism spectrum disorder cohorts, DeepVariant exhibited higher precision and sensitivity for single nucleotide variants (SNVs), while GATK showed advantages in detecting rare variants that are often critical for understanding genetic bases of rare diseases [92].
Somatic Variant Detection Performance
For cancer genomics, DeepSomatic has demonstrated remarkable performance gains, particularly for challenging mutation types. In comprehensive benchmarking using the CASTLE dataset (Cancer Standards Long-read Evaluation), DeepSomatic achieved a 98.3% F1-score for single nucleotide variants (SNVs) on Illumina data, outperforming established tools like MuTect2, Strelka2, and SomaticSniper [93] [90]. The most significant improvement was observed in indel detection, where DeepSomatic reached approximately 90% F1-score on Illumina data compared to 80% for the next-best tool—a substantial advancement given the importance of indels in cancer driver mutations [93].
On long-read sequencing data from PacBio HiFi and Oxford Nanopore platforms, DeepSomatic's advantages were even more pronounced, achieving over 80% F1-score for indels compared to less than 50% for comparator tools [93] [90]. This performance differential highlights the particular strength of deep learning approaches in handling the complex error profiles of emerging sequencing technologies.
Table 2: Performance Metrics Across Variant Types and Technologies
| Variant Type | Sequencing Technology | GATK/MuTect2 Performance | DeepVariant/DeepSomatic Performance |
|---|---|---|---|
| SNVs | Illumina Short-Read | F1-score: 0.9521 [89] | F1-score: 0.983 [93] |
| Indels | Illumina Short-Read | F1-score: ~0.80 [93] | F1-score: ~0.90 [93] |
| SNVs | PacBio HiFi | Moderate performance | High performance, exceeds 80% F1-score [90] |
| Indels | PacBio HiFi | F1-score: <0.50 [93] | F1-score: >0.80 [93] |
| Low-Frequency Variants (1-5% VAF) | Illumina | Recall: 2.7-34.5% [15] | Significantly improved recall [94] |
| FFPE-Derived Variants | Multiple | High false positive rate [95] | Effective artifact suppression [95] [90] |
Performance in Challenging Contexts
AI-based callers demonstrate particular advantages in challenging genomic contexts. For formalin-fixed paraffin-embedded (FFPE) samples—which represent the majority of clinical cancer archives—DeepVariant and DeepSomatic effectively suppress formalin-induced artifacts that traditionally plague variant callers [95] [90]. While FFPE samples show reduced recall (approximately 82% versus 95% on fresh-frozen samples), DeepSomatic maintains clinical utility where other tools struggle with false positives [93]. Similarly, in low-purity tumor samples (below 20% tumor content) and low-coverage sequencing (below 30x), AI-based approaches maintain more robust performance compared to statistical methods [94] [15].
Implementation Guide: Experimental Protocols and Workflows
DeepVariant Germline Variant Calling Protocol
The standard workflow for germline variant calling with DeepVariant consists of the following steps:
Input Preparation:
- Sequence reads in FASTQ format (after base calling)
- Reference genome in FASTA format with corresponding .fai index
Data Preprocessing:
- Read Alignment: Use BWA-MEM2 to align reads to the reference genome
- Sorting and Indexing: Process BAM files with samtools
- Duplicate Marking: Optionally mark duplicates using Picard or samtools
Variant Calling:
- Run DeepVariant: Execute the core calling process
- Post-processing: The output VCF contains quality-calibrated variants ready for analysis without additional filtering
Validation: The Mendelian concordance rate in trio analyses serves as a key validation metric, with expected rates exceeding 96% for well-characterized samples [91].
DeepSomatic Somatic Variant Calling Protocol
For somatic variant detection with DeepSomatic, the workflow incorporates matched tumor-normal pairs:
Input Requirements:
- Tumor and normal BAM files from the same individual
- Reference genome in FASTA format
- Optionally, a panel of normals (PoN) for further artifact suppression
Execution Steps:
- Preprocessing: Follow similar alignment and sorting steps as above for both tumor and normal samples
- Variant Calling: Run DeepSomatic in tumor-normal mode
- Tumor-Only Mode: When normal tissue is unavailable, use tumor-only mode with population frequency filters
Performance Considerations:
- Whole-exome sequencing: 15-30 minutes runtime on 96-core system
- Whole-genome sequencing: 3-6 hours on standard server infrastructure
- Memory requirements: Approximately 384GB RAM for WGS analyses [93]
Figure 1: Complete workflow for AI-based variant calling, illustrating the streamlined pipeline from raw sequencing data to clinical interpretation.
Customization for Specific Sequencing Technologies
A key advantage of DeepVariant and DeepSomatic is their adaptability to different sequencing platforms through model retraining:
Platform-Specific Models:
- Illumina short-read: Default models available
- PacBio HiFi: Requires HiFi-specific model for optimal indel performance
- Oxford Nanopore: ONT-specific model handles characteristic error profiles
- FFPE-derived DNA: Specialized models compensate for formalin damage
Customization Protocol:
- Training Data Curation: Collect validated variant sets for target technology
- Model Retraining: Use transfer learning from existing models
- Validation: Benchmark against gold standard variants
- Deployment: Implement technology-specific models in production
Research Reagent Solutions and Computational Requirements
Table 3: Essential Research Materials and Computational Resources
| Category | Specific Solution | Function/Role in Workflow |
|---|---|---|
| Reference Materials | Genome in a Bottle (GIAB) reference standards | Benchmarking and validation of variant calls |
| CASTLE dataset [90] | Somatic variant training and benchmarking | |
| FFPE reference samples | Optimization of damage-prone sample processing | |
| Sequencing Technologies | Illumina NovaSeq 6000 | High-throughput short-read sequencing |
| PacBio Revio System | Long-read HiFi sequencing for complex variants | |
| Oxford Nanopore PromethION | Ultra-long reads for structural variant detection | |
| Computational Resources | BWA-MEM2 aligner [95] | High-performance read alignment to reference |
| Google Cloud n2-standard-96 instances | Scalable computing infrastructure (96 vCPUs, 384GB RAM) | |
| NVIDIA V100 or A100 GPUs | Accelerated deep learning inference (optional) | |
| Specialized Kits | NEBNext UltraShear FFPE DNA Library Prep Kit [95] | Library preparation from damaged samples |
| Illumina TruSeq PCR-free library prep kit [95] | High-quality library construction for WGS | |
| Analysis Tools | snpEff [95] | Functional annotation of called variants |
| Integrative Genomics Viewer (IGV) | Visual validation of variant calls |
Integration with GATK-Based Somatic Workflows
For research laboratories with established GATK workflows, several integration strategies facilitate the adoption of AI-based callers:
Hybrid Calling Approaches
Many research groups implement parallel calling with both GATK and DeepVariant/DeepSomatic, then intersect the results to generate high-confidence call sets. This approach leverages the complementary strengths of both methodologies—GATK's sensitivity for rare variants and DeepVariant's precision for common polymorphisms [92]. The intersection strategy is particularly valuable for clinical applications requiring maximum specificity.
Quality Control Integration
DeepVariant's well-calibrated quality scores can enhance GATK-based workflows by providing independent validation of variant quality. Research demonstrates that DeepVariant's quality scores show better calibration across different coverage levels and variant types compared to GATK, making them valuable for downstream filtering and prioritization [91].
Specialized Task Delegation
An effective integration strategy deploys AI-based callers for particularly challenging genomic contexts:
- Complex indel detection in repetitive regions
- Variant calling in FFPE-derived samples
- Low-purity tumor samples (<20% tumor content)
- Long-read sequencing data from PacBio or Oxford Nanopore
For these applications, DeepSomatic demonstrates particularly strong advantages over traditional statistical methods [95] [93] [90].
Figure 2: Integration strategy for combining GATK and DeepSomatic in somatic variant detection workflows, highlighting parallel calling and consensus approaches for high-confidence variant sets.
Future Directions and Clinical Translation
The rapid evolution of AI-based variant calling continues to address longstanding challenges in genomics. Several emerging trends warrant attention from the research community:
Multi-Modal Learning
Next-generation variant callers are incorporating additional data types beyond standard sequencing reads, including epigenetic features, chromatin accessibility data, and spatial transcriptomics. This enriched context enables more accurate variant interpretation, particularly for non-coding regions and regulatory elements.
Population-Scale Optimization
As reference datasets expand to include more diverse populations, AI models are being retrained to reduce ancestry-based performance disparities. The integration of pangenome references represents a particularly promising direction for improving variant calling accuracy across global populations [88].
Real-Time Clinical Analysis
Streamlined versions of DeepVariant and DeepSomatic are enabling near-real-time variant calling suitable for clinical diagnostics. Runtime optimizations have reduced whole-genome analysis to under six hours on standard infrastructure, making AI-based calling feasible for time-sensitive clinical applications [93].
Automated Artifact Recognition
Advanced models are developing improved capability to distinguish technical artifacts from biological variants, particularly for challenging sample types like FFPE tissue, circulating tumor DNA, and single-cell sequencing. This capability is essential for expanding the range of clinical samples suitable for genomic analysis.
AI-based variant callers like DeepVariant and DeepSomatic represent a fundamental shift in genomic analysis methodology, offering substantial improvements in accuracy, particularly for challenging variant types and emerging sequencing technologies. Their unique approach—reformulating variant calling as an image classification problem—has demonstrated remarkable performance gains across multiple benchmarking studies. For researchers and drug development professionals working within established GATK workflows, strategic integration of these AI-based tools offers a path to enhanced detection accuracy while maintaining operational continuity. As these tools continue to evolve, they promise to further bridge the gap between genomic research and clinical application, ultimately accelerating therapeutic development and precision medicine implementation.
Integrating Quality Assurance and Regulatory Compliance (ISO 13485, IVDR) for Diagnostic Use
The convergence of advanced bioinformatics pipelines and stringent regulatory frameworks is essential for the development of clinically valid in vitro diagnostic (IVD) devices. This technical guide provides a comprehensive framework for integrating the GATK (Genome Analysis Toolkit) somatic variant calling workflow within a Quality Management System (QMS) compliant with ISO 13485:2016 and the European Union's In Vitro Diagnostic Regulation (IVDR) 2017/746. We demonstrate how research-grade bioinformatics analyses can transition into regulated diagnostic tools through systematic quality controls, risk management, and rigorous documentation practices. The methodologies presented herein enable researchers and drug development professionals to establish robust, reproducible, and legally compliant diagnostic processes that maintain scientific integrity while meeting regulatory requirements for clinical use.
The regulatory landscape for in vitro diagnostic devices in the European Union has undergone significant transformation with the implementation of Regulation (EU) 2017/746 (IVDR), which replaced the previous In Vitro Diagnostic Medical Devices Directive (IVDD) 98/79/EC. Unlike the directive it replaced, IVDR applies uniformly across all EU member states without requiring transposition into national laws, creating a harmonized regulatory framework [96]. This regulation introduces more robust requirements to enhance patient safety, improve transparency, and strengthen traceability throughout the device lifecycle.
ISO 13485:2016 is an international standard that specifies requirements for a quality management system where an organization needs to demonstrate its ability to provide medical devices and related services that consistently meet customer and regulatory requirements [97]. Although ISO 13485 certification does not automatically ensure full compliance with the IVDR's QMS requirements, it provides a foundational framework that manufacturers must build upon to meet regulatory obligations [98]. The EN ISO 13485:2016+A11:2021 is the corresponding harmonized technical standard at the European level, developed within the framework of the Commission's standardization request to provide a voluntary means to comply with IVDR requirements [99].
Regulatory Timelines and Transition Provisions
The IVDR was adopted in May 2017 and was initially set for complete implementation on May 26, 2022. However, due to COVID-19-related disruptions and industry readiness challenges, the transition has been staggered based on device risk classification [96]. The updated implementation deadlines are summarized in Table 1.
Table 1: EU IVDR Implementation Timeline
| Device Classification | Original Deadline | Updated Deadline (Current) |
|---|---|---|
| Class D (highest risk) | 26 May 2022 | Until 26 May 2025 |
| Class C | 26 May 2022 | Until 26 May 2026 |
| Class B and Class A sterile | 26 May 2022 | Until 26 May 2027 |
Manufacturers of legacy devices or IVDs that were certified or self-declared under IVDD before May 26, 2022, may remain on the market if no significant changes to the design or intended use have occurred, provided they implement an IVDR-compliant QMS and fulfill all applicable post-market requirements [96].
IVDR Classification System and Conformity Assessment
Risk-Based Classification
Under IVDR, IVD devices are categorized into four risk-based classes: A, B, C, and D, based on the device's potential impact on individual patients and public health [96]. The classification rules are outlined in Annex VIII of the regulation and are based on the intended use and associated risks.
Table 2: IVDR Risk-Based Classification System
| Class | Patient Risk | Public Health Risk | Example Devices |
|---|---|---|---|
| A | Low | Low | Laboratory instruments, buffer solutions, specimen receptacles |
| B | Moderate | Low | Pregnancy tests, cholesterol tests, urine glucose tests |
| C | High | Moderate | STI tests, cancer markers, human genetic tests |
| D | High | High | Tests for life-threatening transmissible diseases, blood screening for transfusion |
Only Class A non-sterile devices can be self-certified by the manufacturer under IVDR. Devices in classes B, C, and D require Notified Body audit and certification before being placed on the market [96]. This represents a significant shift from the previous IVDD framework, where notified body certification was required for only about 20% of higher-risk IVD devices, whereas under the new regime, this rule will apply to an estimated 80% of all IVD devices [97].
Conformity Assessment Process
The conformity assessment process varies according to the risk classification of the device. Manufacturers must submit documentation on their QMS to the Notified Body in addition to technical documentation and description of procedures [99]. The manufacturer must lodge an application for assessment of its QMS to a Notified Body, providing a clear and detailed overview of how it meets the requirements of the Regulation.
Once certification is obtained, the manufacturer is subjected to ongoing surveillance to ensure continued compliance. Maintenance audits are conducted at least once every 12 months, while every five years, the Notified Body performs surprise audits on the manufacturer or its suppliers [99]. If significant changes concerning the QMS or the device are necessary, the manufacturer must communicate this to the Notified Body, who will evaluate the proposed changes and the need for further audits [99].
Quality Management System Requirements
Essential QMS Elements
According to Article 10 of the IVDR, manufacturers must "establish, document, implement, maintain, keep up to date and continually improve a quality management system that shall ensure compliance with this Regulation in the most effective manner that is proportionate to the risk class and the type of device" [99]. The regulation emphasizes that the QMS needs to be continuously updated to account for changes concerning standards, specifications, internal organization, products/services, and results from continuous improvement processes [99].
The IVDR specifies that the QMS must include at least the following elements [99]:
- A strategy for compliance with the regulations
- Identification of applicable general safety and performance requirements
- Responsibility of the management
- Resource management, including selection and control of suppliers and sub-contractors
- Risk management
- Performance evaluation, including post-market performance follow-up (PMPF)
- Product realization, including planning, design, development, production, and service provision
- Verification of the Unique Device Identification (UDI) assignments
- Setting-up, implementation, and maintenance of a post-market surveillance system
- Handling communication with competent authorities, notified bodies, other economic operators, customers, and other stakeholders
- Processes for reporting of serious incidents and field safety corrective actions
- Management of corrective and preventive actions and verification of their effectiveness
- Processes for monitoring and measurement of output, data analysis, and product improvement
Integrating ISO 13485 with IVDR Requirements
While ISO 13485 serves as a foundational framework for Quality Management Systems for all medical devices, including IVDs, manufacturers must address specific gaps to achieve IVDR compliance [98]. Key considerations for harmonizing these frameworks include:
Risk Management Compliance: IVDR places significant emphasis on risk management and the need to demonstrate a favorable benefit-risk ratio considering the current state of the art. It is highly advised to align with ISO 14971 for risk management practices and ensure this is incorporated into the design control process [98].
Regulatory Compliance Strategy: Manufacturers must develop a comprehensive strategy within their QMS to address new elements such as device classification, conformity assessment routes, economic operator registration in EUDAMED, assigning Unique Device Identification (UDI), and establishing procedures for practical implementation [98].
Roles and Responsibilities: The IVDR introduces specific roles including the Person Responsible for Regulatory Compliance (PRRC) and necessitates clear delineation of responsibilities within the QMS [98] [99].
Technical Documentation Requirements: IVDR Annex II and III outline specific documentation requirements, including provisions for clinical evidence, post-market surveillance, and documentation control that go beyond typical ISO 13485 documentation [98].
Critical Supplier Control: Manufacturers must establish robust processes for controlling critical suppliers, ensuring compliance with IVDR requirements, and facilitating effective risk management [98].
Diagram 1: Integration of ISO 13485 and IVDR Requirements in a QMS
Workflow Components and Best Practices
The Genome Analysis Toolkit (GATK) is the industry standard for identifying SNPs and indels in germline DNA and RNAseq data, with its scope expanding to include somatic short variant calling, copy number variations (CNVs), and structural variations (SVs) [9]. GATK provides best practices workflows for all major classes of variants for genomic analysis in gene panels, exomes, and whole genomes [9] [32].
The somatic short variant discovery workflow in GATK is designed to identify somatic single nucleotide variants (SNVs) and insertions/deletions (indels) in one or more tumor samples from a single individual, with or without a matched normal sample [11]. The workflow involves multiple sophisticated bioinformatics tools and procedures:
Mutect2: This tool calls somatic SNVs and indels via local de-novo assembly of haplotypes in active regions. When Mutect2 encounters a region showing signs of somatic variation, it discards existing mapping information and completely reassembles reads in that region to generate candidate variant haplotypes [11]. Like HaplotypeCaller, Mutect2 aligns each read to each haplotype via the Pair-HMM algorithm to obtain a matrix of likelihoods, then applies a Bayesian somatic likelihoods model to obtain log odds for alleles to be somatic variants versus sequencing errors [11] [12].
Contamination Estimation: Using GetPileupSummaries and CalculateContamination tools, this step emits an estimate of the fraction of reads due to cross-sample contamination for each tumor sample. Unlike other contamination tools, CalculateContamination is designed to work well without a matched normal even in samples with significant copy number variation and makes no assumptions about the number of contaminating samples [11].
Orientation Bias Analysis: The LearnReadOrientationModel tool uses an optional F1R2 counts output of Mutect2 to learn parameters for orientation bias artifacts. This finds prior probabilities of single-stranded substitution errors prior to sequencing for each trinucleotide context, which is particularly important for FFPE tumor samples [11].
Variant Filtering: FilterMutectCalls accounts for correlated errors through several hard filters to detect alignment artifacts and probabilistic models for strand and orientation bias artifacts, polymerase slippage artifacts, germline variants, and contamination [11]. It learns a Bayesian model for the overall SNV and indel mutation rate and allele fraction spectrum of the tumor to refine the log odds emitted by Mutect2, automatically setting a filtering threshold to optimize the F score (harmonic mean of sensitivity and precision) [11].
Variant Annotation: Funcotator adds functional annotation to discovered variants, labeling each variant with one of twenty-three distinct variant classifications and producing gene information including affected gene, predicted variant amino acid sequence, and associations to datasources such as GENCODE, dbSNP, gnomAD, and COSMIC [11].
Experimental Protocols and Methodologies
The GATK somatic variant calling workflow requires specific input data and follows a structured series of processing steps:
Input Requirements: The workflow requires BAM files for each input tumor and normal sample. Input BAMs should be pre-processed as described in the GATK Best Practices for data pre-processing, which includes marking duplicates, base quality score recalibration (BQSR), and indel realignment [11] [100].
Data Pre-processing: Prior to variant calling, BAM files are processed through quality control steps including:
- Local realignment of insertions and deletions using IndelRealigner to locate regions containing misalignments across BAM files
- Base quality score recalibration using BaseRecalibrator to adjust base quality scores based on detectable and systematic errors [100]
Variant Calling Command Structure: The basic command structure for Mutect2 in tumor-normal mode is:
Mutect2 also supports tumor-only mode for cases where matched normal samples are unavailable, and mitochondrial mode for calling variants in mitochondrial DNA with parameters optimized for high-depth sequencing [12].
Validation and Performance Metrics: The workflow generates statistical outputs including contamination estimates, orientation bias metrics, and filtering thresholds that must be documented and monitored as part of the quality management system.
Diagram 2: GATK Somatic Variant Calling Workflow with Quality Control Points
Integration Framework: Bridging Bioinformatics and Regulatory Compliance
Quality Control in Bioinformatics Pipelines
Implementing a QMS for bioinformatics pipelines requires establishing controlled processes throughout the analytical workflow. For somatic variant calling pipelines intended for diagnostic use, the following quality control points must be established and documented:
Input Quality Controls:
- Verification of BAM file integrity and completeness
- Validation of sequencing metrics (coverage, depth, quality scores)
- Confirmation of sample identity and tracking
- Documentation of reference genome version and annotations
Process Quality Controls:
- Monitoring of computational environment and version control
- Tracking of intermediate file integrity
- Validation of algorithm parameters and settings
- Documentation of any deviations from standard protocols
Output Quality Controls:
- Verification of VCF file format compliance
- Validation of variant calling sensitivity and specificity
- Assessment of contamination levels and quality metrics
- Documentation of filtering criteria and results
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Resources for Compliant Somatic Variant Calling
| Resource Category | Specific Tools/Databases | Function in Workflow | Regulatory Considerations |
|---|---|---|---|
| Reference Materials | GRCh38 reference genome, Decoy viral sequences | Alignment and variant calling baseline | Version control, traceability, validation |
| Variant Databases | dbSNP, gnomAD, COSMIC, 1000 Genomes | Filtering common polymorphisms, identifying known somatic variants | Documentation of versions, update procedures |
| Analysis Tools | GATK Mutect2, FilterMutectCalls, Funcotator | Core variant discovery and annotation | Software validation, version control, parameter documentation |
| Quality Metrics | Cross-sample contamination, Orientation bias, Mapping quality | Monitoring analytical performance | Establishment of acceptance criteria, monitoring protocols |
| Panel of Normals | TCGA normal samples, Institutional normal panels | Filtering technical artifacts and germline variants | Composition documentation, version control |
Performance Evaluation and Validation
Under IVDR, manufacturers must conduct performance evaluation studies demonstrating the analytical and clinical performance of their IVD devices [99]. For somatic variant calling workflows, this includes:
Analytical Performance:
- Sensitivity and specificity calculations using reference materials
- Limit of detection studies for variant allele fractions
- Reproducibility across operators, instruments, and sites
- Robustness testing under varying conditions
Clinical Validation:
- Concordance studies with orthogonal methods
- Clinical correlation with patient outcomes
- Database comparisons with established mutations
- Validation against gold standard methodologies
The performance evaluation must be documented in a performance evaluation report that is periodically updated throughout the device lifecycle [99]. Additionally, manufacturers must establish a post-market performance follow-up (PMPFP) plan to proactively collect and review experience gained from devices placed on the market [99].
Documentation Strategies for Regulatory Compliance
Technical Documentation Requirements
Annexes II and III of the IVDR outline specific technical documentation requirements that manufacturers must maintain [98]. For bioinformatics pipelines, this documentation should include:
Device Description and Specification:
- Intended purpose and indications for use
- Principles of operation and scientific rationale
- Target populations and sampling requirements
- Detailed description of all pipeline components
Design and Development Documentation:
- Software requirements specification
- Architecture and design documentation
- Version control and change management records
- Algorithm descriptions and parameter justifications
Verification and Validation Records:
- Installation, operational, and performance qualification protocols
- Test cases, results, and acceptance criteria
- Validation against reference materials and datasets
- User acceptance testing and feedback
Risk Management Documentation:
- Risk management plan and report per ISO 14971
- Hazard analysis and risk control measures
- Residual risk assessment and benefit-risk analysis
- Post-production information monitoring plan
Post-Market Surveillance and Vigilance
IVDR requires manufacturers to establish, implement, and maintain a post-market surveillance system as part of their QMS [99]. For somatic variant calling workflows, this includes:
Post-Market Surveillance Plan:
- Procedures for proactively collecting and reviewing experience
- Methods for gathering user feedback and performance data
- Processes for analyzing emerging trends and issues
- Protocols for updating performance evaluations
Periodic Safety Update Reports (PSUR):
- Summary of post-market surveillance data and results
- Conclusions from post-market performance follow-up
- Assessment of device safety and performance
- Indication of any corrective actions taken
Vigilance Reporting:
- Procedures for reporting serious incidents to competent authorities
- Field safety corrective action plans and implementation
- Communication strategies with users and stakeholders
- Documentation of all incidents and actions taken
Integrating GATK somatic variant calling workflows into a Quality Management System compliant with ISO 13485 and IVDR requires meticulous planning, documentation, and validation. By establishing robust quality controls throughout the bioinformatics pipeline, implementing comprehensive documentation practices, and maintaining rigorous post-market surveillance, manufacturers can develop clinically valuable diagnostic tools that meet regulatory requirements while advancing precision medicine.
The framework presented in this guide provides a structured approach for researchers and drug development professionals to navigate the complex regulatory landscape while maintaining scientific rigor. As genomic technologies continue to evolve and regulatory expectations mature, a proactive approach to quality management and regulatory compliance will be essential for successfully translating bioinformatics research into clinically validated diagnostic applications.
Conclusion
The GATK somatic variant calling workflow, centered on Mutect2, provides a robust, well-documented framework for identifying cancer-associated mutations. Mastery of this pipeline—from foundational concepts and methodological steps to advanced troubleshooting and validation—is crucial for generating reliable, reproducible results in both research and clinical settings. The continuous evolution of the toolkit, alongside emerging AI-based methods, promises even greater accuracy and efficiency. For the future, the seamless integration of these bioinformatic pipelines with regulatory standards and clinical interpretation guidelines will be paramount in accelerating the development of personalized cancer therapies and unlocking the full potential of precision oncology.
References
- 1. Somatic mutations in cancer development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3073190/]
- 2. A comprehensive catalogue of somatic mutations from ... [https://www.nature.com/articles/nature08658]
- 3. Pipeline | Plataforma Andaluza de Medicina... Somatic Variant [https://www.clinbioinfosspa.es/content/somatic-variant-pipeline]
- 4. The Order of Somatic Mutations Influences Cancer Evolution [https://pmc.ncbi.nlm.nih.gov/articles/PMC5378012/]
- 5. Somatic mutation and selection at population scale [https://www.nature.com/articles/s41586-025-09584-w]
- 6. Pipeline: Variant , Tool Comparison, and Accuracy... Calling Workflows [https://bioinfo.cd-genomics.com/resource-variant-calling-workflow-guide-tool-genomic-variants.html]
- 7. Somatic mutations acquired during life: state of the art and ... [https://www.sciencedirect.com/science/article/pii/S0085253825001589]
- 8. The Genome Analysis Toolkit: A MapReduce framework for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2928508/]
- 9. GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us]
- 10. Genome Analysis Toolkit (GATK) the Industry Standard will ... [https://pharmaceuticalintelligence.com/2016/04/06/genome-analysis-toolkit-gatk-the-industry-standard-will-govern-the-new-tools-in-biomedical-research-by-the-collaboration-of-broad-institute-and-intel/]
- 11. Somatic short variant discovery (SNVs + Indels) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035894731-Somatic-short-variant-discovery-SNVs-Indels]
- 12. Mutect2 - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2]
- 13. Go beyond somatic variant calling: GATK SNVs and Somatic ... INDELs [https://www.sevenbridges.com/go-beyond-somatic-variant-calling-gatk-somatic-snvs-and-indels-mutect2-4-1-9-0/]
- 14. Comparison of somatic variant detection algorithms using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6929331/]
- 15. Systematic comparison of somatic variant calling ... [https://www.nature.com/articles/s41598-020-60559-5]
- 16. 7 GATK for Best - Genomics... Practices Somatic Variant Discovery [https://www.oreilly.com/library/view/genomics-in-the/9781491975183/ch07.html]
- 17. (How to) Call somatic mutations using GATK4 Mutect2 – GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035531132--How-to-Call-somatic-mutations-using-GATK4-Mutect2]
- 18. (Notebook) Intro to using Mutect2 for somatic data - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360047232772--Notebook-Intro-to-using-Mutect2-for-somatic-data]
- 19. GitHub - BMurri/ gatk 4- somatic -snvs-indels-azure: Workflows for... [https://github.com/BMurri/gatk4-somatic-snvs-indels-azure]
- 20. About the GATK Best Practices [https://gatk.broadinstitute.org/hc/en-us/articles/360035894711-About-the-GATK-Best-Practices]
- 21. GATK-gCNV enables discovery of rare copy number ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10904014/]
- 22. How To Analyze Whole Genome Sequencing Data For Absolute... [https://ngs101.com/how-to-analyze-whole-genome-sequencing-data-for-absolute-beginners-part-2a-matched-tumor-normal-mutation-calling-with-mutect2/]
- 23. Chapter 2 GATK practice workflow [https://hpc.nih.gov/training/gatk_tutorial/workflow-overview.html]
- 24. Systematic benchmark of state-of-the-art variant calling ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08365-3]
- 25. Recommendations for performance optimizations when using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6842142/]
- 26. germline (GATK Germline Pipeline) [https://docs.nvidia.com/clara/parabricks/latest/Documentation/ToolDocs/man_germline.html]
- 27. What are the Best Practices for Optimizing Server ... - GATK [https://gatk.broadinstitute.org/hc/en-us/community/posts/28062071549595-What-are-the-Best-Practices-for-Optimizing-Server-Performance-When-Running-Large-Scale-GATK-Workflows]
- 28. how to interpret results of gatk Concordance [https://gatk.broadinstitute.org/hc/en-us/community/posts/360072857192-how-to-interpret-results-of-gatk-Concordance]
- 29. Panel of Normals (PON) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035890631-Panel-of-Normals-PON]
- 30. Resource bundle – GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035890811-Resource-bundle]
- 31. Somatic calling is NOT simply a difference between two callsets [https://gatk.broadinstitute.org/hc/en-us/articles/360035890491-Somatic-calling-is-NOT-simply-a-difference-between-two-callsets]
- 32. Best Practices Workflows - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/sections/360007226651-Best-Practices-Workflows]
- 33. Halvade somatic: Somatic variant calling with Apache Spark [https://pmc.ncbi.nlm.nih.gov/articles/PMC8756192/]
- 34. Somatic Small Variant and CNV Discovery Workflow ... [https://documentation.dnanexus.com/science/scientific-guides/somatic-small-variant-and-cnv-discovery-workflow-walkthrough]
- 35. Best practices for variant calling in clinical sequencing [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-020-00791-w]
- 36. Local re-assembly and haplotype determination ... - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360036227612-Local-re-assembly-and-haplotype-determination-HaplotypeCaller-and-Mutect2]
- 37. Evaluating the evidence for haplotypes and variant alleles ... [https://gatk.broadinstitute.org/hc/en-us/articles/360036227632-Evaluating-the-evidence-for-haplotypes-and-variant-alleles-HaplotypeCaller-and-Mutect2]
- 38. Using GATK's HaplotypeCaller or Mutect2 for somatic ... [https://www.biostars.org/p/283291/]
- 39. How to Call somatic mutations using GATK4 Mutect2 [https://sites.google.com/a/broadinstitute.org/legacy-gatk-documentation/tutorials/24057-How-to-Call-somatic-mutations-using-GATK4-Mutect2]
- 40. CalculateContamination - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/30332023942555-CalculateContamination]
- 41. GATK VARIANT CALLING BEST PRACTICE WORKFLOW [https://snakemake-wrappers.readthedocs.io/en/stable/meta-wrappers/bio/gatk_mutect2_calling.html]
- 42. Filtering Mutect output from FFPE based targeted panel ... [https://gatk.broadinstitute.org/hc/en-us/community/posts/7610560664219-Filtering-Mutect-output-from-FFPE-based-targeted-panel-sequencing-data]
- 43. Best Practices for Tumor-Only WES Data Analysis - GATK [https://gatk.broadinstitute.org/hc/en-us/community/posts/29772760093595-Best-Practices-for-Tumor-Only-WES-Data-Analysis-Seeking-Feedback-on-Variant-Calling-Pipeline]
- 44. Benchmarking mouse contamination removing protocols in ... [https://www.nature.com/articles/s41698-025-00902-z]
- 45. FilterMutectCalls - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360036805111-FilterMutectCalls]
- 46. FilterMutectCalls – GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360036856831-FilterMutectCalls]
- 47. Comparative analysis of somatic variant calling on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7336445/]
- 48. Funcotator Information and Tutorial - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035889931-Funcotator-Information-and-Tutorial]
- 49. Funcotator - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360037224432-Funcotator]
- 50. Session 4: Mutation Calling and Analysis [https://nci-iteb.github.io/tumor_epidemiology_approaches/sessions/session_4/practical]
- 51. Funocator chromosome missmatch to somatic variant calling ... [https://gatk.broadinstitute.org/hc/en-us/community/posts/24087106721051-Funocator-chromosome-missmatch-to-somatic-variant-calling-pipeline]
- 52. Mutect2 – GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360042477952-Mutect2]
- 53. Mutect2 - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360036485152-Mutect2]
- 54. Mutect2 – GATK [https://gatk.broadinstitute.org/hc/en-us/articles/30332058799003-Mutect2]
- 55. HaplotypeCaller --max-reads-per-alignment-start - GATK [https://gatk.broadinstitute.org/hc/en-us/community/posts/4418332857243-HaplotypeCaller-max-reads-per-alignment-start]
- 56. HaplotypeCaller - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360037225632-HaplotypeCaller]
- 57. Deep sequencing data is missing variants in M2 called vcf [https://gatk.broadinstitute.org/hc/en-us/articles/360035532212-Deep-sequencing-data-is-missing-variants-in-M2-called-vcf]
- 58. A Novel Affordable and Reliable Framework for Accurate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11311285/]
- 59. ClairS-TO: a deep-learning method for long-read tumor-only ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12579226/]
- 60. Best practices for variant calling in clinical sequencing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7586657/]
- 61. A critical spotlight on the paradigms of FFPE-DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10415133/]
- 62. Enabling whole genome sequencing analysis from FFPE ... [https://www.nature.com/articles/s41467-025-65654-7]
- 63. LearnReadOrientationModel - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/4405451237147-LearnReadOrientationModel]
- 64. Read-orientation filter removes tens of thousands of somatic ... [https://gatk.broadinstitute.org/hc/en-us/community/posts/4416578606875-Read-orientation-filter-removes-tens-of-thousands-of-somatic-variants-after-FilterMutectCalls]
- 65. FAQ for Mutect2 – GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360050722212-FAQ-for-Mutect2]
- 66. Recommendations for performance optimizations when using ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3169-7]
- 67. OVarFlow: a resource optimized GATK 4 based Open source ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8361789/]
- 68. Benchmarking of variant calling software for whole-exome ... [https://www.nature.com/articles/s41598-025-97047-7]
- 69. Reducing false positives in somatic variant calling - GATK [https://gatk.broadinstitute.org/hc/en-us/community/posts/23697608644635-Reducing-false-positives-in-somatic-variant-calling]
- 70. How can I use parallelism to make GATK tools run faster [https://sites.google.com/a/broadinstitute.org/legacy-gatk-documentation/frequently-asked-questions/1975-How-can-I-use-parallelism-to-make-GATK-tools-run-faster]
- 71. Parallelism - Multithreading - Scatter Gather - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035532012-Parallelism-Multithreading-Scatter-Gather]
- 72. Optimizing performance of GATK workflows using Apache ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-07013-y]
- 73. ClairS-TO: a deep-learning method for long-read tumor ... [https://www.nature.com/articles/s41467-025-64547-z]
- 74. Comparative Evaluation of Mutect2, Strelka2, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12650410/]
- 75. Comparative Evaluation of Mutect2, Strelka2, and ... [https://www.mdpi.com/2218-273X/15/11/1532]
- 76. Comprehensive benchmarking and guidelines of mosaic ... [https://www.nature.com/articles/s41592-023-02043-2]
- 77. Evaluation of reference sample type for somatic variant ... [https://pubmed.ncbi.nlm.nih.gov/41123648/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1zG2_xdOOC5VXA_B186mg2wRG5Vierx2syPXCcCUdmU3UrJgnK&fc=None&ff=20251023183004&v=2.18.0.post22+67771e2]
- 78. A Simplified Approach to Somatic Analysis in NGS [https://www.euformatics.com/blog-post/a-simplified-approach-to-somatic-analysis-in-ngs]
- 79. Evaluating Discordant Somatic Calls Across Mutation ... [https://www.sciencedirect.com/science/article/pii/S1525157825001357?dgcid=rss_sd_all]
- 80. Somatic Variant Calling with Mutect2 in Lasergene [https://www.dnastar.com/somatic-variant-calling-with-mutect2-in-lasergene/]
- 81. AMP/ASCO/CAP Standards and Guidelines of Somatic Variant Interpretation and Reporting | AMP [https://educate.amp.org/local/catalog/view/product.php?productid=135]
- 82. AMP 2025 Annual Meeting & Expo [https://amp2025.eventscribe.net/ajaxcalls/SessionInfo.asp?efp=RUFGWU9FR1cyMzQ0MQ&PresentationID=1659682&rnd=0.5924582]
- 83. Somatic Variant Classification: A Welcome Update to the AMP/ASCO/CAP Guidelines - Precision Oncology Solutions | GenomOncology [https://genomoncology.com/blog/somatic-variant-classification-a-welcome-update-to-the-amp-asco-cap-guidelines/]
- 84. Identification of challenges and a framework for implementation of the AMP/ASCO/CAP classification guidelines for reporting somatic variants - PubMed [https://pubmed.ncbi.nlm.nih.gov/32548229/]
- 85. gatk-workflows/gatk4-somatic-snvs-indels [https://github.com/gatk-workflows/gatk4-somatic-snvs-indels]
- 86. Comparison of Somatic Variant Oncogenicity Classification ... [https://www.sciencedirect.com/science/article/pii/S221077622500119X]
- 87. Variant calling from RNA-Seq data reveals allele-specific ... [https://www.nature.com/articles/s43856-025-00901-y]
- 88. DeepVariant Blog - Google [https://google.github.io/deepvariant/]
- 89. DeepSomatic: Accurate somatic small variant discovery for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11370364/]
- 90. Using AI to identify genetic variants in tumors with ... [https://research.google/blog/using-ai-to-identify-genetic-variants-in-tumors-with-deepsomatic/]
- 91. Comparison of GATK and DeepVariant by trio sequencing [https://www.nature.com/articles/s41598-022-05833-4]
- 92. Performance comparison of germline variant calling tools ... [https://pubmed.ncbi.nlm.nih.gov/40913732/]
- 93. DeepSomatic AI: 98% accuracy in cancer mutation detection [https://www.rdworldonline.com/googles-deepsomatic-ai-achieves-98-accuracy-in-cancer-mutation-detection-outperforms-standard-tools/]
- 94. AIVariant: a deep learning-based somatic variant detector ... [https://www.nature.com/articles/s12276-023-01049-2]
- 95. Germline Variant Call Accuracy in Whole Genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12652997/]
- 96. EU IVDR: Definition, Requirements, and Compliance [https://simplerqms.com/eu-ivdr/]
- 97. Journey to Regulatory Compliance for In Vitro Diagnostic ... [https://www.sgs.com/en-es/news/2021/03/journey-to-regulatory-compliance-for-in-vitro-diagnostic-ivd-medical-device-manufacturers]
- 98. Why do I still need to look at my QMS under the IVDR if it is ... [https://www.qbdgroup.com/en/blog/why-do-i-still-need-to-look-at-my-qms-under-the-ivdr-if-it-is-iso-13485-certified]
- 99. IVDR: How to Meet the Quality Management System ... [https://www.greenlight.guru/blog/ivdr-how-to-meet-quality-management-system-requirements-device]
- 100. Bioinformatics Pipeline: DNA-Seq Analysis: Whole Exome and ... [https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/DNA_Seq_Variant_Calling_Pipeline/]