Accurate Gene Prediction in Long-Read Microbial Genomes: Methods, Tools, and Clinical Applications
This article provides a comprehensive guide for researchers and drug development professionals on leveraging long-read sequencing for microbial gene prediction.
Accurate Gene Prediction in Long-Read Microbial Genomes: Methods, Tools, and Clinical Applications
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on leveraging long-read sequencing for microbial gene prediction. It covers foundational principles of long-read assembly, explores integrated bioinformatics platforms and lineage-specific methodologies, addresses common troubleshooting and optimization challenges, and establishes validation frameworks for ensuring prediction accuracy. The content synthesizes current best practices to enable reliable genome annotation, supporting applications in microbial ecology, antibiotic resistance tracking, and therapeutic discovery.
The Rise of Long-Read Sequencing in Microbial Genomics: Foundations and New Frontiers
Why Long-Read Sequencing is a Game-Changer for Microbial Genome Assembly
For years, short-read sequencing (SRS) platforms have been the workhorse of microbial genomics. However, a significant limitation has hindered progress: their inability to accurately resolve repetitive genomic regions and complex structural variants [1] [2]. Assembling a genome from these short snippets, typically a few hundred base pairs long, is like reconstructing a book from countless sentence fragments without any paragraph breaks. This often results in fragmented, incomplete genome assemblies that misrepresent the true biology of the microbe [1] [3].
These gaps are particularly problematic for key genomic features, such as:
- Repetitive elements (e.g., ribosomal RNA operons, transposons).
- Mobile genetic elements (e.g., plasmids, integrons) that facilitate the spread of antimicrobial resistance genes (ARGs) [4].
- Large biosynthetic gene clusters (BGCs) encoding pathways for secondary metabolites, which are crucial for drug discovery [3].
Long-read sequencing (LRS) technologies, primarily from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), have emerged as a transformative solution. By generating sequence reads that are thousands to tens of thousands of bases long, LRS can span these repetitive and complex regions, enabling the routine production of complete, gapless microbial genomes [1] [2]. For researchers focused on gene prediction, this completeness is foundational, as an uninterrupted genomic sequence is essential for the accurate identification and annotation of gene models.
Technical Advantages and Quantitative Comparisons
The core advantage of long-read sequencing lies in its ability to produce reads that are long enough to span repetitive regions, thereby simplifying the assembly process into a much more accurate and complete genomic picture [1]. This capability directly translates into superior outcomes for microbial genomics.
Table 1: A Comparative Overview of Sequencing Technologies for Microbial Genomics
| Feature | Short-Read Sequencing (e.g., Illumina) | Long-Read Sequencing (PacBio HiFi) | Long-Read Sequencing (ONT) |
|---|---|---|---|
| Typical Read Length | 150-300 bp [5] | 15-20 kb [6] | 5-30+ kb (can exceed 1 Mb) [5] [1] |
| Typical Raw Read Accuracy | >99.9% (Q30) [5] | >99.9% (Q30) [6] | ~97-99% (Q12-Q20), improving with new chemistries [5] [1] |
| Primary Assembly Challenge | Highly fragmented assemblies due to repeats [1] | Highly contiguous, often complete assemblies [7] | Highly contiguous, often complete assemblies [8] |
| Ability to Resolve Repetitive Regions | Poor [2] | Excellent [6] | Excellent [4] |
| Plasmid & Mobile Element Reconstruction | Difficult, often misassembled [4] | Accurate, complete reconstruction [4] | Accurate, complete reconstruction [4] |
| Epigenetic Modification Detection | Requires special treatment, degrades DNA [2] | Built-in, native detection [2] | Built-in, native detection [2] |
| Portability / Throughput | Benchtop to high-throughput | Moderate to high-throughput (e.g., Revio system) [6] | Portable (MinION) to ultra-high-throughput (PromethION) [4] [1] |
Table 2: Impact of Long-Read Sequencing on Genome Assembly Quality in Recent Studies
| Study Context | Sequencing Technology | Key Genomic Outcome |
|---|---|---|
| Antimicrobial Resistance (AMR) Research [4] | Nanopore Long-Read Sequencing | Precise identification of the genetic context of ARGs and their location on mobile elements like plasmids. |
| Terrestrial Microbial Diversity [8] | Nanopore Long-Read Sequencing | Recovery of 15,314 previously undescribed microbial species from complex soil samples. |
| Phytopathogen Epidemiology [7] | Nanopore vs. Illumina | Long-read assemblies were more complete than short-read assemblies and contained few sequence errors. |
| Genome Mining for Drug Discovery [3] | PacBio & ONT Long-Reads | Essential for obtaining finished-quality genomes to correctly assemble large NRPS and PKS-I biosynthetic gene clusters. |
Applications in Microbial Research
The shift to long-read sequencing is unlocking new possibilities across multiple areas of microbiology.
Unveiling the Spread of Antimicrobial Resistance (AMR)
LRS uniquely enables researchers to track the horizontal transfer of antimicrobial resistance genes (ARGs). Because long reads can encompass an entire ARG and its surrounding genetic context, they can precisely determine whether the gene is located on a chromosome, plasmid, or other mobile genetic element [4]. This is critical for understanding and containing the spread of multidrug-resistant pathogens in both clinical and environmental settings [4].
Accelerating Natural Product Discovery
Microbial genome mining is a powerful approach for discovering new drugs. Many valuable compounds are synthesized by large, repetitive biosynthetic gene clusters (BGCs), such as those for nonribosomal peptide synthetases (NRPS) and polyketide synthases (PKS). Short-read sequencing routinely fragments and misassembles these BGCs, leading to failed discovery efforts [3]. Finished-quality genomes from LRS are now considered critical for the robust assembly of these clusters, opening up a vast untapped resource for novel antibiotics and therapeutics [3].
Expanding the Microbial Tree of Life
Metagenomic studies of complex environments like soil have been historically challenging due to their immense microbial diversity. Deep long-read sequencing, as demonstrated by the Microflora Danica project, allows for the recovery of high-quality metagenome-assembled genomes (MAGs) directly from environmental samples [8]. This approach has dramatically expanded the known microbial diversity, leading to the discovery of thousands of novel species and genera that had previously eluded detection using short-read methods [8].
Enhancing Pathogen Surveillance and Outbreak Investigation
In microbial epidemiology, long-read sequencing facilitates both accurate genotyping and high-quality genome assembly from a single assay. A 2025 study on phytopathogenic bacteria confirmed that variant calls and genotypes inferred from Nanopore long reads are as accurate as those from short reads when using optimized bioinformatic pipelines [7]. This enables researchers to track transmission chains with high resolution while also obtaining complete genomes to understand virulence and resistance mechanisms.
Experimental Protocols
Below is a generalized workflow for generating a complete microbial genome assembly using long-read sequencing, from DNA extraction to functional annotation.
Sample Preparation and Sequencing
Goal: Obtain high-molecular-weight (HMW), ultra-pure genomic DNA for sequencing.
- Step 1: Cell Lysis. Use gentle, enzyme-based lysis methods (e.g., lysozyme treatment for Gram-positive bacteria) to avoid shearing DNA.
- Step 2: DNA Extraction. Employ HMW DNA extraction kits designed for long-read sequencing. Critical: Assess DNA quality and quantity using a fluorometer (e.g., Qubit) and fragment size using pulsed-field gel electrophoresis (PFGE) or a Femto Pulse system. An ideal input is >150 ng of DNA with fragments >50 kb [1] [3].
- Step 3: Library Preparation. Follow the manufacturer's protocol for your chosen platform. For PacBio, this involves converting DNA into SMRTbell libraries for HiFi sequencing. For ONT, this entails ligating motor protein adapters to the DNA ends. New automated and high-throughput kits have significantly reduced preparation time and cost [6].
Genome Assembly and Quality Control
Goal: Convert raw sequencing reads into a complete, accurate genome sequence.
- Step 1: Base Calling and Read QC. Convert raw signal data (ONT) or movie files (PacBio) into nucleotide sequences. Filter reads by length and quality.
- Step 2: De Novo Assembly. Use long-read-specific assemblers. Common tools include:
- Flye [9]: A fast and efficient assembler that is widely used.
- Canu [9]: Excellent for correcting errors in noisy long reads.
- hifiasm (for PacBio HiFi data): Specialized for highly accurate reads.
- Note: Some advanced pipelines, like the one from MIRRI ERIC, run multiple assemblers and combine the results for the best possible output [9].
- Step 3: Polishing (if required). While HiFi reads typically do not require polishing, traditional ONT or PacBio CLR reads may benefit from a polishing step using the same long reads or with high-accuracy short reads to correct small indels.
- Step 4: Quality Assessment. Evaluate the assembly using:
- Completeness and Contamination: Check with tools like CheckM (for prokaryotes) or BUSCO (for eukaryotes).
- Contiguity: Metrics like N50/L50 (the contig length such that longer contigs cover 50% of the genome; higher is better).
- Circularization: For prokaryotes, check if the chromosome and plasmids have been assembled into single, circular contigs.
The following diagram illustrates the core bioinformatics workflow from raw data to an annotated genome.
Diagram 1: Core bioinformatics workflow for long-read genome assembly, from raw data to an annotated genome.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Long-Read Microbial Genome Sequencing
| Item | Function / Application | Examples / Notes |
|---|---|---|
| HMW DNA Extraction Kits | Gentle isolation of long, intact DNA fragments. | Kits from Qiagen, MagAttract, or similar, optimized for microbes. |
| PacBio SMRTbell Prep Kits | Library preparation for PacBio HiFi sequencing. | SMRTbell prep kit 3.0; enables multiplexing of microbial genomes [6]. |
| ONT Ligation Sequencing Kits | Library preparation for Nanopore sequencing. | Ligation Sequencing Kit (e.g., SQK-LSK114); compatible with various flow cells. |
| Long-Range PCR Kits | Target enrichment for specific genes or regions. | Not always required but useful for amplifying low-abundance BGCs or resistance genes. |
| Flow Cells | The consumable where sequencing occurs. | PacBio SMRT Cells (8M, 25M); ONT Flongle, MinION (R10.4.1), PromethION (R10.4.1). |
| Bioinformatics Platforms | For assembly, gene prediction, and annotation. | MIRRI ERIC platform [9], Galaxy, CLAWS, or custom Snakemake/CWL workflows. |
Long-read sequencing has unequivocally transformed microbial genome assembly from a challenging puzzle into a streamlined process for generating complete, reference-quality genomes. Its ability to resolve complex genomic landscapes provides an accurate foundation for all downstream analyses, most critically for precise gene prediction and functional annotation. As the technology continues to evolve, with costs decreasing and accuracy and throughput increasing [6], long-read sequencing is poised to become the new gold standard in microbial genomics. For researchers and drug development professionals, adopting this technology is no longer a niche choice but a strategic imperative to drive discovery in antimicrobial resistance, natural products, and our fundamental understanding of the microbial world.
The advent of long-read sequencing technologies has revolutionized microbial genomics, enabling researchers to generate unprecedented amounts of raw genomic data. However, transforming this data into meaningful biological insights presents significant computational and analytical challenges [9] [10]. This application note examines the key bioinformatics hurdles in gene prediction from long-read assembled microbial genomes and presents integrated solutions currently bridging the gap between raw data and biological understanding, with particular relevance for drug development targeting microbial pathogens.
Key Challenges in Microbial Genome Analysis
The journey from raw sequencing data to assembled, annotated genomes involves multiple critical steps where challenges can compromise final results. The table below summarizes these primary challenges and their implications for downstream analysis.
Table 1: Key Bioinformatics Challenges in Microbial Genome Analysis
| Challenge Category | Specific Challenges | Impact on Biological Insight |
|---|---|---|
| Computational Demands | Genome reconstruction and annotation remain computationally demanding and technically complex [9] [10]. | Limits accessibility for researchers without advanced computational skills or HPC access. |
| Data Integration | Few platforms integrate assembly, gene prediction, and annotation for both prokaryotes and eukaryotes [9]. | Hinders comprehensive analysis of host-pathogen systems relevant to drug development. |
| Workflow Reproducibility | Combining multiple tools into reproducible, scalable workflows requires significant bioinformatics expertise [9]. | Reduces reliability and transparency of results for critical applications like therapeutic target identification. |
| Genome Complexity | Repetitive elements, heterozygosity, and extreme GC-content complicate assembly and annotation [11]. | Can obscure important genomic features such as virulence factors or drug resistance genes. |
Integrated Platforms Addressing Analytical Challenges
The MIRRI-ERIC Bioinformatics Platform
The Italian node of the Microbial Resource Research Infrastructure (MIRRI ERIC) has developed a specialized bioinformatics platform to address these challenges through a unified workflow [9] [10]. This service provides a comprehensive solution for analyzing both prokaryotic and eukaryotic genomes, from assembly to functional protein annotation, specifically designed for long-read sequencing data.
The platform's architecture employs a hybrid computational infrastructure, integrating cloud computing for user interaction and High-Performance Computing (HPC) for accelerated analysis execution [9]. This design effectively addresses the computational demands highlighted in Table 1 while maintaining accessibility for non-bioinformatics specialists.
Comparative Analysis of Genomic Workflows
Table 2: Comparison of Genomic Analysis Workflows and Platforms
| Platform/Workflow | Supported Assemblers | Gene Prediction Tools | Functional Annotation | Key Limitations |
|---|---|---|---|---|
| MIRRI-ERIC Platform [9] [10] | Canu, Flye, wtdbg2 | BRAKER3 (eukaryotes), Prokka (prokaryotes) | InterProScan | Newer platform with growing adoption |
| Galaxy Europe [9] | CANU, Flye | Prokka, BRAKER3 | Various tools | Lacks integrated workflow for both genomic domains |
| CLAWS [9] | Flye, NextDenovo | None | None | No gene prediction or functional annotation |
| MGnify [9] | Multiple | Multiple | Multiple | Focused on metagenomics rather than isolated microbes |
Experimental Protocol: From Sequencing to Annotation
Complete Microbial Genome Analysis Workflow
The following protocol outlines the comprehensive workflow for microbial genome analysis using the MIRRI-ERIC platform, which integrates state-of-the-art tools within a reproducible, scalable framework built on Common Workflow Language (CWL) and containerized with Docker [9] [10].
Phase 1: Genome Assembly
Input Requirements: Pacific Biosciences (PacBio) HiFi or Oxford Nanopore Technologies (ONT) long-read sequencing data. DNA quality is crucial for successful long-read sequencing, requiring High Molecular Weight (HMW) DNA with both chemical purity and structural integrity [11].
Assembly Tools and Parameters:
- Canu: Specialized for noisy long reads, performs correction, trimming, and assembly [9]
- Flye: Designed for de novo assembly using repetitive graphs [9]
- wtdbg2: Efficient assembly without error correction [9]
Protocol Notes: The platform executes all three assemblers in parallel, then integrates their outputs to enhance performance, completeness, and accuracy [9]. Users specify their sequencing technology (Nanopore, PacBio, or PacBio HiFi) through the graphical interface.
Phase 2: Assembly Evaluation
Quality Metrics:
- Standard metrics: N50 and L50 statistics [9]
- Evolutionarily informed metrics: BUSCO analysis to assess gene content completeness using near-universal single-copy orthologs [9]
Validation: This phase ensures the assembly quality before proceeding to computationally intensive annotation steps, crucial for reliable downstream analysis.
Phase 3: Gene Prediction
Organism-Specific Tools:
- Prokaryotic genomes: Prokka for rapid annotation [9] [10]
- Eukaryotic genomes: BRAKER3 for automated eukaryotic genome annotation [9] [10]
Implementation: The workflow automatically routes data through the appropriate prediction tool based on the organism type specified by the user.
Phase 4: Functional Annotation
Primary Tool: InterProScan for comprehensive protein classification, identifying domains, families, and functional sites [9] [10].
Output: Annotated genomic features with functional predictions, enabling biological interpretation and insight generation for downstream applications such as drug target identification.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Computational Tools for Long-Read Microbial Genomics
| Category | Specific Tool/Reagent | Function/Purpose | Application Context |
|---|---|---|---|
| Sequencing Technologies | PacBio HiFi | Generates highly accurate long reads (>99% accuracy) [12]. | Ideal for variant calling and assembly where accuracy is prioritized. |
| Oxford Nanopore (ONT) | Produces long reads with additional functionality for methylation analysis [12]. | Suitable when detecting epigenetic modifications or adaptive sampling is needed. | |
| Assembly Tools | Canu | Performs assembly optimized for noisy long reads [9]. | Microbial genome assembly from noisy long-read data. |
| Flye | Enables de novo assembly using repetitive graphs [9]. | Assembling genomes with significant repetitive content. | |
| Gene Prediction | BRAKER3 | Automated eukaryotic genome annotation [9] [10]. | Gene prediction in fungal pathogens such as Candida auris. |
| Prokka | Rapid prokaryotic genome annotation [9] [10]. | Annotation of bacterial genomes like Klebsiella pneumoniae. | |
| Functional Annotation | InterProScan | Classifies proteins into families, predicts domains and functional sites [9] [10]. | Functional characterization of predicted genes. |
| Quality Assessment | BUSCO | Assesses genome completeness using evolutionary-informed expectations [9]. | Evaluation of assembly and annotation completeness. |
Implementation Considerations for Research and Drug Development
Technical Infrastructure Requirements
Successful implementation of microbial genome analysis workflows requires appropriate computational resources. The MIRRI-ERIC platform utilizes a hybrid infrastructure with both cloud computing and High-Performance Computing (HPC) components [9]:
- Cloud subsystem: Built on OpenStack, hosting the web-based component for data upload, parameter configuration, and result visualization
- HPC subsystem: Orchestrated by BookedSlurm, comprising multiple nodes with varied architectures (Intel and ARM) for parallel processing of computationally intensive tasks
- Storage systems: Utilizing BeeGFS and LUSTRE configured in all-flash setups for rapid data access
Data Management and FAIR Principles
Effective data management is crucial in genomic research. Researchers should:
- Submit assembled genomes and annotations to public repositories such as INSDC (GenBank, ENA, or DDBJ) [11]
- Apply FAIR (Findable, Accessible, Interoperable, and Reusable) principles to ensure data sustainability and reuse [11]
- Document workflow parameters and software versions to ensure reproducibility [9] [10]
This application note has detailed the key bioinformatics challenges in deriving biological insights from long-read sequencing data of microbial genomes and presented integrated solutions through platforms like MIRRI-ERIC. The comprehensive workflow from assembly to functional annotation, when properly implemented with appropriate computational resources and quality controls, enables researchers and drug development professionals to reliably characterize microbial genomes. This capability is particularly valuable for identifying potential therapeutic targets in pathogenic species, advancing both basic research and applied drug discovery efforts. The continuous evolution of long-read technologies and analytical methods promises to further enhance our ability to extract meaningful biological insights from microbial genomic data.
The vast majority of microorganisms on Earth have never been cultivated in a laboratory, representing a vast reservoir of unexplored biological diversity known as "microbial dark matter" (MDM) [13]. Traditional metagenomic studies, relying on short-read sequencing technologies, have struggled to assemble complete genomes from complex environmental samples due to challenges in resolving repetitive regions and distinguishing between closely related strains [1]. The emergence of high-throughput, long-read DNA sequencing has fundamentally transformed this landscape, enabling researchers to recover microbial genomes from environmental samples at an unprecedented scale and quality [8] [14]. This application note details how long-read sequencing technologies and associated bioinformatic workflows are expanding the known microbial diversity, providing researchers with powerful tools to access this untapped source of biodiversity for drug discovery and basic research.
Quantitative Advances in Microbial Diversity Discovery
Recent large-scale studies demonstrate the profound impact of long-read sequencing on cataloging microbial diversity. The Microflora Danica project, which performed deep long-read Nanopore sequencing of 154 complex terrestrial samples, exemplifies this progress [8] [14].
Table 1: Genome Recovery from the Microflora Danica Project
| Metric | Value | Significance |
|---|---|---|
| Total high-quality MAGs | 6,076 | Meet MIMAG high-quality standards |
| Total medium-quality MAGs | 17,767 | Useful for diversity assessments |
| Previously undescribed species | 15,314 | Substantial expansion of known diversity |
| Previously uncharacterized genera | 1,086 | 8% expansion of prokaryotic tree of life |
| Total sequencing data generated | 14.4 Tbp | Deep coverage enables high MAG recovery |
| Median MAGs per sample | 154 | Effective for complex terrestrial habitats |
The taxonomic novelty of these discoveries is striking, with 97.9% of the recovered metagenome-assembled genomes (MAGs) representing previously undescribed microbial genera or species [8]. This expansion is not merely quantitative but also functional, as the long-read assemblies enabled the recovery of thousands of complete ribosomal RNA operons, biosynthetic gene clusters, and CRISPR-Cas systems, providing valuable insights into the functional capabilities of these novel microorganisms [8] [14].
Table 2: Impact on Taxonomic Classification
| Database Enhancement | Improvement | Application |
|---|---|---|
| Incorporation into public databases | Substantially improved species-level classification | Better interpretation of soil and sediment metagenomes |
| Recovery of complete rRNA operons | Improved phylogenetic resolution | More accurate taxonomic placement |
| Biosynthetic gene cluster discovery | Identification of novel natural product pathways | Drug discovery and biotechnology |
Experimental Protocols for Genome-Resolved Metagenomics
Sample Collection and DNA Extraction
The success of long-read metagenomics begins with appropriate sample handling. The Microflora Danica project analyzed 125 soil, 28 sediment, and 1 water sample, selected from over 10,000 samples to represent 15 distinct habitat types [8]. For optimal results:
- Sample Preservation: Immediately freeze samples at -80°C or use specialized preservation buffers to prevent nucleic acid degradation.
- DNA Extraction: Use amplification-free, high-quality DNA extraction protocols. Long-read sequencing typically requires 150 ng to 1 μg of high-molecular-weight DNA, with minimal fragmentation [1].
- Quality Assessment: Verify DNA integrity using pulsed-field gel electrophoresis or similar methods to ensure sufficient fragment length.
Sequencing Platform Considerations
The two dominant long-read sequencing platforms offer complementary advantages for metagenomic studies:
Table 3: Sequencing Platform Comparison for Metagenomics
| Parameter | PacBio HiFi | Oxford Nanopore Technologies (ONT) |
|---|---|---|
| Read accuracy | >99.5% [15] | 97-99% [1] |
| Average read length | 15-18 kb [1] | 13-20 kb (up to 4 Mb) [1] |
| DNA input requirement | 150 ng-1 μg [1] | 150 ng-1 μg [1] |
| Throughput | 90 Gb per SMRT Cell (Revio) [1] | Up to 120 Gb (PromethION) [1] |
| Methylation detection | Native detection without bisulfite conversion [15] | Direct detection of modifications [12] |
| Cost considerations | Higher perGb cost [1] | Lower initial investment for portable units |
For the Microflora Danica project, Nanopore sequencing was selected, generating a median of 94.9 Gbp per sample with a read N50 of 6.1 kbp [8]. The total output of 14.4 Tbp demonstrates the scalability of this approach for large biodiversity surveys.
The mmlong2 Bioinformatics Workflow
The computational recovery of genomes from complex metagenomes requires specialized bioinformatic workflows. The custom mmlong2 workflow developed for the Microflora Danica project incorporates multiple optimizations for recovering prokaryotic MAGs from extremely complex datasets [8]:
Figure 1: The mmlong2 bioinformatic workflow for recovering MAGs from complex metagenomes. The workflow integrates multiple assembly and binning strategies to maximize genome recovery from long-read data.
Key computational steps include:
- Metagenome Assembly: Use long-read assemblers such as Canu or Flye to generate contigs from raw reads.
- Polishing and Eukaryotic Contig Removal: Refine assemblies and filter eukaryotic sequences based on taxonomic classification.
- Circular MAG Extraction: Identify circular contigs representing potential complete genomes or plasmids.
- Differential Coverage Binning: Incorporate read mapping information from multi-sample datasets to distinguish populations.
- Ensemble Binning: Apply multiple binning algorithms (e.g., MetaBAT2, MaxBin2) to the same metagenome and consolidate results.
- Iterative Binning: Perform multiple rounds of binning, progressively refining the metagenome.
This workflow recovered 3,349 (14.0%) additional MAGs through iterative binning that would have been missed with standard approaches [8].
Advantages of Long-Read Over Short-Read Metagenomics
Long-read technologies provide distinct advantages for exploring microbial dark matter:
Figure 2: Comparative advantages of long-read versus short-read sequencing for metagenomic studies. Long reads enable more complete genome reconstruction and additional layers of analysis.
- Resolution of Repetitive Regions: Long reads can span repetitive elements and highly homologous regions that confound short-read assembly, enabling complete genome reconstruction [12] [1].
- Strain-Level Differentiation: Read lengths covering multiple variants enable precise discrimination of microbial strains, which is crucial as strain-level differences often determine functional capabilities and host interactions [16].
- Complete Gene and Operon Recovery: Long reads facilitate the assembly of complete biosynthetic gene clusters, ribosomal RNA operons, and other multi-gene elements, providing better insights into functional potential [8].
- Epigenetic Profiling: Both PacBio and ONT technologies can natively detect DNA methylation patterns during sequencing, providing additional layers of regulatory information without requiring specialized library preparation [12] [15].
Table 4: Key Research Reagents and Computational Tools for Long-Read Metagenomics
| Category | Tool/Resource | Function | Application Notes |
|---|---|---|---|
| Wet Lab | AMPure XP beads | DNA size selection and cleanup | Critical for obtaining high-molecular-weight DNA |
| Nanopore Ligation Sequencing Kit | Library preparation for ONT | Rapid protocol (1-2 hours) [1] | |
| PacBio SMRTbell Prep Kit | Library preparation for HiFi | Longer protocol (minimum 7 hours) [1] | |
| Sequencing Platforms | PacBio Revio | High-throughput HiFi sequencing | 360 Gb in 24 hours [1] |
| ONT PromethION | High-throughput nanopore sequencing | ~120 Gb per flow cell [1] | |
| ONT MinION | Portable sequencing | Enables field sequencing [1] | |
| Bioinformatics | mmlong2 [8] | MAG recovery from complex samples | Custom workflow for terrestrial metagenomes |
| hifiasm [15] | De novo assembly | Optimized for PacBio HiFi reads | |
| minimap2/pbmm2 [12] [15] | Read alignment | Foundation for variant detection | |
| Dorado [12] | Basecalling for ONT | Converts raw signal to nucleotide sequences | |
| Databases | GTDB (Genome Taxonomy Database) [17] | Taxonomic classification | Standardized microbial taxonomy |
| IMG/M [18] | Genome data management and analysis | Includes statistical analysis tools |
Implications for Gene Prediction and Functional Annotation
The application of long-read sequencing to microbial dark matter has profound implications for gene prediction in assembled genomes:
- Improved Gene Model Accuracy: Continuous sequence information across full coding sequences enables more accurate prediction of gene start and stop sites, particularly for genes with repetitive domains or complex structures.
- Discovery of Novel Gene Families: The expanded genomic diversity revealed through long-read metagenomics has led to the identification of previously unknown protein families and functional domains [8].
- Metabolic Pathway Reconstruction: Complete genome assemblies enable more reliable reconstruction of metabolic pathways, revealing the functional potential of uncultivated microorganisms [17].
- Connection to Expression Data: The generation of complete gene models facilitates integration with metatranscriptomic data, enabling researchers to distinguish which genes are actively expressed in different environments [16].
Long-read sequencing technologies have fundamentally transformed our ability to explore microbial dark matter, moving from fragmented genomic glimpses to complete genome reconstruction for thousands of previously unknown microorganisms. The combination of advanced sequencing platforms with specialized bioinformatic workflows like mmlong2 enables researchers to efficiently recover high-quality microbial genomes from even the most complex terrestrial environments. These advances are rapidly expanding the known microbial tree of life and providing unprecedented access to the genomic potential of uncultivated microorganisms, opening new frontiers for drug discovery, biotechnology, and fundamental understanding of microbial evolution and ecology.
Within gene prediction research for long-read assembled microbial genomes, the initial quality of the genome assembly is paramount. A fragmented or incomplete assembly will inevitably lead to fragmented and incomplete gene models, compromising all downstream biological interpretation [9]. For drug development professionals investigating microbial secondary metabolites or virulence factors, accurate identification of these often repetitive genomic features is entirely dependent on a high-quality, contiguous assembly [19]. This guide details the essential metrics and standardized protocols for evaluating the completeness and accuracy of microbial genome assemblies, providing a critical foundation for reliable gene prediction and annotation.
Core Concepts and Metric Definitions
The quality of a de novo genome assembly is evaluated through multiple lenses, primarily focusing on contiguity, completeness, and accuracy [20]. Contiguity measures how much of the assembly is reconstructed into large, uninterrupted sequences. Completeness assesses whether the entire expected genomic content, particularly genes, is present. Accuracy evaluates the correctness of the assembled nucleotide sequence.
Key Metric Definitions
- N50/L50: The N50 is the length of the shortest contig or scaffold at which 50% of the entire assembly is contained [20]. The L50 is the minimal number of contigs or scaffolds whose length sum makes up 50% of the genome size [20]. A higher N50 and a lower L50 indicate a more contiguous assembly.
- BUSCO (Benchmarking Universal Single-Copy Orthologs): This metric assesses genome completeness by searching for a set of evolutionarily conserved, single-copy orthologs that are expected to be present in a single copy in a given lineage [9] [20]. The result is presented as the percentage of these orthologs found as complete (and whether single or duplicated), fragmented, or missing.
- QV (Quality Value): A logarithmic measure of assembly accuracy, calculated as QV = -10 × log₁₀(error rate). For example, a QV of 30 corresponds to an error rate of 1 in 1,000 bases [20]. This can be calculated using k-mer based tools like Merqury.
- k-mer Completeness: This metric, derived from tools like Merqury, evaluates what percentage of the k-mers from the original sequencing reads are present in the final assembly, indicating how well the assembly represents the raw data [20].
Table 1: A summary of key assembly quality metrics, their descriptions, and ideal targets for microbial genomes.
| Metric Category | Specific Metric | Description | Ideal Target (Microbial Genomes) |
|---|---|---|---|
| Contiguity | Number of Contigs | Total number of contigs in the assembly. | As low as possible; approaching the number of chromosomes/plasmids. |
| N50 / L50 | N50: Length of the shortest contig at 50% of total genome length. L50: The number of contigs at the N50 size [20]. | Higher N50, Lower L50. | |
| Total Assembly Length | Total number of base pairs in the assembly, including 'N's. | Should match the expected genome size for the organism. | |
| Completeness | BUSCO Score | Percentage of conserved, single-copy orthologs found complete in the assembly [9] [20]. | >95% for a high-quality draft. |
| k-mer Completeness | Percentage of unique k-mers from sequencing reads found in the assembly [20]. | >99%. | |
| Accuracy | QV (Quality Value) | Logarithmic measure of base-level accuracy [20]. | QV > 40 (error rate < 1/10,000) is excellent. |
| GC Content | Percentage of Guanine and Cytosine bases. | Should be consistent with known biology of closely related species. |
Experimental Protocols for Quality Assessment
Implementing a standardized workflow is crucial for consistent and comprehensive assembly evaluation. The following protocols are widely adopted in the field.
A Standardized Workflow for Assembly QC
The diagram below outlines a generalized workflow for genome assessment, integrating the key tools and metrics described in this guide.
Protocol 1: Contiguity and Basic Statistics with QUAST
Principle: This protocol uses QUAST (Quality Assessment Tool for Genome Assemblies) to generate a comprehensive report on assembly contiguity and basic statistics, which are fundamental for initial quality screening [20].
Materials:
- Genome assembly file in FASTA format.
- A reference genome (optional, for comparative analysis).
- QUAST software (v5.0.2 or higher).
Method:
- Execute QUAST: Run QUAST from the command line. If you have a reference genome, include it for a more detailed analysis.
- Interpret Results: Open the generated
report.htmlfile. Key metrics to examine include:- Total number of contigs: Fewer is better.
- Largest contig: Larger is better.
- N50 & L50: Primary indicators of contiguity.
- Total length: Check against expected genome size.
- GC (%): Verify it is consistent with the organism.
Protocol 2: Gene-Completeness Assessment with BUSCO
Principle: BUSCO assesses the completeness of a genome assembly based on expected gene content. It works by searching for universal single-copy orthologs that should be present in a specific evolutionary lineage [9] [20].
Materials:
- Genome assembly file in FASTA format.
- BUSCO software (v5.4.6 or higher).
- Appropriate BUSCO lineage dataset (e.g.,
bacteria_odb10for prokaryotes).
Method:
- Run BUSCO: Execute BUSCO with the appropriate lineage.
- Analyze Output: The key results are in
short_summary.json:- Complete Single-Copy: The percentage of BUSCO genes found exactly once. This is the primary measure of completeness.
- Complete Duplicated: A high percentage here may indicate haplotypic duplication or collapse of repeats.
- Fragmented: BUSCOs found only partially.
- Missing: BUSCOs entirely absent from the assembly.
Protocol 3: k-mer Based Accuracy and Completeness with Merqury
Principle: Merqury evaluates assembly quality by comparing the k-mers present in the assembly to those in the original high-quality sequencing reads (e.g., PacBio HiFi or Illumina). This provides independent measures of accuracy (QV) and completeness without a reference genome [20].
Materials:
- Genome assembly file in FASTA format.
- The raw sequencing reads used for assembly (in
fastqsanger.gzformat). - Merqury software (v1.3 or higher).
- Meryl for k-mer counting.
Method:
- Build k-mer Database: First, count k-mers from the raw reads using Meryl.
- Run Merqury: Use the k-mer database to evaluate the assembly.
- Review Key Metrics:
- QV: The overall base-level quality value.
- k-mer Completeness: The proportion of read k-mers found in the assembly.
- Spectra CN Plot: Visualizes k-mer multiplicity to help identify assembly artifacts like false duplications.
The Scientist's Toolkit
Table 2: Essential software and databases for evaluating genome assembly quality.
| Tool / Resource | Function | Application in Quality Control |
|---|---|---|
| QUAST | Genome assembly quality assessment [20]. | Calculates contiguity metrics (N50, L50) and compares assembly versions. |
| BUSCO | Assessment of genome completeness [9] [20]. | Quantifies the presence of universal single-copy orthologs to benchmark completeness. |
| Merqury | k-mer based evaluation of accuracy and completeness [20]. | Provides QV and k-mer completeness scores without a reference genome. |
| BUSCO Lineage Datasets | Curated sets of orthologs for specific taxonomic groups. | Serves as the reference for BUSCO analysis; critical to select the correct lineage (e.g., bacteria, fungi). |
| Meryl | Efficient k-mer counting and database management [20]. | Creates the k-mer databases required for Merqury analysis. |
| LongReadSum | Quality control for long-read sequencing data [12] [21]. | Assesses raw read quality prior to assembly, which impacts final assembly quality. |
Rigorous evaluation of assembly completeness and accuracy is a non-negotiable step in any research pipeline involving long-read microbial genomes. For gene prediction in particular, a high BUSCO score ensures that the full gene repertoire is present for annotation, while a high QV and contiguity metrics ensure that the gene models themselves are accurately reconstructed and not fragmented. By adhering to the standardized metrics and protocols outlined in this guide, researchers and drug development scientists can establish a robust foundation for their genomic studies, ensuring that subsequent discoveries in gene function, virulence, and drug discovery are built upon reliable data.
Integrated Workflows and Advanced Tools for Precision Gene Prediction
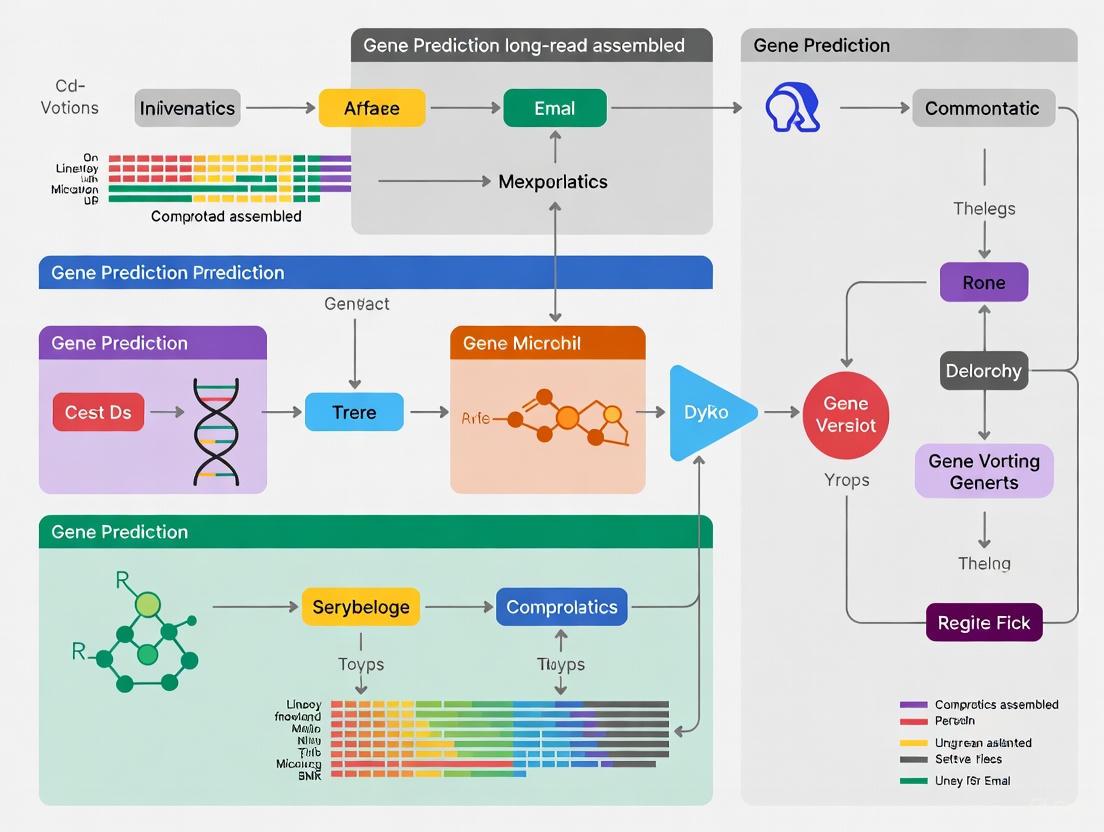
The advent of long-read sequencing technologies has significantly enhanced our ability to generate high-quality microbial genome assemblies, providing more complete and contiguous genomic sequences. However, transforming these raw sequencing data into biologically meaningful insights remains a formidable challenge for many researchers. The process requires the integration of multiple sophisticated tools for genome assembly, gene prediction, and functional annotation, demanding advanced computational skills and access to powerful computing infrastructure that may not be readily available to all research groups [9] [10].
To address these challenges, the Italian node of the Microbial Resource Research Infrastructure (MIRRI ERIC) has developed a comprehensive bioinformatics platform specifically designed for long-read microbial sequencing data. This service provides an end-to-end solution for analyzing both prokaryotic and eukaryotic genomes, making advanced genomic analysis accessible to researchers without extensive computational expertise while maintaining the reproducibility and rigor required for scientific research [9]. Developed as part of the SUS-MIRRI.IT project, this platform represents a significant advancement toward user-centered scalable bioinformatics services for microbial research [9] [10].
The MIRRI platform employs a modular architecture that operates on a hybrid computational infrastructure, seamlessly integrating both cloud computing and High-Performance Computing (HPC) resources. This design ensures both accessibility and computational power for demanding bioinformatics workflows [9] [10].
The system is structured around two primary components. The web-based component operates on virtual machines within a cloud infrastructure and is responsible for user interactions, including data upload, configuration of analysis parameters, and visualization of results. It features an intuitive user interface designed to ensure seamless interaction for users with varying levels of computational expertise. The computing component manages the execution of data analysis workflows on HPC infrastructure, processing user data in parallel across multiple HPC nodes and returning results to the web interface [9].
This service is integrated into the broader Italian Collaborative Working Environment (ItCWE), serving as the primary access point for SUS-MIRRI.IT services. The platform stands out for its three key innovative aspects: (1) ease of use through an intuitive web application, (2) transparent leveraging of HPC infrastructure to accelerate analysis, and (3) ensuring reproducibility through Common Workflow Language (CWL) and Docker containers [9] [10].
Table 1: Computational Infrastructure Supporting the MIRRI Platform
| Infrastructure Component | Specifications | Role in Platform |
|---|---|---|
| Cloud System (OpenStack) | 2,400+ physical cores, 60 TB RAM, 120 GPUs, 25 Gb/s networking | Hosts web-based component for user interaction |
| HPC Subsystem (BookedSlurm) | 68 Intel nodes (36 cores, 128 GB RAM each), 4 ARM nodes (80 cores, 512 GB RAM each) | Executes computationally intensive analysis workflows |
| Storage Systems | BeeGFS and LUSTRE, configured in all-flash setup | Manages large genomic datasets and interim results |
Application Notes: Workflow and Implementation
End-to-End Analysis Workflow
The platform implements a comprehensive workflow for microbial genome analysis that encompasses four main phases: assembly, evaluation, gene prediction, and functional annotation. This workflow is designed to be flexible, supporting data from various long-read sequencing technologies including Nanopore, PacBio, and PacBio HiFi [10].
The following diagram illustrates the complete analysis workflow implemented by the platform:
Diagram 1: The four-phase workflow for microbial genome analysis, showing the pathway from raw sequencing data to functional annotation. The process begins with multiple assemblers operating in parallel to enhance assembly quality, followed by comprehensive evaluation, domain-specific gene prediction, and concluding with functional characterization.
Phase 1: Assembly Phase Protocol
The assembly phase employs multiple state-of-the-art assemblers to enhance the completeness and accuracy of genome reconstructions. The protocol begins with quality assessment of raw long-read sequencing data, followed by simultaneous processing through three assemblers [9] [10].
Materials and Reagents:
- Input: Long-read sequencing data (Nanopore, PacBio, or PacBio HiFi)
- Canu assembler (v2.0 or higher)
- Flye assembler (v2.9 or higher)
- wtdbg2 assembler (v2.5 or higher)
- Computational resources: Minimum 128 GB RAM, 36 cores per assembler instance
Step-by-Step Procedure:
- Data Preprocessing: Upload raw FASTQ files through the web interface. The platform accepts compressed (.gz) or uncompressed formats.
- Parameter Configuration: Select sequencing technology used (Nanopore, PacBio, or PacBio HiFi) via the graphical interface. Default parameters are optimized for each technology but can be customized.
- Parallel Assembly Execution: Launch simultaneous assembly jobs using Canu, Flye, and wtdbg2. The platform automatically distributes these across available HPC nodes.
- Output Generation: Each assembler produces a draft genome assembly in FASTA format. The platform retains all outputs for evaluation in the next phase.
Technical Notes: Using multiple assemblers improves the probability of obtaining a high-quality assembly, as different algorithms may perform better depending on the specific characteristics of the dataset and organism [9].
Phase 2: Assembly Evaluation Protocol
The evaluation phase assesses the quality of the generated assemblies using both standard metrics and evolutionarily informed assessments of gene content.
Materials and Reagents:
- Input: Draft genome assemblies from Phase 1
- BUSCO software (v5 or higher) with appropriate lineage datasets
- QUAST or similar assembly metrics tool
- Reference genome (optional, for comparative analysis)
Step-by-Step Procedure:
- Contiguity Metrics Calculation: Compute standard assembly statistics including N50, L50, total assembly size, and number of contigs.
- BUSCO Analysis: Run BUSCO analysis using the appropriate lineage dataset (bacteria, fungi, etc.) to assess completeness based on evolutionarily informed expectations of gene content.
- Comparative Assessment: Compare results across assemblies from different tools to select the highest quality assembly for downstream analysis.
- Report Generation: Compile a comprehensive quality assessment report highlighting strengths and weaknesses of each assembly.
Technical Notes: The platform automatically selects the best assembly based on a weighted score incorporating both contiguity metrics and BUSCO completeness scores. However, users can manually override this selection based on their specific research needs [9] [10].
Phase 3: Gene Prediction Protocol
This phase employs specialized tools for gene prediction based on the genomic domain of the organism—prokaryotic or eukaryotic.
Materials and Reagents:
- Input: Selected genome assembly from Phase 2
- Prokka (v1.14 or higher) for prokaryotic genomes
- BRAKER3 (v3.0 or higher) for eukaryotic genomes
- Reference protein sequences (optional, for homology-based prediction)
Step-by-Step Procedure:
- Domain Specification: Indicate whether the organism is prokaryotic or eukaryotic through the web interface.
- Tool Execution:
- For prokaryotes: Run Prokka with default parameters or customized settings
- For eukaryotes: Execute BRAKER3 pipeline incorporating both gene model prediction and evidence-based annotation
- Output Processing: Extract predicted gene models, protein sequences, and functional assignments
- Format Standardization: Generate standard GFF3 and GBK files containing annotation information
Technical Notes: BRAKER3 incorporates GeneMark-EP+ and AUGUSTUS supported by protein database information, significantly improving prediction accuracy for eukaryotic genomes [9].
Phase 4: Functional Annotation Protocol
The final phase focuses on characterizing the functional elements of predicted genes through protein domain analysis and functional classification.
Materials and Reagents:
- Input: Predicted protein sequences from Phase 3
- InterProScan (v5.0 or higher)
- Functional databases: Pfam, PROSITE, PRINTS, PANTHER, Gene3D, SUPERFAMILY
Step-by-Step Procedure:
- Protein Sequence Submission: Submit predicted protein sequences in FASTA format to InterProScan
- Domain Analysis: Execute simultaneous searches against multiple protein signature databases
- Integration: Combine results from all databases to generate comprehensive functional annotations
- Enrichment: Map functional annotations to Gene Ontology (GO) terms, metabolic pathways, and other functional classifications
Technical Notes: The platform provides enriched annotations by querying multiple external repositories, facilitating biological interpretation and extraction of meaningful insights from analysis outcomes [9].
Table 2: Bioinformatics Tools Integrated in the MIRRI Platform
| Tool | Version | Function | Organism Type |
|---|---|---|---|
| Canu | 2.0+ | Long-read assembly | Prokaryotes & Eukaryotes |
| Flye | 2.9+ | Long-read assembly | Prokaryotes & Eukaryotes |
| wtdbg2 | 2.5+ | Long-read assembly | Prokaryotes & Eukaryotes |
| BUSCO | 5.0+ | Assembly evaluation | Prokaryotes & Eukaryotes |
| Prokka | 1.14+ | Gene prediction | Prokaryotes |
| BRAKER3 | 3.0+ | Gene prediction | Eukaryotes |
| InterProScan | 5.0+ | Functional annotation | Prokaryotes & Eukaryotes |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of genomic analyses requires both computational tools and biological materials. The following table details essential research reagent solutions for researchers working with the MIRRI platform or similar bioinformatics workflows.
Table 3: Essential Research Reagents and Materials for Microbial Genome Analysis
| Reagent/Material | Function/Application | Specifications |
|---|---|---|
| Microbial Culturing Media | Isolation and propagation of pure microbial cultures | Composition varies by microbial type (e.g., LB for bacteria, PDA for fungi) |
| DNA Extraction Kits | High-molecular-weight DNA isolation suitable for long-read sequencing | Must yield DNA >20 kb fragment size (e.g., Qiagen Genomic-tip, Nanobind CBB) |
| Long-read Sequencing Kits | Library preparation for sequencing platforms | Oxford Nanopore Ligation Sequencing Kit or PacBio SMRTbell Prep Kit 3.0 |
| Quality Control Reagents | Assessment of DNA quality and quantity prior to sequencing | Fluorometric assays (Qubit dsDNA HS), fragment analyzers (Femto Pulse) |
| Reference Genomes | Comparative analysis and validation | Species-specific complete genomes from NCBI RefSeq |
| BUSCO Lineage Datasets | Assessment of genome completeness | Specific to taxonomic group (e.g., bacteriaodb10, fungiodb10) |
| Functional Annotation Databases | Protein domain identification and functional classification | InterPro-integrated databases (Pfam, PROSITE, Gene3D, etc.) |
Validation and Case Studies
The utility of the MIRRI platform has been demonstrated through case studies involving three microorganisms of clinical and environmental significance from the TUCC culture collections: Scedosporium dehoogii MUT6599, Klebsiella pneumoniae TUCC281, and Candida auris TUCC287 [9] [10].
For each organism, the platform successfully generated high-quality genome assemblies, accurate gene predictions, and biologically meaningful functional annotations. The integration of multiple assemblers proved particularly valuable, as different tools performed variably across organisms, allowing selection of the optimal assembly for each species. The platform's ability to handle both prokaryotic (K. pneumoniae) and eukaryotic (S. dehoogii and C. auris) genomes demonstrated its versatility across microbial domains [9].
The case studies validated the platform's performance in producing reliable, biologically meaningful insights, positioning it as a valuable tool for both routine genome analysis and advanced microbial research. The automated evaluation metrics provided objective assessment of assembly and annotation quality, while the user-friendly interface made these advanced analyses accessible to researchers without specialized bioinformatics training [9] [10].
The MIRRI ERIC Italian node's bioinformatics platform represents a significant advancement in microbial genome analysis, providing an end-to-end solution that bridges the gap between raw long-read sequencing data and biologically meaningful insights. By integrating state-of-the-art tools within a reproducible, scalable workflow and providing access through an intuitive web interface, the platform addresses critical challenges in computational microbiology.
The platform's modular architecture, leveraging both cloud computing for accessibility and HPC for computational intensity, makes advanced genomic analyses accessible to a broader research community. Its support for both prokaryotic and eukaryotic organisms, combined with rigorous quality assessment at each analysis phase, ensures reliable results suitable for diverse research applications from basic microbiology to drug development.
As long-read sequencing technologies continue to evolve and become more widely adopted, comprehensive platforms like this will play an increasingly important role in accelerating microbial genomics research and translating genomic information into biological understanding with potential applications in health, biotechnology, agriculture, and environmental science.
In the field of microbial genomics, accurate gene prediction is a cornerstone for understanding gene function, evolutionary dynamics, and biotechnological potential. Traditional gene prediction tools often assume a standard genetic code and uniform gene structure, an approach that fails to account for the remarkable diversity of genetic codes used by different microbial lineages. This limitation is particularly acute when analyzing complex metagenomic samples or long-read assembled genomes encompassing organisms from multiple domains of life (Bacteria, Archaea, Eukarya, and Viruses), each with their own distinct genetic architectures. Lineage-specific prediction has emerged as a critical solution, leveraging the taxonomic assignment of genetic sequences to apply optimized, lineage-appropriate gene-finding tools and parameters. This paradigm shift enables a more accurate and comprehensive exploration of the functional potential encoded within microbial genomes [22].
The advent of long-read sequencing technologies has significantly improved the quality of microbial genome assemblies by producing longer contiguous sequences (contigs). However, transforming these high-quality assemblies into biologically meaningful annotations requires sophisticated computational workflows that respect biological diversity. Lineage-specific prediction addresses this by ensuring that the correct genetic code is used for different taxa, that incomplete protein predictions are filtered out, and that the prediction of small proteins is optimized. This is especially vital for the growing field of protein ecology, which studies the distribution and association of proteins, rather than just taxonomic markers, to understand their ecological importance and relationship with host health [22].
Comparative Analysis of Gene Prediction Approaches
A comparative analysis reveals the significant quantitative and qualitative advantages of a lineage-specific workflow over a one-size-fits-all approach. The core improvement lies in using taxonomic classification to select the most appropriate gene prediction tools for each contig, rather than applying a single tool to all data.
Quantitative Workflow Output Comparison
The table below summarizes a performance comparison between a standard single-tool approach and an integrated lineage-specific workflow, applied to a large-scale human gut microbiome dataset comprising 9,677 metagenomes [22].
Table 1: Performance comparison of gene prediction approaches on human gut microbiome data
| Performance Metric | Standard Approach (Pyrodigal only) | Lineage-Specific Workflow | Change |
|---|---|---|---|
| Total Genes Predicted | 737,874,876 | 846,619,045 | +14.7% |
| Protein Clusters (90% similarity) | Not Available | 29,232,514 | +210.2% vs. UHGP* |
| Singleton Protein Clusters | Not Available | 14,043,436 | - |
| Expressed Singletons (via metatranscriptomics) | Not Available | 39.1% | - |
| Bacterial Contig Proteins | Dominant | 58.4% ± 18.9% | - |
| Eukaryotic Contig Proteins | Underrepresented | 0.03% ± 1.31% | - |
*UHGP: Unified Human Gastrointestinal Protein catalogue, used as a reference benchmark [22].
The lineage-specific workflow identified over 108 million additional genes, substantially expanding the known protein landscape of the human gut. Crucially, metatranscriptomic validation confirmed that a significant proportion of the rare "singleton" proteins are expressed, confirming they are not computational artifacts but real, functionally relevant components of the microbiome [22].
Tool Selection by Taxonomic Domain
The effectiveness of the lineage-specific approach depends on using the optimal combination of gene prediction tools for each taxonomic group. The following table details the tool selection based on systematic benchmarking.
Table 2: Lineage-specific tool selection and key parameters for gene prediction
| Taxonomic Group | Recommended Tool Combination | Key Rationale and Parameters |
|---|---|---|
| Bacteria | Pyrodigal, MetaGeneMark, FragGeneScan+ | Optimized for prokaryotic gene structure; uses translation table 11 [22]. |
| Archaea | Pyrodigal, MetaGeneMark, FragGeneScan+ | Adapted for archaeal genetic codes and gene structures [22]. |
| Eukaryotes | AUGUSTUS, SNAP, GeneMark-ES | Critical for predicting multi-exon genes with introns; Pyrodigal is not suitable [22]. |
| Viruses | Pyrodigal, MetaGeneMark, PHANOTATE | Tailored for compact viral genomes and alternative genetic codes [22]. |
| Unknown/Unassigned | Pyrodigal, MetaGeneMark, FragGeneScan+ | Applies a conservative prokaryotic-leaning model for contigs without taxonomic assignment [22]. |
Benchmarking showed that while using multiple tools per domain slightly increases spurious predictions, the benefit of capturing a much larger set of real proteins outweighs this cost. For eukaryotic genes, the combination of tools is particularly important due to the inability of prokaryotic-centric tools like Pyrodigal to handle introns [22].
Integrated Protocol for Lineage-Specific Gene Prediction
This section provides a detailed, executable protocol for implementing lineage-specific gene prediction, from genome assembly to functional annotation.
The diagram below illustrates the complete integrated workflow for long-read microbial genome assembly and lineage-specific gene prediction.
Step-by-Step Experimental Protocol
Phase 1: Genome Assembly and Evaluation
Step 1.1: Multi-Tool Assembly
- Objective: Generate a high-quality, contiguous genome assembly from long-read data.
- Procedure: Execute at least two of the following assemblers in parallel on your long-read data (e.g., Oxford Nanopore or PacBio):
- Canu:
canu -p [output_prefix] -d [output_dir] genomeSize=[size] useGrid=false -nanopore [input.fastq] - Flye:
flye --nano-raw [input.fastq] --out-dir [out_dir] --threads [threads] - wtdbg2:
wtdbg2 -x ont -g [size] -i [input.fastq] -t [threads] -o [output_prefix]
- Canu:
- Rationale: Using multiple assemblers and generating a consensus improves assembly completeness and accuracy [9] [10].
Step 1.2: Assembly Evaluation
- Objective: Assess the quality and completeness of the assembled genome.
- Procedure:
- Run BUSCO to assess gene content completeness against a near-universal single-copy ortholog dataset:
busco -i [assembly.fasta] -l [lineage_dataset] -o [busco_output] -m genome - Calculate standard assembly metrics (N50, L50) using tools like Quast.
- Run BUSCO to assess gene content completeness against a near-universal single-copy ortholog dataset:
- Note: A high-quality microbial assembly should typically have a BUSCO completeness score >90% and a high N50 value, indicating contiguity [9] [10].
Phase 2: Taxonomic Classification and Gene Prediction
Step 2.1: Taxonomic Classification of Contigs
- Objective: Determine the taxonomic origin of each contig in the assembly to inform tool selection.
- Procedure: Use Kraken 2 with a standard database (e.g., RefSeq) to classify contigs:
kraken2 --db [kraken_db] --threads [threads] --report [report.txt] --output [output.txt] [assembly.fasta][22].
Step 2.2: Lineage-Specific Gene Calling
- Objective: Accurately predict protein-coding genes on each contig using domain-appropriate tools.
- Procedure: Parse the Kraken 2 output and process contigs based on their taxonomic assignment using the tool combinations specified in Table 2.
- For Bacterial/Archaeal/Viral/Unknown contigs: Execute the recommended three-tool combination (e.g., Pyrodigal, MetaGeneMark, FragGeneScan+). Merge the results, prioritizing overlapping predictions.
- For Eukaryotic contigs: Execute AUGUSTUS, SNAP, and GeneMark-ES. Use evidence-based approaches where possible to reconcile multi-exon gene predictions.
- Critical Parameter: Ensure the correct genetic code (e.g., translation table) is specified for each tool based on the lineage [22].
Phase 3: Functional Annotation and Downstream Analysis
Step 3.1: Functional Protein Annotation
- Objective: Assign biological functions to the predicted protein sequences.
- Procedure: Run InterProScan on the final set of predicted protein sequences:
interproscan.sh -i [proteins.faa] -f tsv -o [output.tsv] --goterms --pathways[9] [10]. For prokaryotic-focused analyses, Prokka can provide a rapid, integrated annotation [9].
Step 3.2: Protein Ecology Analysis
- Objective: Study the ecological distribution of proteins and their association with host parameters.
- Procedure: For large-scale metagenomic studies, dereplicate proteins into clusters (e.g., at 90% identity using CD-HIT). Use tools like InvestiGUT to correlate protein cluster prevalence and abundance with host metadata (e.g., disease state, diet) [22].
This section catalogs the key software tools and computational resources essential for implementing the lineage-specific prediction protocol.
Table 3: Essential resources for lineage-specific gene prediction workflows
| Resource Name | Type | Primary Function | Key Application Note |
|---|---|---|---|
| Flye / Canu | Software Tool | De novo genome assembly from long reads. | Used in the initial assembly phase to generate contigs from raw sequencing data [9] [10]. |
| Kraken 2 | Software Tool | Taxonomic classification of sequence contigs. | Determines the lineage of each contig, directing it to the appropriate gene prediction tools [22]. |
| Pyrodigal | Software Tool | Gene prediction in prokaryotic and viral sequences. | Fast and accurate; a core tool for bacterial, archaeal, and viral contigs [22]. |
| AUGUSTUS | Software Tool | Gene prediction in eukaryotic sequences. | Critical for predicting complex, multi-exon genes in fungal and other microbial eukaryotic contigs [22]. |
| BRAKER3 | Software Tool | Eukaryotic gene prediction with RNA-seq data integration. | An alternative for eukaryotic gene prediction that can leverage transcriptomic evidence [9]. |
| InterProScan | Software Tool | Functional annotation of protein sequences. | Scans against multiple databases to assign protein families, domains, and functional sites [9] [10]. |
| Prokka | Software Tool | Rapid prokaryotic genome annotation. | Provides a streamlined pipeline for functional annotation of bacterial and archaeal genomes [9]. |
| MIRRI-IT Platform | Web Service | Integrated online platform for microbial analysis. | Provides a user-friendly, HPC-powered implementation of a similar long-read assembly-to-annotation workflow [9]. |
| InvestiGUT | Software Tool | Protein ecology analysis. | Enables association studies between protein prevalence from metagenomes and host parameters [22]. |
Workflow Logic of Lineage-Specific Gene Prediction
The core logical process of the lineage-specific prediction step (Phase 2) is detailed in the following diagram.
Within the framework of gene prediction research on long-read assembled microbial genomes, the selection of an appropriate annotation tool is a critical determinant of success. High-quality genome assemblies from technologies like Oxford Nanopore or PacBio provide the foundation, but accurate gene structural annotation transforms this sequence into biologically meaningful information [9] [10]. For microbial genomics, which encompasses both prokaryotic and eukaryotic organisms, this process is not one-size-fits-all. The choice of tool must be guided by the fundamental biological distinctions between these cellular life forms, primarily the presence of a nucleus and complex gene architecture in eukaryotes.
This Application Note provides a structured comparison between two established annotation tools: Prokka, optimized for prokaryotic genomes, and BRAKER3, designed for eukaryotic genomes. We detail their operational principles, provide validated protocols for their use with long-read data, and contextualize their application within a broader microbial genomics research pipeline. The platform developed by the Italian MIRRI ERIC node demonstrates the integration of both tools (Prokka for prokaryotes and BRAKER3 for eukaryotes) into a unified, reproducible workflow for long-read microbial data, highlighting their complementary roles in comprehensive microbial research [9] [10].
Tool Comparison and Selection Criteria
The table below summarizes the core characteristics of Prokka and BRAKER3 to guide tool selection.
Table 1: Key Comparison between Prokka and BRAKER3
| Feature | Prokka | BRAKER3 |
|---|---|---|
| Primary Domain | Bacteria, Archaea, Viruses [23] | Eukaryotes [24] [25] |
| Core Prediction Method | Integration of evidence-based tools (e.g., Prodigal) | Combination of GeneMark-ETP and AUGUSTUS [24] |
| Evidence Integration | Protein homology (e.g., UniProt) [23] | RNA-Seq alignments and/or protein homology [24] [25] |
| Typical Inputs | Assembled genome (FASTA) [23] | Assembled genome (FASTA), plus RNA-Seq (BAM) and/or protein sequences (FASTA) [24] [25] |
| Key Strength | Speed, standardization of output for prokaryotes [23] | High accuracy by leveraging extrinsic evidence and combining multiple gene finders [24] |
| Considerations | Less suitable for genomes with atypical features without manual curation [26] | Computationally intensive; requires evidence data for optimal performance [24] |
Rationale for Domain-Specific Tool Selection
The divergence in tool design is driven by fundamental biological differences. Prokaryotic genes are relatively simple, lacking introns and being densely packed on the genome. Prokka leverages this by using fast, ab initio predictors like Prodigal and aligning sequences to protein databases for functional annotation [23]. In contrast, eukaryotic genes contain introns, making their prediction more complex. BRAKER3 addresses this by employing a sophisticated pipeline that first trains GeneMark-ETP and AUGUSTUS using extrinsic evidence from RNA-Seq or protein homologs. This evidence is crucial for accurately identifying exon-intron boundaries [24] [25]. Using Prokka on a eukaryotic genome would fail to predict spliced genes, while using BRAKER3 on a prokaryote would be unnecessarily complex and resource-intensive.
Experimental Protocols for Long-Read Assembled Genomes
The following protocols assume you have a high-quality, long-read genome assembly. Using a repeat-masked genome is highly recommended, especially for eukaryotes, as it prevents the prediction of false positive gene structures in repetitive regions [24].
Protocol 1: Rapid Prokaryotic Genome Annotation with Prokka
This protocol is designed for annotating a bacterial or archaeal genome assembly using Prokka.
Materials and Reagents
- Input Data: A high-quality prokaryotic genome assembly in FASTA format.
- Computational Environment: A Unix-based system (Linux/macOS) with Prokka installed. Installation can be performed via Conda:
conda install -c conda-forge -c bioconda -c defaults prokka[23]. - Optional: A custom protein database (in FASTA format) for improved homology-based annotation, if annotating a non-model organism.
Step-by-Step Procedure
- Database Setup (First-time use): Run
prokka --setupdbto index the default databases [23]. - Basic Annotation Command: Execute Prokka with a minimal command.
This will create a directory
my_genome_annotationcontaining all output files with the prefixmy_bacterium[23]. - Advanced Annotation (Recommended): For more accurate and compliant annotation, specify the genus and enable the
--addgenesflag. For public submission, use--compliant. - Output Analysis: Key output files include:
.gff: The master annotation file in GFF3 format..gbk: A standard GenBank format file..faa: Protein FASTA file of the translated CDS sequences..txt: Summary statistics of the annotated features [23].
Protocol 2: Evidence-Driven Eukaryotic Genome Annotation with BRAKER3
This protocol describes annotating a eukaryotic genome using BRAKER3 with RNA-Seq and protein evidence.
Materials and Reagents
- Input Data:
genome.fasta: The repeat-masked eukaryotic genome assembly.rnaseq.bam: RNA-Seq reads aligned to the genome using a splice-aware aligner (e.g., HISAT2, STAR with--outSAMstrandField intronMotifoption) [25].protein_db.fasta: A database of protein sequences from a related species (e.g., a subset of UniProt/SwissProt) [24] [25].
- Computational Environment: BRAKER3 is available as a command-line tool and on public platforms like Galaxy Europe (
usegalaxy.eu) [24] [25].
Step-by-Step Procedure
- Data Preparation: Ensure the BAM file is sorted and indexed. The genome file should have simple scaffold names (e.g.,
>scaffold_1) [24]. - Run BRAKER3 with Combined Evidence: Execute the pipeline using both RNA-Seq and protein evidence. This is the most robust mode for BRAKER3.
--species: A unique name for the training species.--prg=exonerate: Specifies the tool for protein-to-genome alignment.--gff3: Produces output in GFF3 format [24].
- Run BRAKER3 with Proteins Only: If no RNA-Seq data is available, run using protein evidence alone. This requires a database of protein families for optimal performance (e.g., OrthoDB) [24].
- Output Analysis: The primary output is
braker/annotations.gff3. This file contains the final gene predictions combining results from both GeneMark-ETP and AUGUSTUS, filtered for high support from extrinsic evidence [24] [25]. Visual inspection of the results in a genome browser like UCSC or JBrowse is highly recommended [24].
The logical relationship and data flow for the BRAKER3 protocol with combined evidence is illustrated below.
BRAKER3 Evidence Integration Flow
The Scientist's Toolkit: Research Reagents and Materials
The table below lists essential materials and their functions for the gene prediction workflows described.
Table 2: Essential Research Reagents and Computational Tools
| Item Name | Function / Application Notes |
|---|---|
| High-Quality Genomic DNA | Source material for long-read sequencing. For challenging samples like plants, a sorbitol wash may be required to remove polysaccharides before extraction [27]. |
| Long-read Sequencer (Nanopore/PacBio) | Generates long sequencing reads, enabling high-contiguity genome assemblies that are crucial for accurate annotation [9] [8]. |
| Prokka Software Suite | Integrated annotation tool for prokaryotes. Rapidly produces standard-compliant GFF and GenBank files [23]. |
| BRAKER3 Pipeline | Eukaryotic gene prediction pipeline. Uses RNA-Seq and/or protein evidence to train and run GeneMark-ETP and AUGUSTUS [24]. |
| RNA-Seq Data (Paired-end) | Provides direct evidence of transcribed regions and splice sites for training and guiding eukaryotic gene prediction in BRAKER3 [24] [25]. |
| Curated Protein Database (e.g., UniProt/SwissProt) | Provides protein homology evidence for both Prokka and BRAKER3. Using high-quality, curated sequences is critical for accuracy [24] [25]. |
| High-Performance Computing (HPC) Infrastructure | Essential for managing computationally demanding tasks, especially BRAKER3 and long-read genome assembly [9] [10]. |
The accurate annotation of microbial genomes is a critical step in translating sequence data into biological discovery. The choice between Prokka for prokaryotes and BRAKER3 for eukaryotes is dictated by the fundamental biology of the organism under study. Prokka offers a fast, efficient, and standardized solution for bacterial and archaeal genomes. In contrast, BRAKER3 provides a powerful, evidence-driven approach capable of handling the complexity of eukaryotic gene structures. By following the detailed protocols and leveraging the integrated toolkit outlined in this guide, researchers can confidently apply these tools to their long-read assembled genomes, ensuring high-quality annotations that serve as a reliable foundation for downstream functional and comparative genomic studies within a thesis or broader research project.
Functional annotation is a critical step following gene prediction in microbial genomics, transforming raw nucleotide sequences into biologically meaningful information. For research on long-read assembled microbial genomes, this process reveals the putative roles of predicted genes within metabolic pathways, cellular components, and biological processes, thereby enabling hypothesis generation about the organism's ecological role or biotechnological potential [9]. InterProScan stands as a cornerstone tool in this domain, providing a unified interface to multiple protein signature databases through a single analysis [28].
This protocol details the application of InterProScan for the comprehensive functional annotation of protein sequences derived from microbial genomes. By integrating results from databases such as Pfam, PANTHER, and Gene Ontology (GO), InterProScan facilitates the transfer of functional knowledge from well-characterized proteins to novel sequences identified in genomic studies [29] [28]. The following sections provide a structured workflow, from data preparation to advanced analysis, tailored for researchers annotating microbial genomes.
Experimental Protocols
Installation and Setup
Local Installation on a Computing Cluster For large-scale projects, such as annotating an entire microbial genome, a local installation of InterProScan is recommended for performance and flexibility [29].
- Download and Install: Follow the official installation instructions from the EMBL-EBI website. Ensure your system meets the requirements, notably an updated version of Java (above 1.7) and GCC libraries [29].
- Add Proprietary Algorithms: InterProScan does not include all algorithms by default. To add Phobius, SignalP, and TMHMM, request the compiled binaries and place them in the respective subfolders within the
bindirectory of your InterProScan installation [29]. - Configure Environment Variables: For tools like SignalP, you must set environment variables to point to the installation directory. Add a line similar to the following to your configuration to ensure proper library loading:
- Verify Installation: Run
./interproscan.shwithout arguments. The tool will list all successfully loaded analysis algorithms. Check this list to ensure no critical algorithms failed to load [29].
Using the Web-Based REST Service For smaller datasets or users without access to a high-performance computing cluster, the InterProScan REST service provides a accessible alternative, though it is limited to 30 sequences per job [29] [30].
- Access Method: The REST service can be accessed programmatically using provided client libraries for Python 2 or 3. The service is also integrated into user-friendly platforms like the Geneious Prime bioinformatics software via a dedicated plugin [31].
Input Data Preparation
The standard input for InterProScan is a FASTA file containing protein sequences. These sequences are typically the output from a structural annotation tool (e.g., Prodigal for prokaryotes or BRAKER3 for eukaryotes) applied to your microbial genome assembly [9].
Batch Processing for Large Datasets When dealing with thousands of sequences from a microbial genome, it is efficient to split the main FASTA file into smaller batches for parallel processing [29].
- Simple Split: If your FASTA file is formatted with exactly two lines per entry (header and sequence), use the
splitcommand: This creates multiple files, each containing 500 sequences (1000 lines) [29]. - BioPython Parsing: For typically formatted FASTA files (sequences wrapped at 60-80 characters), a more robust method uses BioPython to create a "filtered" file with one line per sequence, which can then be split [29].
Execution of InterProScan
Basic Command-Line Usage The core command for a local InterProScan run is:
Table 1: Key InterProScan Command-Line Parameters
| Parameter | Function |
|---|---|
-i, --input |
Specifies the input protein FASTA file. |
-f, --formats |
Defines output formats (e.g., TSV, XML, GFF3). |
--goterms |
Enables retrieval of Gene Ontology (GO) terms. |
--iprlookup |
Includes InterPro metadata in the output. |
--pathways |
Annotates proteins with metabolic pathway information. |
-dp, --disable-precalc |
Turns off pre-calculated match lookup, useful for novel sequences not in public databases [29]. |
Submission to a Computing Cluster For a high-performance computing (HPC) environment using a job scheduler like SLURM, you would create a job submission script. The following is a template that can be adapted [29].
Performance and Benchmarking
Performance varies based on sequence length, number of sequences, and available computing resources. As a reference point, an analysis of 49,598 Lotus japonicus protein sequences was completed by processing batches of 1,000 sequences on nodes equipped with 12 cores and 24 GB of RAM. The average real time per job was 2.50 ± 0.28 hours [29]. This benchmark can help in planning computational resources for microbial genome annotation projects.
Data Interpretation and Analysis
Understanding the Output
InterProScan generates output in multiple formats. The Tab-Separated Values (TSV) file is particularly useful for downstream analysis and database import. Each row represents a single domain or motif found in a protein.
Table 2: Structure of the InterProScan TSV Output File
| Column | Content | Significance |
|---|---|---|
| 1 | Protein Accession | Identifier of the query protein sequence. |
| 3 | Signature Accession | Identifier of the matched signature (e.g., a Pfam ID). |
| 4 | Signature Database | Source database of the match (e.g., Pfam, PANTHER, SMART). |
| 5 | Signature Description | Name of the protein family, domain, or site. |
| 6 | Start - End | Positional coordinates of the match within the protein. |
| 7 | E-value | Statistical significance of the match. |
| 8 | Status | Annotation status. |
| 12 | InterPro Accession | Cross-referenced InterPro identifier (e.g., IPR036859). |
| 13 | InterPro Description | Functional description from the InterPro database. |
| 14 | GO Annotations | Associated Gene Ontology terms [28]. |
Integration with Downstream Analyses
The functional annotations obtained from InterProScan can be directly used for several advanced biological interpretations:
- Functional Enrichment Analysis: GO terms from InterProScan can be used to test for statistically significant enrichment of specific functions within a set of proteins (e.g., differentially expressed proteins) using a Fisher's exact test [32].
- Metabolic Pathway Reconstruction: Identifiers such as KEGG Orthology (KO) terms, often retrieved via tools like EggNOG-mapper, can be mapped to metabolic pathways to infer the metabolic capabilities of a microorganism [33] [28]. The completeness of specific KEGG modules can be calculated to assess the presence of entire metabolic pathways [33].
- Database Integration: The TSV output can be easily imported into a relational database (e.g., MySQL) for integration with other transcriptomic or genomic metadata, facilitating complex queries and data management [29].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for Functional Annotation
| Item | Function in the Protocol |
|---|---|
| InterProScan | Core analysis tool that scans protein sequences against multiple databases to identify domains, families, and functional sites [29] [28]. |
| High-Performance Computing (HPC) Cluster | Essential computational resource for processing large microbial genome datasets in a reasonable time frame [29] [9]. |
| EggNOG-mapper | Complementary tool for functional annotation that provides KEGG and COG annotations, often used alongside InterProScan for comprehensive coverage [28]. |
| Geneious Prime | Commercial graphical software with an InterProScan plugin, suitable for users preferring a point-and-click interface over the command line [31]. |
| BioPython | A suite of Python tools for computational molecular biology, useful for parsing and manipulating FASTA files during data preparation [29]. |
| SLURM Workload Manager | Job scheduler for managing and submitting batch jobs to a computing cluster [29]. |
| MySQL Database | Relational database system for storing, managing, and querying large-scale annotation results [29]. |
Workflow Visualization
The following diagram summarizes the complete computational protocol for functional annotation using InterProScan within the context of a long-read microbial genome analysis project.
Figure 1. Functional Annotation Workflow
This protocol outlines a robust and scalable approach for the functional annotation of protein sequences using InterProScan, specifically framed within long-read microbial genome research. By following the detailed methods for installation, data preparation, execution, and data interpretation, researchers can systematically assign biological meaning to gene predictions. The integration of InterProScan outputs with downstream analytical frameworks, such as enrichment analysis and metabolic pathway mapping, provides a powerful foundation for generating biologically significant insights into the functions of microbial genes and genomes.
The advent of high-throughput sequencing has led to an exponential growth in microbial genome data, necessitating computational pipelines that are not only comprehensive but also scalable for large-scale genomic surveillance and research [34]. The challenge is particularly acute in the context of long-read assembled microbial genomes, where the goal is to move from raw sequence data to biologically meaningful insights, such as gene prediction and annotation, in a efficient and reproducible manner [9] [10]. While several pipelines exist, many require high-end computational infrastructure and do not scale efficiently for collections numbering thousands of genomes, making analysis prohibitively time-consuming [34] [35].
AMRomics addresses these challenges as an optimized, open-source microbial genomics pipeline, explicitly designed for speed and scalability when working with large datasets [34] [36]. Its capability to handle diverse data types, including long-read sequencing technologies, and its innovative approach to pangenome analysis and variant calling, make it a powerful tool for research centered on gene prediction in microbial genomes. This protocol details the application of AMRomics for the comprehensive analysis of large genome collections within a research framework focused on long-read assembled genomes.
The AMRomics pipeline is structurally divided into two consecutive stages: (1) the single-sample analysis stage, where each genome in a collection is processed individually, and (2) the collection analysis stage, where a comparative pangenomic analysis is performed across all samples [34]. This modular design allows for the efficient processing of new samples by leveraging existing analysis results, a key feature for managing growing genome collections. The workflow integrates state-of-the-art tools for each analytical step, ensuring best practices while maintaining flexibility for user-specific preferences [34] [35].
The following diagram illustrates the complete workflow, from raw data input to the final comparative analysis and visualization.
Detailed Experimental Protocol
Single-Sample Analysis Stage
The initial stage processes each microbial genome individually, standardizing the data and generating foundational genomic information.
Procedure:
Input Data Preparation:
- Create a tab-separated values (TSV) file listing all samples in the collection, one sample per line. Include metadata such as phenotypic information if available [35].
- Input for each sample can be raw sequencing reads (FASTQ format), pre-assembled genomes (FASTA format), or existing annotations (GFF3 format). AMRomics supports Illumina, PacBio, and Oxford Nanopore sequencing technologies [34] [35].
Quality Control and Assembly:
- For Illumina Short Reads: Quality control, adaptor trimming, and read pruning are performed using
fastp. The pre-processed reads are then assembled into a genome assembly usingSKESA(default for speed) orSPAdes(optional for improved assembly quality) [34]. - For Long Reads (PacBio/Oxford Nanopore): The genome is assembled directly using
Flye[34] [35]. The assembly step is skipped if the user provides a pre-assembled genome in FASTA format.
- For Illumina Short Reads: Quality control, adaptor trimming, and read pruning are performed using
Genome Annotation and Specialized Scanning:
- The genome assembly is annotated using
Prokkato identify protein-coding genes, tRNA sequences, and their functions. Contig names and sample IDs are standardized to ensure uniqueness within the collection [34]. - The annotated assembly is subjected to a series of analyses using BLAST against specific databases:
- Multi-locus Sequence Typing (MLST): Performed against the
pubMLSTdatabase for bacterial strain typing [34] [35]. - Antibiotic Resistance Gene Identification: Conducted using
AMRFinderPlusdatabase [34]. - Virulence Gene Identification: Screened against the Virulence Factor Database (
VFDB) [34]. - Plasmid Detection: Analyzed with the
plasmidfinderdatabase [34].
- Multi-locus Sequence Typing (MLST): Performed against the
- All results from the single-sample analysis are organized into standardized file structures and locations for the subsequent collection analysis.
- The genome assembly is annotated using
Collection Analysis Stage
This stage performs a comparative analysis across the entire genome collection, focusing on pangenome dynamics and phylogeny.
Procedure:
Pangenome Construction:
- The annotations (GFF format) of all genomes are loaded into a pangenome inference module. The primary tool for this is
PanTA, chosen for its speed and scalability with large collections. Users can optionally selectRoaryas an alternative [34]. - Gene clusters are classified into:
- Core genes: Present in at least 95% of the genomes (default, user-adjustable).
- Shell genes: Present in a certain percentage of genomes (default 25%, user-adjustable).
- Accessory genes: Present in a smaller subset of genomes [34].
- The annotations (GFF format) of all genomes are loaded into a pangenome inference module. The primary tool for this is
Multiple Sequence Alignment and Phylogenetics:
- For each identified gene cluster, protein sequences are aligned using
MAFFTto create a multiple sequence alignment (MSA). The nucleotide MSA is then inferred from this protein alignment [35]. - Phylogenetic trees for individual gene families are constructed from these MSAs using either
FastTree 2orIQ-TREE 2[34]. - A core-genome phylogeny for the entire collection is built from a concatenated MSA of all core genes using the selected tree-building method [34] [35].
- For each identified gene cluster, protein sequences are aligned using
Pan-SNPs and Variant Analysis:
- AMRomics introduces a reference-free variant calling method known as "pan-SNPs." A pan-reference genome is built, comprising the representative gene from each gene cluster (by default, the gene from the first genome in the collection list) [34].
- Variants for all genes in a cluster are identified directly from the MSA against the representative gene. The complete variant profile for each sample is reported in a VCF file, providing a comprehensive view of genetic variation across the entire pangenome rather than just against a single reference genome [34] [35].
Visualization and Data Management with AMRViz
The AMRViz toolkit provides a web-based platform for interactive visualization and management of the analysis results generated by the AMRomics pipeline [35] [37].
Procedure:
Platform Initialization:
Visualization and Interpretation:
- Single-Sample View: For individual samples, inspect assembly statistics, genome structure via a circular Circos plot or a linear genome browser, and detailed tables of resistance and virulence genes. The genome browser can be zoomed to the location of a selected gene for visual inspection of its genomic context [35].
- Collection View: For the entire collection, explore the pangenome statistics (core/accessory gene pie charts), a searchable table of all gene families, and the core-genome phylogeny. The phylogeny can be colored based on sample metadata. Selecting a gene family displays its multiple sequence alignment and gene tree, allowing for comparison with the core-genome phylogeny [35].
- Heatmap Analysis: Generate a heatmap showing the presence/absence of resistance and virulence genes across the collection, which can be visually correlated with phenotypic metadata to identify potential associations [35].
Key Research Reagents and Computational Tools
The following table catalogues the essential software tools and databases integrated into the AMRomics pipeline, which constitute the key "research reagents" for conducting a scalable genomic analysis.
Table 1: Essential Research Reagents and Computational Tools in AMRomics
| Tool/Database Name | Category/Function | Specific Role in the Workflow |
|---|---|---|
| SKESA/SPAdes [34] [35] | Genome Assembly | De novo assembly of Illumina short-read sequencing data. |
| Flye [34] [35] | Genome Assembly | De novo assembly of long-read data from PacBio or Oxford Nanopore technologies. |
| Prokka [34] [35] | Genome Annotation | Rapid annotation of microbial genomes, identifying protein-coding genes and other features. |
| pubMLST [34] [35] | Strain Typing | Database for Multi-locus Sequence Typing (MLST) to classify bacterial strains. |
| AMRFinderPlus [34] | Gene Identification | Database and tool for identifying antibiotic resistance genes. |
| VFDB [34] | Gene Identification | Virulence Factor Database for identifying bacterial virulence genes. |
| PlasmidFinder [34] | Gene Identification | Database for identifying plasmid replicons. |
| PanTA [34] | Pangenome Construction | Primary tool for fast and scalable pangenome clustering of gene families. |
| Roary [34] [35] | Pangenome Construction | Alternative tool for pangenome analysis. |
| MAFFT [34] [35] | Sequence Alignment | Generating multiple sequence alignments of protein and nucleotide sequences for gene clusters. |
| FastTree 2 / IQ-TREE 2 [34] [35] | Phylogenetics | Inference of phylogenetic trees from multiple sequence alignments. |
Performance and Comparative Analysis
AMRomics is designed for high performance on large datasets. Benchmarking tests demonstrate that it can generate results comparable to other established pipelines like Nullarbor, Bactopia, and ASA3P, but with significantly improved speed and lower computational resource requirements, making the analysis of thousands of genomes feasible on regular desktop computers [34].
A critical differentiator is its handling of genetic variants. Unlike traditional methods that rely on a single reference genome (limiting analysis to genes present in that reference), AMRomics' pan-SNPs approach calls variants against the entire pangenome reference. This provides a more comprehensive view of genetic diversity, especially valuable for diverse collections where no single genome is representative [34]. The following table summarizes a comparison of key features.
Table 2: Feature Comparison of AMRomics with Other Microbial Genomics Pipelines
| Feature | AMRomics | Nullarbor [34] | Bactopia [34] | ASA3P [34] |
|---|---|---|---|---|
| Input Formats | Reads (Illumina, PacBio, Nanopore), Assembly, Annotations [34] | Illumina paired-end reads only [34] | Reads (Illumina, PacBio, Nanopore) [34] | Reads (Illumina, PacBio, Nanopore) [34] |
| Assembly Support | SKESA/SPAdes (Illumina), Flye (Long reads) [34] | Specific for short reads | Various | Various |
| Variant Analysis | Pan-SNPs (reference-free, against pangenome) [34] | SNP alignment against a user-provided reference [34] | SNP alignment against a user-provided reference [34] | SNP alignment against a user-provided reference [34] |
| Phylogeny | Core-genome alignment [34] | SNP-based or 16S-based [34] | SNP-based or 16S-based [34] | SNP-based or 16S-based [34] |
| Scalability | High (optimized for large collections) [34] | Limited with large collections [34] | Moderate [34] | Moderate [34] |
| Progressive Analysis | Yes (new samples can be added without re-analyzing the entire collection) [34] | No | No | No |
| Integrated Visualization | Yes (via AMRViz) [35] [37] | Limited | Limited | Limited |
Overcoming the Hurdles: Optimization Strategies for Complex Microbial Genomes
Within the context of gene prediction in long-read assembled microbial genomes, the assumption of a universal and static genetic code presents a significant risk of generating spurious predictions. The genetic code, once thought to be immutable, is now known to exhibit substantial flexibility in microorganisms, including variations such as codon reassignment, ambiguous decoding, and natural genetic code expansion [38]. These deviations from the standard code can lead to systematic errors in automated gene annotation pipelines, causing mis-annotation of start/stop sites and incorrect amino acid assignments, which ultimately compromises downstream functional analyses and metabolic models. This application note provides a structured framework, including quantitative measures and detailed protocols, to identify and mitigate these errors, ensuring more accurate genomic interpretation for research and drug development.
The challenge of genetic code diversity is compounded by the technical complexities of long-read genome assembly and annotation. The tables below summarize core aspects of this challenge and the quantitative measures available to manage it.
Table 1: Common Genetic Code Variations in Microbes and Their Impact on Gene Prediction
| Variation Type | Description | Example in Microorganisms | Potential for Spurious Prediction |
|---|---|---|---|
| Stop Codon Reassignment | A stop codon is redefined to encode an amino acid. | UGA encoding tryptophan in yeast mitochondria [38] | High; premature truncation of predicted proteins. |
| Sense Codon Reassignment | A sense codon is reassigned to a different amino acid. | CUN codons assigned to threonine instead of leucine [38] | High; incorrect amino acid sequence in predicted proteins. |
| Natural Genetic Code Expansion | Incorporation of non-canonical amino acids via recoding. | UAG codon reassigned to pyrrolysine (Pyl) in some archaea and bacteria [38] | Medium; can be mis-annotated as a stop codon. |
| Ambiguous Decoding | The same codon is decoded by more than one amino acid. | Misacylation of tRNAs under stress conditions [38] | Medium; leads to heterogeneous protein sequences. |
Table 2: Quantitative Measures for Annotation Management and Comparison
| Metric Name | Calculation/Definition | Interpretation | Utility in Managing Code Diversity |
|---|---|---|---|
| Annotation Edit Distance (AED) | A value between 0 and 1 quantifying the discrepancy between a gene prediction and a reference annotation based on exon-intron structure [39]. | AED=0: perfect match; AED=1: no overlap. Identifies structurally problematic annotations for manual review. | Highlights gene models that may be erroneous due to atypical coding rules. |
| Annotation Turnover | Tracks the addition and deletion of gene annotations between successive releases of a genome annotation [39]. | High turnover can indicate instability and previous spurious predictions. | Flags genomic regions where annotation is inconsistent, potentially due to code ambiguity. |
| Splice Complexity | Quantifies the complexity of alternative splicing patterns for a gene [39]. | Higher complexity indicates more transcript isoforms. | Less relevant for most microbes but critical for eukaryotic microbial annotations. |
| BUSCO Score | Assesses the completeness of a genome assembly and annotation based on the presence of universal single-copy orthologs [40]. | Reported as % of complete, fragmented, or missing orthologs. | A low score can indicate widespread gene prediction errors, potentially from unaccounted code variations. |
Experimental Protocols for Validation
Protocol 1: De Novo Genome Annotation with MAKER2
This protocol outlines a standardized pipeline for annotating a de novo microbial genome assembly, incorporating steps that enhance the detection of non-standard genetic codes [40].
Masking Repetitive Elements
- Construct species-specific repetitive elements using RepeatModeler.
- Mask the genome assembly using RepeatMasker with the constructed species-specific repeats and the RepBase library.
Training Ab Initio Gene Predictors
- Train AUGUSTUS using BUSCO to create a species-specific gene prediction model.
The
--longparameter enables optimization mode for non-model organisms [40]. - Train SNAP. Run the MAKER pipeline initially with expressed sequence tag (EST) or protein evidence to generate a set of high-quality gene models. Use these models to train SNAP, iterating this process three times for optimal performance [40].
Executing the MAKER2 Pipeline
- Generate control files:
maker -CTL - Edit
maker_opts.ctlto specify the following key parameters: - Execute the pipeline:
mpirun -n <number_of_cores> maker
Protocol 2: Computational Identification of Gene Expansion
Gene expansions can be a source of annotation error and may co-occur with genetic code variations. This protocol details their identification and validation [40].
Identifying Gene Family Expansion
- Run CAFE5. Use the installed CAFE5 software to analyze gene family phylogeny and identify significantly expanded gene families across a given species tree.
Validating Expanded Genes
- Validate with GeneWise. Compare the annotated gene models against curated protein sequences from databases like UniProtKB/Swiss-Prot to verify the integrity of the gene structure.
- Experimental Validation with RNA-seq. Use Kallisto to quantify transcript abundance across different tissues or conditions, providing evidence for the expression of expanded gene copies.
Visualization of Workflows
The following diagram illustrates the integrated workflow for long-read genome assembly and robust gene annotation, highlighting key steps to prevent spurious predictions.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Genomic Analysis
| Tool / Resource | Category | Function in Analysis | Relevance to Preventing Spurious Predictions |
|---|---|---|---|
| Flye / Canu | Genome Assembler | Assembles long-read sequencing data into contiguous sequences (contigs) [9] [10]. | Produces high-quality assemblies that are crucial for accurate prediction of full-length genes. |
| BRAKER3 | Gene Predictor | Predicts gene structures in eukaryotic genomes using RNA-seq and protein data [9] [10]. | Leverages multiple evidence sources to improve prediction accuracy where genetic code may be atypical. |
| Prokka | Annotation Pipeline | Rapidly annotates prokaryotic genomes [9] [10]. | A standard tool whose default parameters may need adjustment for organisms with genetic code variations. |
| MAKER2 | Annotation Pipeline | Integrates evidence from ESTs, proteins, and ab initio predictors into a unified annotation [40]. | Its evidence-driven approach helps flag and correct spurious ab initio predictions. |
| RepeatMasker | Repeat Analysis | Identifies and masks repetitive elements in the genome [40]. | Prevents spurious gene predictions in repetitive regions, a common source of error. |
| BUSCO | Quality Assessment | Benchmarks genome and annotation completeness using universal single-copy orthologs [40]. | A low BUSCO score can indicate systemic gene prediction failures, prompting investigation into causes like code diversity. |
| CAFE5 | Evolutionary Analysis | Analyzes gene family evolution and identifies significant expansions/contractions [40]. | Identifies gene expansions that require careful annotation to distinguish between true copies and artifacts. |
| Apollo | Annotation Viewer | Web-based tool for manual collaborative curation of genomic annotations [40]. | Essential for expert review and correction of automated annotations flagged by metrics like AED. |
Within the field of microbial genomics, the advent of long-read sequencing technologies has revolutionized our ability to reconstruct complex microbial genomes from environmental samples [8] [12]. However, transforming these long-read data into accurate gene predictions and functional annotations presents substantial computational challenges. The enormous data volumes, often reaching 100 gigabytes per individual genome, and the complexity of bioinformatics workflows demand a sophisticated approach to computational resource management [41]. High-Performance Computing (HPC) and cloud infrastructure have emerged as critical enablers for researchers conducting gene prediction on long-read assembled microbial genomes, allowing them to overcome limitations of traditional computing environments and accelerate scientific discovery [10] [42].
This application note provides detailed protocols for leveraging HPC and cloud infrastructure within the specific context of microbial genome research. We frame these resources within a comprehensive gene prediction workflow, from initial long-read data processing to final functional annotation, with specific guidance on computational strategies that enhance efficiency, reduce costs, and maximize the biological insights derived from complex microbial datasets.
HPC and Cloud Infrastructure Fundamentals for Genomic Analysis
High-Performance Computing (HPC) refers to the practice of aggregating computing resources to achieve performance greater than that of a single workstation or server [42]. In genomic analysis, HPC typically takes the form of either on-premises computer clusters or cloud-based resources from providers such as AWS, Google Cloud, and Microsoft Azure. These systems are characterized by three main components: compute (processors), network (interconnects), and storage (data systems) [42].
For microbial genomics applications, understanding the distinction between tightly coupled and loosely coupled workloads is essential for appropriate resource allocation [42]. Tightly coupled workloads, such as genome assembly, require frequent communication between computing nodes and benefit from low-latency networking. Loosely coupled workloads, including many variant calling and gene prediction tasks, can be executed in parallel across multiple nodes with minimal inter-process communication, making them ideal for distributed cloud computing environments [42].
Cloud HPC offers particular advantages for microbial genomics research through its flexibility and scalability. Researchers can access specialized resources on-demand without substantial capital investment in physical infrastructure [41]. This elasticity allows computational capacity to align with project requirements, scaling resources up during intensive processing phases and down during analysis or interpretation phases. A properly designed cloud implementation can reduce processing times dramatically, as demonstrated by Theragen Bio's migration to AWS, which reduced standard data processing time from 40 hours to just 4 hours – a tenfold performance improvement [41].
Application Notes: HPC Strategies for Gene Prediction from Long-Read Microbial Genomes
Integrated Bioinformatics Workflow for Long-Read Data
Gene prediction from long-read assembled microbial genomes involves a multi-stage process with distinct computational requirements at each step. The following workflow integrates best practices for long-read analysis with appropriate HPC resource management strategies.
Workflow: From Sequencing to Gene Prediction
Figure 1: Computational workflow for gene prediction from long-read microbial genomic data, with HPC resource recommendations for each stage.
Computational Resource Requirements by Workflow Stage
Table 1: Computational profiles and resource recommendations for key workflow stages
| Workflow Stage | Computational Profile | Recommended HPC Resources | Tools & Technologies |
|---|---|---|---|
| Basecalling | GPU-accelerated, high memory | GPU nodes (NVIDIA T4/V100/A40); 32+ GB RAM | Dorado (ONT), CCS (PacBio) [12] |
| Genome Assembly | Tightly coupled, high CPU & memory | High-memory CPU nodes; 128+ GB RAM; low-latency networking | Canu, Flye, wtdbg2 [10] [8] |
| Gene Prediction | Loosely coupled, moderate CPU | Standard CPU nodes; 64-128 GB RAM | BRAKER3 (eukaryotes), Prokka (prokaryotes) [10] |
| Functional Annotation | Loosely coupled, high I/O | Standard CPU nodes; parallel file systems | InterProScan, custom databases [10] |
Implementation Protocol: Large-Scale Microbial Genome Analysis
Objective: Process 150-200 complex terrestrial metagenomic samples through complete assembly and gene prediction pipeline.
Experimental Background: Recent research demonstrates that long-read sequencing enables recovery of high-quality microbial genomes from highly complex ecosystems, which remain an untapped source of biodiversity [8]. The mmlong2 workflow, specifically designed for complex metagenomic datasets, incorporates differential coverage binning, ensemble binning, and iterative binning to maximize MAG (metagenome-assembled genome) recovery from terrestrial samples [8].
Computational Protocols:
Workflow Orchestration:
- Implement the analytical workflow using Common Workflow Language (CWL) or Nextflow for enhanced reproducibility and portability between HPC and cloud environments [10].
- Containerize tools using Docker or Singularity to ensure consistent execution environments across distributed systems.
Resource Management Strategy:
- Utilize workload managers (Slurm, PBS) for efficient job scheduling and resource allocation [43].
- Implement dynamic resource allocation policies based on job requirements:
- Assembly jobs: Request 36-80 cores, 128GB-1TB RAM, and high-speed interconnects.
- Gene prediction jobs: Request 16-32 cores and 64-128GB RAM.
- Functional annotation: Request 8-16 cores and 64GB RAM.
Data Management Plan:
- Utilize parallel file systems (Lustre, BeeGFS) for intermediate data storage during assembly and analysis [10].
- Implement a tiered storage strategy:
HPC Performance Optimization and Cost Management Strategies
Key Performance Indicators for Genomic HPC Workloads
Table 2: Key Performance Indicators for monitoring and optimizing HPC resources in genomic analysis
| KPI Category | Specific Metric | Target Range | Optimization Strategies |
|---|---|---|---|
| Compute Efficiency | CPU Utilization | >85% during active processing | Implement job arrays for parallel samples; Right-size resource requests |
| Storage Performance | I/O Throughput | >5 GB/s for assembly steps | Use parallel file systems; Implement data staging |
| Cost Management | Cost per Sample | 30-50% below on-demand rates | Use spot instances; Auto-scaling policies |
| Workflow Efficiency | Job Queue Time | <2 hours for standard priority | Implement fair-share scheduling; Pre-emption policies |
Cloud Cost Optimization Protocol
Objective: Execute large-scale gene prediction workflow while minimizing cloud computing costs.
Procedure:
- Instance Selection: Use compute-optimized instances (C-series) for assembly steps and memory-optimized instances (M-series) for gene prediction.
- Purchasing Model: Implement hybrid instance strategy:
- Spot instances for fault-tolerant workloads (75% of capacity)
- On-demand instances for critical path workloads (25% of capacity)
- Storage Optimization:
- Apply lifecycle policies to automatically transition raw data to cold storage (e.g., Amazon S3 Glacier) after 30 days.
- Use distributed caching for frequently accessed reference databases.
- Monitoring and Adjustment:
- Implement budget alerts at 50%, 75%, and 90% of allocated budget.
- Use cloud cost management tools (AWS Cost Explorer, Azure Cost Management) to identify underutilized resources [43].
Expected Outcomes: Proper implementation can reduce cloud computing costs per run by 50-60% while maintaining performance standards [41].
The Scientist's Toolkit: Essential Research Reagents & Computational Solutions
Table 3: Essential research reagents and computational solutions for HPC-enabled microbial gene prediction
| Category | Item | Specification/Function | Implementation Notes |
|---|---|---|---|
| Sequencing Technologies | Oxford Nanopore | Long-read sequencing; enables access to repetitive regions | Requires GPU resources for basecalling [12] |
| PacBio HiFi | Highly accurate long reads; circular consensus sequencing | Lower computational requirement for basecalling [12] | |
| Bioinformatics Tools | Flye, Canu | Long-read assemblers | High memory requirement (>128GB for complex metagenomes) [10] [8] |
| BRAKER3 | Eukaryotic gene prediction | Combines RNA-seq and protein evidence [10] | |
| Prokka | Prokaryotic gene prediction | Rapid annotation of bacterial genomes [10] | |
| InterProScan | Functional annotation of predicted genes | Parallelize across multiple samples [10] | |
| Computational Infrastructure | HPC Cluster | On-premises computing resource | Typical configuration: 36-80 cores/node, 128GB-1TB RAM [10] |
| Cloud HPC (AWS, GCP, Azure) | Scalable, on-demand computing | Use spot instances for cost-sensitive projects [41] [42] | |
| Workflow Management | CWL, Nextflow | Workflow reproducibility and portability | Essential for multi-step genomic analyses [10] |
Effective computational resource management is fundamental to advancing research in gene prediction from long-read assembled microbial genomes. The integration of HPC systems - whether on-premises, cloud-based, or in a hybrid configuration - provides the necessary foundation to handle the massive computational demands of long-read data analysis. By implementing the protocols and strategies outlined in this application note, researchers can significantly accelerate their genomic analyses while optimizing costs and maximizing the scientific return from their microbial genomics projects. As long-read technologies continue to evolve and decrease in cost, these computational approaches will become increasingly vital for unlocking the functional potential hidden within microbial genomes.
Eukaryotic genomes are characterized by a remarkable complexity that presents significant challenges for accurate gene prediction, especially within the context of modern long-read sequencing technologies. Unlike the relatively compact and gene-dense genomes of prokaryotes, eukaryotic genomes contain vast amounts of non-coding DNA, including introns, repetitive elements, and spacer sequences that can comprise the majority of the genomic material [44]. This biological complexity is compounded by technical limitations in sequencing and computational prediction methods, often resulting in error-prone genome annotations that can impact downstream biological interpretations [45].
The advent of long-read sequencing technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) has revolutionized genomic studies by producing reads that are thousands to tens of thousands of bases long [12] [46]. These technologies enable researchers to span repetitive regions and complete complex gene structures in a single read, offering unprecedented potential for resolving eukaryotic gene models. However, fully leveraging this potential requires understanding both the architectural challenges of eukaryotic genes and developing specialized bioinformatic approaches to address them [9] [10].
This application note examines the fundamental challenges in eukaryotic gene prediction, quantitative assessments of current error rates, and provides detailed protocols for generating accurate gene models from long-read sequencing data, with a specific focus on microbial eukaryotes of relevance to drug discovery and development.
The Complex Landscape of Eukaryotic Genes
Architectural Complexity and Its Implications
Eukaryotic genes exhibit a split structure where protein-coding segments (exons) are separated by non-coding intervening sequences (introns). This fundamental architectural feature has profound implications for gene prediction:
- Intron-Exon Organization: The number and size of introns within genes can vary dramatically. While some genes like histone genes contain no introns, others such as the human factor VIII gene contain 26 introns spanning approximately 186 kb of genomic DNA, with only about 9 kb representing exonic sequences [44]. This means that over 95% of the gene consists of non-coding intronic sequence.
- Gene Families and Pseudogenes: Eukaryotic genomes often contain multiple copies of genes organized in families, such as the α and β globin genes, with different members expressed at various developmental stages [44]. These families arise from gene duplication events followed by divergence, and sometimes result in pseudogenes - nonfunctional copies that can confuse gene prediction algorithms.
- Repetitive DNA Elements: A substantial portion of eukaryotic genomes consists of highly repeated noncoding DNA sequences, sometimes present in hundreds of thousands of copies [44]. These simple-sequence or satellite DNAs can account for 10-20% of the DNA of higher eukaryotes and further complicate genome assembly and annotation.
Prevalence of Gene Prediction Errors
The complex architecture of eukaryotic genes directly contributes to high error rates in computational gene prediction. A comprehensive study of primate proteomes revealed that approximately 47% of protein sequences in public databases contain at least one error [45]. These errors are categorized as:
Table 1: Types and Frequencies of Gene Prediction Errors in Primate Proteomes
| Error Type | Description | Frequency | Impact on Downstream Analyses |
|---|---|---|---|
| Internal Deletions | Missing internal protein sequences | 29,045 errors | Truncated protein models; potential loss of functional domains |
| Internal Insertions | Additional amino acids inserted | 12,436 errors | Frameshifts; potential introduction of non-functional segments |
| Mismatched Segments | Correct sequence replaced with erroneous sequence | 11,015 errors | Significant impact on structural/functional annotations; altered evolutionary analyses |
| N-terminal Extensions | Additional sequence at protein start | 10,280 errors | Potential disruption of start codon and localization signals |
| N-terminal Deletions | Missing sequence at protein start | 10,264 errors | Loss of initiation codon and regulatory sequences |
| C-terminal Extensions | Additional sequence at protein end | 4,573 errors | Disruption of stop codon and protein termination |
| C-terminal Deletions | Missing sequence at protein end | 4,672 errors | Truncated proteins; potential loss of functional domains |
The distribution of these errors across primate species is not uniform, with some species showing significantly higher error rates than others, independent of their evolutionary distance from the reference human genome [45]. This suggests that database-specific curation practices and sequencing quality contribute substantially to annotation accuracy.
Experimental Protocols for Accurate Eukaryotic Gene Prediction
Comprehensive Workflow for Long-Read Sequencing and Analysis
Accurate gene prediction in eukaryotic microbes requires an integrated approach that combines optimized laboratory protocols with sophisticated computational analyses. The following workflow has been specifically validated for eukaryotic microorganisms including fungi and microbial parasites:
Table 2: Wet-Lab Protocol for Long-Read Sequencing of Microbial Eukaryotes
| Step | Procedure | Critical Parameters | Quality Assessment |
|---|---|---|---|
| DNA Extraction | Use high-molecular-weight DNA extraction kits with extended lysis | DNA integrity number (DIN) >7; minimum fragment size >20 kb | Pulse-field gel electrophoresis to confirm DNA size |
| Library Preparation | Prepare SMRTbell libraries without fragmentation | Input DNA: 5 µg; precise quantification via Qubit fluorometry | Fragment size distribution analysis on FemtoPulse or TapeStation |
| Sequencing | Perform HiFi sequencing on PacBio Revio or Sequel IIe systems | >20 CCS passes per molecule; 30× intended genome coverage | Real-time monitoring of loading efficiency and polymerase binding |
| RNA Extraction | Isolate RNA from same culture conditions for transcriptome integration | RNA integrity number (RIN) >8.5; poly-A selection for mRNA | Bioanalyzer trace to confirm absence of degradation |
| cDNA Preparation | Generate full-length cDNA using SMARTer or similar kits | PCR cycle optimization to minimize amplification bias | Size selection for >2 kb transcripts to enrich for full-length molecules |
Integrated Computational Analysis Pipeline
The wet-lab procedures must be coupled with a robust computational workflow to maximize prediction accuracy:
Figure 1: Integrated computational workflow for eukaryotic gene prediction from long-read data
Protocol: BRAKER3-Mediated Gene Prediction
The following detailed protocol implements the BRAKER3 pipeline [9] [10], which combines protein homology evidence and RNA-seq data to generate accurate gene models:
Input Preparation
- Genome assembly in FASTA format (preferably contig-level or chromosome-level)
- RNA-seq alignments in BAM format (sorted and indexed)
- Protein homology data from closely related species in FASTA format
Execution Commands
Critical Parameters
--eponymous: Use this flag to name output files with the species prefix--softmasking: Essential for properly handling repetitive regions--gff3: Produce GFF3 output for compatibility with downstream tools--cores: Utilize multiple cores to accelerate processing
Output Validation
- Check the number of predicted genes matches biological expectations
- Verify BUSCO scores show high completeness (>90% for most eukaryotes)
- Confirm proper splicing patterns through comparison with RNA-seq coverage
Error Detection and Correction Methods
To address the high rate of gene prediction errors documented in public databases [45], implement the following validation protocol:
Multiple Alignment Validation
- Generate alignments of predicted proteins against trusted reference datasets
- Identify regions with unexpected insertions, deletions, or frameshifts
- Use HMMER profiles to detect domain architecture anomalies
Transcriptomic Evidence Integration
- Map RNA-seq reads to the genome assembly using splice-aware aligners
- Compare exon boundaries between computational predictions and transcript evidence
- Resolve discrepancies through manual review in genome browsers
Common Error Patterns and Solutions
- Undefined genomic regions: Mask regions with excessive 'N' characters before prediction
- Short introns: Filter introns shorter than 30 nucleotides, which may represent prediction artifacts
- Frameshifts: Verify through transcript evidence; may represent sequencing errors
- Fragmented genes: Use long-range scaffolding to join disparate contigs
Successful eukaryotic gene prediction requires both wet-lab reagents and computational resources:
Table 3: Essential Research Reagents and Computational Tools for Eukaryotic Gene Prediction
| Category | Specific Tool/Reagent | Function | Application Context |
|---|---|---|---|
| Wet-Lab Reagents | PacBio SMRTbell Prep Kit | Preparation of sequencing libraries | Construction of high-molecular-weight libraries for HiFi sequencing |
| Nanopore Ligation Sequencing Kit | Library preparation for ONT | Preparation of native DNA libraries for ultra-long reads | |
| MagAttract HMW DNA Kit | High-molecular-weight DNA extraction | Isolation of intact DNA fragments >50 kb | |
| RNAstable | RNA stabilization | Preservation of RNA integrity for transcriptome studies | |
| Computational Tools | BRAKER3 [9] [10] | Gene prediction | Automated training of gene predictors using multiple evidence types |
| Flye [9] [10] | Genome assembly | De novo assembly of long reads into contigs | |
| Canu [9] [10] | Genome assembly | Error correction and assembly of noisy long reads | |
| InterProScan [9] [10] | Functional annotation | Domain architecture and functional motif identification | |
| BUSCO [9] | Assembly evaluation | Assessment of genome completeness using universal single-copy orthologs | |
| Database Resources | UniProt Knowledgebase [45] | Protein sequence database | Source of curated protein sequences for homology evidence |
| Pfam [45] | Protein family database | Domain-based functional annotation of predicted proteins | |
| GTDB [8] | Taxonomic database | Taxonomic classification of microbial eukaryotes |
Discussion and Future Perspectives
Long-read sequencing technologies have fundamentally transformed our ability to resolve complex eukaryotic gene structures, yet accurate gene prediction remains challenging. The high error rates documented in public databases (approximately 47% of proteins containing errors) highlight the critical need for the integrated experimental and computational approaches described in this application note [45].
Future methodological developments will likely focus on several key areas:
- Deep learning approaches that better integrate multiple evidence types during gene prediction
- Single-molecule sequencing of both DNA and RNA to directly observe splicing patterns
- Long-range scaffolding methods to properly assemble complex genomic regions
- Standardized benchmarking against experimentally validated gene models
For researchers in drug development, accurate gene prediction is particularly critical for identifying potential drug targets, understanding resistance mechanisms, and characterizing biosynthetic pathways in eukaryotic microbes. The protocols outlined here provide a foundation for generating high-quality genomic annotations that can support these discovery efforts.
As long-read technologies continue to evolve, with both PacBio and Oxford Nanopore achieving read accuracies exceeding 99% [12] [46], the bioinformatic challenges will shift from assembly quality to optimal evidence integration during annotation. The community-based development of standardized evaluation metrics and benchmark datasets will be essential for advancing the field of eukaryotic genome annotation.
The accurate reconstruction of microbial genomes from long-read sequencing data is a cornerstone of modern genomic research, with direct implications for understanding drug targets, resistance mechanisms, and pathogen evolution [4]. While individual genome assemblers are powerful, each employs distinct algorithms that can produce assemblies with varying strengths and weaknesses in continuity, accuracy, and completeness [9] [10]. Consequently, integrating outputs from multiple assemblers has emerged as a critical strategy to overcome the limitations of any single tool and produce optimal, high-quality genomic scaffolds for downstream gene prediction and functional annotation.
This application note details practical methodologies for combining multiple assembler outputs within the context of microbial genomics research. We focus on techniques that leverage High-Performance Computing (HPC) infrastructure to generate biologically meaningful insights, which are essential for applications in drug development and clinical microbiology [9].
Comparative Analysis of Long-Read Assemblers
Selecting appropriate assemblers is the first critical step in a multi-assembler workflow. The table below summarizes the key characteristics of three state-of-the-art long-read assemblers commonly integrated in modern microbial genomics pipelines.
Table 1: Key Long-Read Assemblers for Microbial Genomes
| Assembler | Underlying Algorithm | Strengths | Considerations for Integration |
|---|---|---|---|
| Canu [9] | Overlap-Layout-Consensus (OLC) | Effective for noisy reads; includes error correction | Computationally demanding; longer runtimes |
| Flye [9] [10] | Repeat graph | Efficient assembly of repetitive regions; faster than Canu | May require careful parameter tuning for complex genomes |
| wtdbg2 [9] [10] | de Bruijn graph (fuzzy) | Extremely fast assembly speed | May produce more fragmented assemblies for some datasets |
Integrated Workflow for Assembly and Evaluation
A robust workflow for combining assembler outputs integrates parallel execution, rigorous quality assessment, and a decision point for selecting the best assembly. The following diagram and subsequent sections detail this process.
Parallel Assembly Execution
The initial phase involves the simultaneous execution of selected assemblers. This process is computationally intensive and benefits significantly from HPC infrastructure, which allows jobs to be distributed across multiple computing nodes [9] [10]. A scalable workflow management system, such as those built using the Common Workflow Language (CWL), is recommended to ensure reproducibility and portability across different computing environments [9]. In this setup, the same set of long-read data (from Oxford Nanopore Technologies or PacBio) is processed in parallel by Canu, Flye, and wtdbg2, generating multiple draft genomes for evaluation.
Multi-Metric Assembly Evaluation
Each generated assembly must be rigorously evaluated using a combination of standardized metrics before selection [9]. The key metrics include:
- Contiguity Metrics: N50 and L50 statistics provide a primary measure of assembly continuity. A higher N50 indicates a more contiguous assembly.
- Completeness Assessment: Tools like BUSCO (Benchmarking Universal Single-Copy Orthologs) assess assembly completeness by searching for a set of evolutionarily conserved, near-universal single-copy genes that should be present in a full genome [9] [10].
- Accuracy Evaluation: Quality value (QV) and k-mer analysis can help identify the assembly with the highest base-level accuracy.
This multi-faceted evaluation prevents over-reliance on a single metric and provides a holistic view of assembly quality, guiding the subsequent selection process.
Experimental Protocol: Implementing a Multi-Assembler Pipeline
This section provides a detailed, actionable protocol for implementing the workflow described above, based on the platform developed by the Italian MIRRI ERIC node [9] [10].
Computational Infrastructure and Setup
- Hardware: The workflow is designed for a hybrid cloud-HPC infrastructure. The web-based component (for data upload and results visualization) operates on cloud virtual machines, while the computing component (for assembly execution) runs on a dedicated HPC cluster [9].
- Software Environment: Tools are containerized using Docker to ensure consistency and reproducibility. The workflow itself is defined using Common Workflow Language (CWL) for portability [9].
- Job Orchestration: In the HPC environment, a job scheduler like Slurm (e.g., via an extension like BookedSlurm) manages the distribution of assembly jobs across multiple nodes [9]. The described infrastructure can leverage dozens of nodes, each with 36-80 cores and 128 GB to 1 TB of RAM.
Step-by-Step Procedure
Data Input and Preparation:
Workflow Submission and Parallel Execution:
- The workflow manager submits three separate assembly jobs to the HPC cluster—one for each assembler (Canu, Flye, wtdbg2). This leverages parallel computing resources to minimize total runtime [9].
- Example Slurm job submission script for Flye:
Automated Quality Assessment:
- Upon completion of all assemblers, the workflow automatically runs quality assessment tools on each output assembly.
- This includes calculating N50/L50 and running BUSCO. The following command is an example of how BUSCO can be run:
Selection and Downstream Processing:
- The results from all quality reports are compiled. The assembly that best balances contiguity (highest N50), completeness (highest BUSCO score), and accuracy is automatically selected for the next stage [9].
- The selected assembly is then passed to the gene prediction and functional annotation phase of the pipeline, using tools like Prokka for prokaryotes or BRAKER3 for eukaryotes [9].
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Category/Item | Function in the Workflow |
|---|---|
| Computational Infrastructure | |
| HPC Cluster with Slurm | Manages and executes parallel assembly jobs across multiple compute nodes [9]. |
| Core Bioinformatics Tools | |
| Canu | Generates a draft genome assembly, performing read correction and consensus calling [9]. |
| Flye | Generates a competing draft genome assembly, specializing in resolving repeats [9] [10]. |
| wtdbg2 | Generates a draft genome assembly rapidly using a fuzzy Bruijn graph approach [9] [10]. |
| BUSCO | Evaluates the completeness of the assembled genome based on conserved single-copy orthologs [9] [10]. |
| Workflow & Environment | |
| Common Workflow Language (CWL) | Defines the multi-assembler workflow for maximum reproducibility and portability [9]. |
| Docker/Singularity | Containerizes tools to ensure a consistent software environment across different systems [9]. |
In genomic research, particularly in gene prediction from long-read assembled microbial genomes, the ability to reproduce computational analyses is a fundamental requirement for scientific validity and cumulative knowledge building. Workflow reproducibility ensures that bioinformatics tools yield consistent results across technical replicates, a concept critically defined as the capacity to maintain consistent outcomes when analyzing genomic data from different sequencing runs using fixed experimental protocols [47]. The combination of containerization with Docker and the Common Workflow Language (CWL) has emerged as a powerful framework to address the multifaceted challenges of reproducibility. This framework provides the technological foundation for standardized, portable, and verifiable computational analyses in microbial genomics, enabling researchers to accurately reconstruct and annotate microbial genomes while ensuring that results remain consistent across different computing environments and over time.
Theoretical Foundation
The Reproducibility Challenge in Genomics
Genomic reproducibility faces challenges at two pivotal stages: during pre-sequencing and sequencing where technical variability may emerge, and during computational analysis where stochastic algorithms can introduce uncertainties [47]. In the context of gene prediction from long-read assembled microbial genomes, these challenges manifest distinctly. Technical variability can arise from the use of diverse sequencing platforms and differences between individual flow cells [47]. Even with identical sequencing protocols across multiple runs, experimental variation still occurs due to random sampling variance of the sequencing process and variations in library preparation [47]. Bioinformatics tools must accommodate this experimental variation to generate consistent results across different sequencing runs and library preparations.
Conceptual Framework of Containerization and CWL
The theoretical underpinning of using Docker and CWL rests on creating computational isolation and process standardization. Docker containers provide encapsulated environments that ensure all software dependencies, versions, and system libraries remain consistent across executions. This directly addresses the problem of algorithmic biases and stochastic variations in bioinformatics tools that can compromise genomic reproducibility [47]. CWL adds a layer of process transparency by providing a standardized description of analysis workflows, making both the tools and their execution methodology explicitly defined and repeatable. Together, they create what Goodman et al. define as "methods reproducibility" – the ability to precisely replicate computational procedures using the same data and tools to yield identical results [47].
Table 1: Types of Reproducibility in Genomic Research
| Type | Definition | Application in Microbial Genomics |
|---|---|---|
| Methods Reproducibility | Ability to replicate experimental and computational procedures using same data and tools [47] | Re-running identical gene prediction pipeline on same genomic data |
| Genomic Reproducibility | Capacity of bioinformatics tools to maintain consistent results across technical replicates [47] | Consistent gene predictions across different sequencing runs of same microbial sample |
| Results Reproducibility | Obtaining same results when independent studies on different datasets are conducted with similar procedures [47] | Reproducing gene finding results across different microbial strains using same workflow |
Technical Specifications
Common Workflow Language (CWL) Components
CWL provides a standardized framework for describing analysis workflows and tools in a portable and scalable manner. The reference implementation, cwltool, is the primary Python module containing the reference implementation and console executable [48]. For optimal functionality, CWL requires specific dependencies:
- Python 3.9-3.13: cwltool is written and tested for these Python versions [48]
- Container Engine: Docker, Podman, Singularity/Apptainer, or udocker [48]
- Node.js: For evaluating CWL Expressions quickly (optional but recommended) [48]
The CWL ecosystem includes two primary packages: The cwltool package is the primary Python module, while the cwlref-runner package provides an additional entry point under the implementation-agnostic name cwl-runner [48]. Installation can be performed through multiple channels:
Docker Containerization Requirements
Docker provides the containerization backbone for ensuring consistent execution environments. Key requirements include:
- Platform Consistency: On Apple Silicon (arm64) systems, export
DOCKER_DEFAULT_PLATFORM=linux/amd64for amd64-only containers [48] - Storage Configuration: Proper mounting of directories accessible to Docker containers, particularly when using virtual machines like boot2docker [48]
- Resource Allocation: Adequate CPU, memory, and storage resources based on microbial genome analysis requirements
Table 2: Essential Software Components for Reproducible Microbial Genomics
| Component | Function | Example Tools/Implementations |
|---|---|---|
| Workflow Language | Standardized description of analysis steps | CWL (Common Workflow Language) [9] [48] |
| Container Platform | Environment and dependency isolation | Docker, Singularity, Podman [48] |
| Workflow Engine | Execution of described workflows | cwltool, Nextflow, Toil [48] [49] |
| Resource Manager | HPC cluster job scheduling | Slurm, Grid Engine, Torque/PBS [50] [51] |
Experimental Protocols
Protocol 1: Implementing a Reproducible Gene Prediction Workflow
This protocol outlines the complete process for implementing a reproducible gene prediction workflow for long-read assembled microbial genomes using CWL and Docker, based on the MIRRI ERIC Italian node platform [9].
Workflow Design and Description
The foundational step involves designing a CWL workflow that integrates all components from genome assembly to functional annotation. The workflow should incorporate multiple assemblers to enhance performance, completeness, and accuracy of genome assemblies [9]. A robust implementation includes:
- Multi-assembler Integration: Combine outputs from Canu, Flye, and wtdbg2 to improve assembly quality [9]
- Modular Tool Description: Define each bioinformatics tool as a separate CWL
CommandLineToolwith precise Docker container requirements - Data Flow Specification: Explicitly define inputs and outputs for each step to enable proper dependency tracking and potential restartability
Below is the DOT language visualization of the complete microbial genome analysis workflow:
Containerization Strategy
Each tool in the workflow requires a specific Docker image to ensure version consistency and dependency management:
- Tool-Specific Images: Use curated Docker images for each bioinformatics tool (e.g., Canu, Flye, Prokka, BRAKER3)
- Version Pinning: Explicitly specify Docker image versions in CWL tool definitions
- Registry Management: Maintain a private Docker registry if custom tool modifications are necessary
Example Docker image specifications for core tools:
Execution and Monitoring
Execute the workflow using a CWL runner with appropriate resource allocation:
Monitor workflow execution through:
- Real-time Logging: Capture standard output and error streams for each tool
- Resource Utilization: Track computational resources for optimization
- Intermediate Results: Validate outputs at each workflow stage
Protocol 2: Validation and Reproducibility Testing
This protocol describes methods for validating workflow reproducibility using technical replicates and synthetic datasets specifically designed for microbial genomics applications.
Technical Replicate Analysis
Technical replicates are obtained from the same biological sample sequenced multiple times using identical experimental and computational procedures [47]. They assess variability arising from the experimental process itself. For microbial genome analysis:
- Replicate Generation: Sequence the same microbial isolate across multiple library preparations and sequencing runs
- Workflow Application: Process each technical replicate through the identical CWL/Docker workflow
- Output Comparison: Quantify consistency in gene predictions, functional annotations, and assembly metrics
Reproducibility Metrics
Establish quantitative measures to assess reproducibility:
- Gene Prediction Consistency: Percentage of identical gene calls across replicates
- Assembly Metric Stability: Variation in N50, L50, and BUSCO scores across replicates
- Annotation Concordance: Consistency in functional assignments and domain predictions
Table 3: Reproducibility Assessment Metrics for Microbial Gene Prediction
| Metric Category | Specific Metrics | Acceptable Variance Threshold |
|---|---|---|
| Assembly Quality | N50, L50, contig count | <5% coefficient of variation |
| Gene Content | Number of predicted genes, BUSCO completeness | <3% difference between replicates |
| Functional Annotation | COG categories, EC numbers, GO terms | >95% concordance between replicates |
| Variant Detection | SNP/indel calls in conserved genes | >98% concordance for high-confidence calls |
Visualization and Data Representation
Workflow Architecture Diagram
The system architecture for implementing reproducible workflows integrates multiple components from user interface to computational execution:
Reproducibility Assessment Workflow
The process for assessing and ensuring reproducibility throughout the analysis pipeline:
The Scientist's Toolkit
Research Reagent Solutions for Reproducible Microbial Genomics
Table 4: Essential Computational Tools for Reproducible Microbial Genome Analysis
| Tool/Resource | Function | Implementation in CWL/Docker |
|---|---|---|
| Canu | Long-read genome assembler for noisy sequences [9] | Docker image with version pinning; CWL tool definition specifying inputs/outputs |
| Flye | De novo assembler for long reads using repeat graphs [9] | Version-controlled Docker container; CWL description with parameters |
| BRAKER3 | Eukaryotic gene prediction tool using RNA-seq and protein evidence [9] | Containerized environment; CWL tool with evidence data inputs |
| Prokka | Rapid prokaryotic genome annotation pipeline [9] | Docker image with dependency resolution; CWL wrapper for workflow integration |
| InterProScan | Functional analysis of proteins by classifying them into families [9] | Containerized execution; CWL tool definition for domain annotation |
| BUSCO | Assessment of genome completeness using universal single-copy orthologs [9] | Version-specific Docker image; CWL component for quality metrics |
| cwltool | Reference implementation of CWL for executing workflow descriptions [48] | Core execution engine; integrates with Docker daemon for container execution |
| Docker | Containerization platform for packaging tools and dependencies [9] [48] | Runtime environment ensuring consistency across compute infrastructures |
Advanced Implementation Considerations
High-Performance Computing Integration
The MIRRI ERIC platform demonstrates that reproducible workflows can leverage high-performance computing (HPC) infrastructure transparently to accelerate analysis while maintaining reproducibility [9]. Key implementation strategies include:
- Hybrid Infrastructure: Deploying web-based components on cloud infrastructure while executing computationally intensive workflows on HPC systems [9]
- Container Compatibility: Ensuring Docker images (or Singularity equivalents) are compatible with HPC scheduling systems like Slurm, Grid Engine, or Torque/PBS [51]
- Parallel Execution: Configuring CWL workflows to leverage parallel processing where possible through CWL's built-in scattering capabilities
Workflow Portability and Scaling
For microbial genomics applications that may span from individual bacterial genomes to large-scale pan-genome analyses, workflow scalability is essential:
- Resource Profiling: Document computational requirements for each workflow step to enable appropriate resource allocation
- Adaptive Execution: Implement workflow logic that adjusts processing parameters based on input data characteristics
- Multi-platform Support: Configure workflows to execute on diverse computing environments from local servers to cloud infrastructure
The integration of Docker containerization with Common Workflow Language establishes a robust foundation for reproducible gene prediction in long-read assembled microbial genomes. This approach directly addresses the critical challenge of genomic reproducibility by ensuring that bioinformatics tools maintain consistent results across technical replicates [47]. The methodological framework presented here, encompassing workflow design, containerization strategies, validation protocols, and assessment metrics, provides researchers with a comprehensive toolkit for implementing reproducible computational analyses. As microbial genomics continues to advance toward more precise and clinically relevant applications, maintaining rigorous standards of reproducibility through technologies like CWL and Docker becomes increasingly essential for generating trustworthy, verifiable, and biologically meaningful insights. The protocols and implementations detailed in this document serve as both a practical guide and a conceptual foundation for advancing reproducible research practices in the field of microbial genomics.
Benchmarking for Impact: Validation Frameworks and Clinical Translation
In the context of gene prediction on long-read assembled microbial genomes, the accuracy of downstream biological interpretations is fundamentally dependent on the quality of the initial genome reconstruction. Draft genomes, by their nature, are fragmented and incomplete, presenting significant challenges for comprehensive gene prediction and functional annotation [52]. The establishment of rigorous, multi-faceted validation standards is therefore not merely a procedural formality but a critical step to ensure that subsequent analyses—from identifying metabolic pathways to inferring ecological roles—are based on a reliable genomic foundation.
The advent of long-read sequencing technologies has dramatically improved our ability to reconstruct microbial genomes, producing assemblies with greater contiguity and more complete gene representation [9] [12]. However, these technological advances do not eliminate the need for systematic quality assessment. Instead, they necessitate more sophisticated validation approaches that can quantify improvements in assembly quality and identify residual limitations. Within a research thesis focused on gene prediction, establishing these validation standards provides the essential framework for evaluating methodological successes, interpreting functional capacities with appropriate caution, and comparing results across different studies or microbial systems.
This protocol details the implementation of three complementary validation metrics—BUSCO, N50 statistics, and completeness/contamination assessments—that together provide a comprehensive picture of genome assembly quality. By integrating these standards into the analytical workflow, researchers can make informed decisions about which gene sets are suitable for specific analyses, identify potential artifacts in their assemblies, and communicate the reliability of their findings with greater scientific rigor.
Metric Definitions and Theoretical Foundations
N50, L50, and Related Contiguity Metrics
N50 is a weighted median statistic that describes assembly contiguity by identifying the sequence length for which all contigs of that length or longer contain at least 50% of the total assembly length [53] [54]. Unlike a simple mean or median contig length, N50 gives greater weight to longer contigs, providing a more robust assessment of how well an assembly represents large, continuous genomic regions. To calculate N50, contigs are first sorted by length from longest to shortest. The cumulative length is then calculated by successively adding contig lengths until the sum reaches or exceeds 50% of the total assembly size. The length of the contig that contributes to reaching this threshold is the N50 value [53].
L50 is a related statistic representing the minimal number of contigs whose combined length reaches the 50% genome coverage threshold [53]. For example, an L50 value of 5 indicates that half of the assembled genome is contained in just five contigs. Lower L50 values generally indicate more contiguous assemblies, as fewer contigs are needed to represent half the genome.
NG50 is a variant that adjusts for situations where the assembly size differs from the expected genome size. Instead of using 50% of the assembly size, NG50 uses 50% of the estimated or known genome size, enabling more meaningful comparisons between assemblies of different sizes [53]. This is particularly valuable for microbial genomes where assembly size may vary significantly due to technical artifacts or biological differences.
Table 1: Key Contiguity Metrics for Genome Assembly Quality Assessment
| Metric | Definition | Interpretation | Optimal Range |
|---|---|---|---|
| N50 | Length of shortest contig at 50% of total assembly length | Higher values indicate greater contiguity | Microbial genomes: >50-100 kbp |
| L50 | Number of contigs covering 50% of assembly | Lower values indicate greater contiguity | Ideally <10-20 for microbial genomes |
| NG50 | N50 relative to expected genome size | Enables cross-assembly comparisons | Dependent on genome size |
| N90 | Length of shortest contig at 90% of total assembly length | Measure of base-level assembly quality | Higher values preferred |
| Total Contigs | Total number of contigs in assembly | Lower values indicate less fragmentation | Minimize while maintaining completeness |
BUSCO (Benchmarking Universal Single-Copy Orthologs)
BUSCO assesses genome completeness and gene content by searching for evolutionarily informed expectations of universal single-copy orthologs [9] [55]. These are genes that are expected to be present in single copies in virtually all members of a specific phylogenetic lineage. The percentage of these conserved genes that are successfully identified in an assembly provides a quantitative measure of completeness, while duplicated hits can indicate assembly artifacts or contamination [55].
The power of BUSCO lies in its phylogenetic approach; different sets of conserved genes are available for various taxonomic groups, allowing researchers to select appropriate references for their specific organisms. For bacterial genomes, BUSCO utilizes sets of conserved genes that are nearly universal within particular phyla or broader taxonomic groups.
Completeness and Contamination Assessment
Completeness estimates what percentage of an organism's genome has been successfully assembled, typically assessed using single-copy core genes (SCCGs) that are expected to be present in most bacteria [52]. Contamination measures the percentage of genes that appear to originate from different organisms, often indicated by the presence of multiple copies of typically single-copy genes [56].
Tools like CheckM and CheckM2 implement this approach by identifying lineage-specific marker genes and assessing their presence and copy numbers [56]. These metrics are particularly crucial for metagenome-assembled genomes (MAGs), where incomplete separation of closely related species can lead to contaminated assemblies [52].
Experimental Protocols and Implementation
Workflow for Comprehensive Assembly Validation
The following workflow diagram illustrates the integrated process for applying validation standards to long-read assembled microbial genomes:
Figure 1: Workflow for comprehensive validation of long-read assembled microbial genomes, integrating contiguity, completeness, and gene content assessments.
Protocol 1: Contiguity Assessment with N50 and Related Metrics
Materials and Software Requirements
- Genome assembly in FASTA format
- QUAST (Quality Assessment Tool for Genome Assemblies) [55]
- Perl/Python environment (for running QUAST)
- Reference genome (optional, for NG50 calculation)
Step-by-Step Procedure
Prepare Assembly File: Ensure your genome assembly is in FASTA format. The file should contain all contigs or scaffolds from your assembly process.
Run QUAST Analysis:
The
--min-contigparameter filters out contigs smaller than 500 bp, which are often assembly artifacts.Interpret Results: QUAST generates a comprehensive report (report.txt) containing:
- N50, L50, N90, L90 statistics
- Total number of contigs
- Total assembly length
- Largest contig size
- GC percentage
Calculate NG50 (if reference genome size is known):
Replace
5000000with your estimated microbial genome size.Quality Thresholding: For microbial genomes, aim for:
- N50 > 50-100 kbp (depending on organism and sequencing technology)
- Contig count significantly lower than gene count expected for organism
- Assembly length within expected range for similar organisms
Protocol 2: Completeness and Contamination Assessment with CheckM
Materials and Software Requirements
- CheckM or CheckM2 [56]
- Python 3.7+ environment
- Genome assembly in FASTA format
- Computational resources: CheckM can be memory-intensive for large datasets
Step-by-Step Procedure
Install CheckM and required databases:
Run CheckM Analysis:
Alternative CheckM2 Approach (machine learning-based, faster):
Interpret Key Outputs:
- Completeness: Percentage of expected marker genes found
- Contamination: Percentage of marker genes found in multiple copies
- Strain heterogeneity: Indicator of multiple strains in assembly
Quality Thresholding for downstream gene prediction:
Protocol 3: Gene Content Assessment with BUSCO
Materials and Software Requirements
- BUSCO (latest version) [55]
- Genome assembly or gene predictions in FASTA format
- Appropriate lineage dataset (e.g., bacteria_odb10)
Step-by-Step Procedure
Download and Install BUSCO:
Select Appropriate Lineage Dataset: Choose the most specific applicable lineage:
Run BUSCO Analysis:
Interpret Key Outputs:
- Complete BUSCOs: Percentage of conserved genes found as single copies (ideal)
- Fragmented BUSCOs: Partial matches to conserved genes
- Duplicated BUSCOs: Multiple copies of typically single-copy genes (potential red flag)
Quality Assessment:
- >95% complete BUSCOs indicates excellent gene content completeness
- <5% duplicated BUSCOs suggests minimal redundancy or contamination
- Fragmented BUSCOs may indicate assembly fragmentation or gene fragmentation
Integration and Interpretation of Multi-Metric Validation
Comparative Analysis of Validation Metrics
Table 2: Interpretation of Combined Validation Metrics for Microbial Genomes
| Metric Combination | Interpretation | Recommendation for Gene Prediction |
|---|---|---|
| High N50, High Completeness, High BUSCO | High-quality, contiguous assembly | Suitable for comprehensive gene prediction and comparative genomics |
| Low N50, High Completeness, High BUSCO | Fragmented but gene-complete assembly | Gene prediction possible but may miss regulatory regions and gene clusters |
| High N50, Low Completeness, Low BUSCO | Contiguous but incomplete assembly | Gene prediction will miss significant portions of genome; use with caution |
| High Completeness with High Contamination | Mixed assembly from multiple organisms | Requires binning or separation before gene prediction |
| High BUSCO with High Duplication | Potential assembly artifacts or polyploidy | Investigate potential misassembly before gene prediction |
Impact of Assembly Quality on Gene Prediction
The relationship between genome completeness and functional inference is well-established in microbial genomics. Research has demonstrated that genome completeness has a significant impact on the recovered functional signal across all domains of metabolic functions [52]. When a genome is estimated to be 70% complete, it is probable that many functions encoded in the actual genome will not be captured, leading to underestimation of functional capacity.
Studies examining the relationship between estimated genome completeness and metabolic function fullness have found a positive correlation for 94% of metabolic modules, spanning all functional domains and levels of complexity [52]. Overall, increasing completeness from 70% to 100% is associated with a 15 ± 10% increase in module fullness. This relationship remains constant across the completeness gradient, indicating that the problem persists even when considering 'high quality' (>90%) genomes.
Research Reagent Solutions and Computational Tools
Table 3: Essential Tools for Genome Assembly Validation
| Tool | Function | Application in Validation Pipeline | Key Outputs |
|---|---|---|---|
| QUAST | Assembly quality assessment | Contiguity metrics calculation | N50, L50, NG50, total contigs |
| CheckM/CheckM2 | Completeness/contamination assessment | Quality filtering of assemblies | Completeness %, contamination %, strain heterogeneity |
| BUSCO | Gene content completeness assessment | Evolutionary-informed quality control | % Complete, fragmented, duplicated genes |
| GTDB-Tk | Taxonomic classification | Contamination detection and classification | Taxonomic assignment, marker-based quality |
| FastANI | Genome similarity assessment | Cross-assembly comparisons | Average Nucleotide Identity (ANI) values |
| BBTools | Sequence manipulation | Data preprocessing and quality control | Sequencing depth, coverage statistics |
The establishment of comprehensive validation standards using BUSCO, N50, and completeness metrics provides an essential framework for ensuring the reliability of gene prediction in long-read assembled microbial genomes. By implementing the protocols outlined in this document, researchers can quantitatively assess assembly quality, identify potential limitations before proceeding to downstream analyses, and make informed decisions about the suitability of specific genomic data for particular research questions.
The integration of these validation metrics is particularly crucial in the context of drug development and functional genomics, where incomplete or contaminated assemblies can lead to erroneous conclusions about metabolic capabilities or potential drug targets. As long-read technologies continue to evolve and become more accessible, these validation standards will play an increasingly important role in ensuring that the biological insights derived from genomic data are built upon a foundation of technical rigor and reproducibility.
Moving forward, researchers should consider these validation metrics not as optional add-ons but as integral components of the genome assembly and annotation workflow. By establishing minimum quality thresholds—such as >90% completeness, <5% contamination, and N50 values appropriate for their specific microbial system—the scientific community can enhance the reliability and comparability of genomic findings across studies, ultimately accelerating discoveries in microbial ecology, evolution, and biotechnology.
The accuracy of gene prediction is a cornerstone of microbial genomics, directly influencing downstream interpretations of gene function, evolutionary biology, and biotechnological potential. While long-read sequencing technologies have revolutionized genome assembly by producing more complete and contiguous genomes, the subsequent step of gene prediction on these assemblies presents unique challenges and opportunities. This application note frames the critical task of gene prediction within a broader thesis on long-read assembled microbial genomes. We present a detailed case study validating a comprehensive bioinformatics platform, demonstrating its utility for generating reliable, biologically meaningful insights from both clinical and environmental microorganisms. The platform's performance is quantified through its application to three microbes of significance: Scedosporium dehoogii MUT6599, Klebsiella pneumoniae TUCC281, and Candida auris TUCC287 [9] [10].
The bioinformatics service used in this validation is designed as a comprehensive solution for long-read microbial sequencing data. Developed as part of the Italian MIRRI ERIC node, it provides an end-to-end workflow from raw sequence assembly to functional protein annotation, supporting both prokaryotic and eukaryotic organisms [9]. A key innovation of this platform is its integration of multiple state-of-the-art tools within a reproducible, scalable framework built on the Common Workflow Language (CWL) and accelerated via high-performance computing (HPC) infrastructure [9] [10]. This architecture ensures that the computationally demanding processes of assembly and annotation are both efficient and reliable.
The following diagram illustrates the core computational architecture and the staged workflow that forms the basis for the subsequent case study analyses.
Diagram Title: Computational architecture and workflow for microbial genome analysis.
The workflow is structured into four distinct, sequential phases [10]:
- Assembly Phase: Utilizes multiple assemblers (e.g., Canu, Flye) to reconstruct genomic sequences from long-read data.
- Assembly Evaluation Phase: Employs metrics like N50 and BUSCO to assess the completeness and accuracy of the assembly.
- Gene Prediction and Annotation Phase: Uses specialized tools (BRAKER3 for eukaryotes, Prokka for prokaryotes) to identify and annotate genes.
- Functional Protein Annotation Phase: Leverages InterProScan to assign functional information to the predicted genes.
Case Study: Experimental Design and Microbial Selection
To validate the platform, three microorganisms with high clinical and environmental relevance were selected from the TUCC culture collections [9] [10]. This selection tests the platform's capability to handle genetically diverse organisms from different domains of life.
- Scedosporium dehoogii MUT6599: A environmental fungus noted for its role in bioremediation and as an opportunistic human pathogen [9].
- Klebsiella pneumoniae TUCC281: A prokaryotic bacterium of clinical importance, known for antibiotic resistance and healthcare-associated infections.
- Candida auris TUCC287: A eukaryotic fungal pathogen recognized as a major global health threat due to its multi-drug resistance and rapid transmission in healthcare settings.
Detailed Experimental Protocol
The validation followed a standardized protocol for each microbe, as implemented by the platform [9] [10].
Step 1: Sample Preparation and Sequencing
- DNA Extraction: Obtain high-molecular-weight genomic DNA from pure microbial cultures using standardized extraction kits.
- Library Preparation: Prepare sequencing libraries according to the manufacturer's protocols for long-read technologies (PacBio or Oxford Nanopore).
- Sequencing: Perform deep sequencing on the prepared libraries to generate raw long-read data in FASTQ format.
Step 2: Computational Genome Assembly & Evaluation
- Input: Raw long-read FASTQ files.
- Assembly Execution: Execute the assembly phase of the workflow using multiple, integrated assemblers (Canu, Flye, wtdbg2) to improve consensus and accuracy [9].
- Quality Assessment: Run the assembly evaluation phase to generate key metrics. This includes standard assembly metrics (N50, L50) and evolutionarily-informed assessments of gene content using BUSCO (Benchmarking Universal Single-Copy Orthologs) [9] [10].
- Output: A high-quality, curated genome assembly in FASTA format.
Step 3: Gene Prediction and Functional Annotation
- Organism-Specific Prediction: Direct the assembly to the appropriate prediction branch:
- Functional Analysis: Execute the functional annotation phase using InterProScan to identify protein domains, families, and functional sites [9].
- Output: Comprehensive files (GFF, GBK) containing predicted gene models, their coordinates, and functional annotations.
Results and Data Analysis
The platform successfully processed the sequencing data for all three case study organisms, generating complete genome assemblies and annotations. The quantitative results from the assembly evaluation phase are summarized in the table below.
Table 1: Genome Assembly Metrics for Clinical and Environmental Microbes
| Microbial Strain | Domain | Relevance | Key Assembly Metric (N50) | Gene Content Metric (BUSCO) |
|---|---|---|---|---|
| Scedosporium dehoogii MUT6599 [9] | Eukaryote | Environmental & Clinical | Provided by platform evaluation | Provided by platform evaluation |
| Klebsiella pneumoniae TUCC281 [9] | Prokaryote | Clinical | Provided by platform evaluation | Not Applicable (Prokaryote) |
| Candida auris TUCC287 [9] | Eukaryote | Clinical | Provided by platform evaluation | Provided by platform evaluation |
The platform's integrated approach allowed for the recovery of high-quality genomic data. The use of multiple assemblers enhanced the completeness and accuracy of the genome assemblies, providing a robust foundation for the downstream gene prediction steps [9]. Furthermore, the tailored gene prediction using BRAKER3 for eukaryotes and Prokka for prokaryotes ensured that the unique gene structures of each domain were accurately captured.
The functional annotation pipeline successfully provided biologically meaningful insights by linking predicted genes to protein families and domains. This step is critical for moving from a raw gene model to a hypothesis about its biological function, enabling researchers to identify potential virulence factors, antibiotic resistance genes, or novel metabolic pathways of interest [9].
The Scientist's Toolkit
The following table details key research reagents and computational tools essential for executing the genomic analyses described in this application note.
Table 2: Essential Research Reagents and Computational Tools
| Category / Item | Function / Application | Specifications / Notes |
|---|---|---|
| Bioinformatics Platform | ||
| MIRRI-IT Platform [9] [10] | Integrated, user-friendly web service for end-to-end microbial genome analysis. | Built on Common Workflow Language (CWL); uses Docker for containerization; leverages HPC. |
| Genome Assemblers | ||
| Canu [9] | Long-read assembler for noisy, single-molecule sequencing reads. | Used for initial assembly; robust to high error rates. |
| Flye [9] | De novo assembler for long reads; can reconstruct repeat regions. | Used for assembly; effective with complex genomes. |
| Gene Prediction Tools | ||
| BRAKER3 [9] | Eukaryotic gene prediction tool using evidence from RNA-Seq and protein homology. | Applied to S. dehoogii and C. auris. |
| Prokka [9] | Rapid annotation software for prokaryotic genomes. | Applied to K. pneumoniae. |
| Functional Annotation | ||
| InterProScan [9] | Scans protein sequences against multiple databases to classify them into families/domains. | Provides Gene Ontology terms, pathways, and other functional data. |
| Quality Assessment | ||
| BUSCO [10] | Assesses genome completeness based on universal single-copy orthologs. | Critical for evaluating eukaryotic assembly quality. |
Concluding Remarks
This application note demonstrates the successful validation of a comprehensive bioinformatics platform for gene prediction on long-read assembled microbial genomes. Through a carefully designed case study on clinically and environmentally significant microbes, we have shown that an integrated workflow—from multi-tool assembly to domain-specific gene prediction and functional annotation—can produce reliable, high-quality genomic data. The platform's design, which emphasizes ease of use, reproducibility, and leveraging of high-performance computing, effectively lowers the barrier for researchers to perform advanced microbial genomics [9]. The methodologies and results detailed herein provide a robust framework for future genomic studies aimed at unlocking the functional potential encoded within microbial DNA.
Within microbial genomics, the selection of a sequencing platform is a foundational decision that profoundly influences the quality of genome assemblies and the accuracy of subsequent gene predictions. While short-read sequencing has been the workhorse of genomic studies for years, long-read technologies have emerged as powerful alternatives capable of resolving complex genomic regions. This Application Note provides a detailed comparative analysis of these approaches, focusing on their performance in microbial genome assembly and gene prediction. We present structured quantitative data, detailed experimental protocols, and analytical workflows to guide researchers in selecting and implementing the most appropriate sequencing strategy for their specific research objectives, particularly within the context of gene prediction on long-read assembled microbial genomes.
Technical Comparison of Sequencing Technologies
Performance Characteristics and Error Profiles
The fundamental differences between short-read and long-read sequencing technologies impart distinct advantages and limitations for microbial genomics applications. Table 1 summarizes the key technical specifications and performance metrics of both approaches.
Table 1: Technical comparison of short-read and long-read sequencing technologies
| Parameter | Short-Read Sequencing | Long-Read Sequencing |
|---|---|---|
| Read Length | 50-300 bases [57] | 5,000-30,000 bases; up to 1 Mb+ for Nanopore [5] |
| Primary Technologies | Illumina, Ion Torrent, Element Biosciences AVITI, MGI DNBSEQ [5] | PacBio SMRT, Oxford Nanopore [58] [5] |
| Typical Error Rate | <0.1% (Illumina) [57] | PacBio HiFi: <0.1% [58] [5]; Nanopore: ~5% raw, improvable with correction [58] [59] |
| Error Type | Substitution errors [57] | PacBio: Stochastic errors [58]; Nanopore: Systematic errors in homopolymer regions [58] |
| DNA Input Requirements | Low to moderate [57] | Varies by protocol; can be higher for some long-read applications |
| Library Preparation Time | Moderate to extensive [5] | Generally faster with fewer amplification steps [5] |
| Cost per Sample | Lower [57] [5] | Higher, though decreasing [5] |
Short-read technologies excel in applications requiring high accuracy for small variant detection and when working with limited DNA quantities [57]. Their main limitation lies in resolving repetitive regions, structural variants, and haplotyping due to the limited read length [57] [5]. In contrast, long-read technologies fundamentally address these challenges by spanning repetitive elements and complex regions, providing complete context for genomic analysis [5]. The development of PacBio's HiFi (High Fidelity) reads through circular consensus sequencing has significantly improved accuracy while maintaining long read lengths [58] [5], making this technology particularly suitable for microbial genome assembly and gene prediction.
Error Profiles and Correction Strategies
Understanding the distinct error profiles of each technology is crucial for optimizing data quality and selecting appropriate bioinformatic processing tools.
PacBio sequencing errors are primarily stochastic, arising from limitations in fluorescence signal detection during real-time sequencing [58]. The HiFi mode addresses this through circular consensus sequencing (CCS), where the same DNA molecule is sequenced multiple times to generate a highly accurate consensus read [58]. This approach reduces the initial error rate from approximately 15% to less than 0.1% [58], making PacBio HiFi reads suitable for high-precision genome assembly and variant detection.
Nanopore errors are predominantly systematic, with the most significant challenges occurring in homopolymer regions (e.g., consecutive A/T bases) where current signal recognition biases can lead to inaccurate base calling [58]. Recent hardware improvements, such as the R10 chip with its dual-reader head design, have significantly enhanced accuracy in these problematic regions [58]. Additionally, deep learning algorithms (e.g., Bonito, Guppy) and consensus-based tools (e.g., Medaka) have improved base calling and error correction capabilities [58].
Applications in Microbial Genomics
Genome Assembly and Metagenome-Assembled Genomes (MAGs)
Long-read sequencing has demonstrated remarkable capabilities in recovering high-quality microbial genomes from complex environmental samples, addressing what has been termed the "grand challenge" of terrestrial metagenomics [8]. A recent large-scale study employing deep long-read Nanopore sequencing of 154 complex environmental samples recovered 23,843 MAGs, of which 15,314 represented previously undescribed microbial species [8]. This work expanded the phylogenetic diversity of the prokaryotic tree of life by 8%, demonstrating the profound impact of long-read technologies in exploring microbial dark matter [8].
The superiority of long reads in genome assembly stems from their ability to span repetitive regions that typically fragment short-read assemblies. This results in dramatically improved contiguity, with one study reporting a median contig N50 of 79.8 kbp for long-read assemblies of complex soil samples compared to typically <10-20 kbp for short-read assemblies [8]. This enhanced contiguity directly benefits gene prediction by providing more complete gene contexts and reducing fragmentation of coding sequences.
Table 2: Comparison of genome assembly outcomes for short-read and long-read approaches
| Assembly Metric | Short-Read Assembly | Long-Read Assembly |
|---|---|---|
| Contiguity (N50) | Typically lower (often <20 kbp) | Significantly higher (median 79.8 kbp reported) [8] |
| Completeness | Often fragmented, missing repetitive regions | More complete, spans repetitive elements |
| MAG Recovery from Complex Samples | Challenging, requires deep sequencing [8] | High yield (median 154 MAGs/sample from soil) [8] |
| Strain Resolution | Limited | Enabled by long contextual information |
| Plasmid Recovery | Often missed | Frequently recovered as circular contigs |
For microbial whole-genome sequencing, PacBio's HiFi sequencing enables complete bacterial genomes to be obtained, often resulting in fully closed chromosomes and plasmids, while simultaneously capturing epigenetic modifications such as methylation patterns [60].
Gene Prediction and Functional Annotation
The improved contiguity of long-read assemblies directly enhances gene prediction accuracy by providing complete gene contexts and reducing fragmentation. A bioinformatics platform specifically designed for long-read microbial data provides integrated workflows for both prokaryotic and eukaryotic gene prediction, incorporating tools such as BRAKER3 for eukaryotes and Prokka for prokaryotes [10]. The platform demonstrates that long-read assemblies enable more complete gene models, particularly for genes containing repetitive elements or multiple exons [10].
Functional annotation also benefits from long-read sequencing. Comparative studies have shown that approximately twice the proportion of long reads can be assigned functional annotations compared to short reads [61]. Furthermore, long-read metagenomic approaches yield richer functional information and enable more precise profiling of species, which is crucial for understanding microbial community functions and interactions [61] [60].
Hybrid Approaches for Enhanced Variant Detection
Emerging evidence suggests that hybrid approaches combining both short- and long-read technologies can maximize variant detection accuracy while potentially reducing overall costs. A recent innovative approach demonstrated that a hybrid DeepVariant model, jointly processing Illumina and Nanopore data, could match or surpass the germline variant detection accuracy of state-of-the-art single-technology methods [62]. This strategy is particularly promising for clinical applications, as shallow hybrid sequencing (e.g., 15× ONT + 15× Illumina coverage) can achieve performance comparable to deep sequencing with a single technology, potentially lowering costs for large-scale screenings [62].
Experimental Protocols
Microbial Whole Genome Sequencing Using Long-Read Technologies
Protocol 1: PacBio HiFi Microbial Whole Genome Sequencing
This protocol describes the recommended workflow for generating high-quality complete microbial genomes using PacBio HiFi sequencing [60].
Materials:
- PacBio HiFi plex prep kit 96
- Sequel IIe or Revio system
- DNA extraction kit (magnetic bead-based recommended)
- Qubit dsDNA HS Assay Kit
- Femto Pulse system (for quality control)
Procedure:
DNA Extraction and Quality Control
- Extract high-molecular-weight (HMW) DNA using a protocol optimized for microbial cells. Mechanical shearing should be minimized.
- Quantify DNA using fluorometric methods (Qubit).
- Assess DNA quality and size distribution using pulse-field capillary electrophoresis (Femto Pulse). DNA fragments should average >20 kbp.
Library Preparation
- Use the HiFi plex prep kit 96 according to manufacturer's instructions.
- Perform DNA repair and end-repair steps.
- Ligate SMRTbell adapters to create circular templates.
- Purify the library using size-selective binding to remove fragments <1 kbp.
- Quantify the final library and assess size distribution.
Sequencing
- Bind the SMRTbell library to DNA polymerase.
- Load the complex onto SMRT Cells for sequencing.
- Sequence on Revio or Sequel IIe system using appropriate sequencing chemistry.
- Typical run: 1-4 SMRT Cells per microbiome sample, depending on complexity.
Data Analysis
- Process raw data using SMRT Link software to generate HiFi reads.
- Perform genome assembly using Flye or Canu assemblers.
- Polish assemblies if necessary using tools like Medaka.
- Annotate genomes using Prokka or similar annotation pipelines.
Expected Outcomes: Reference-grade, complete microbial genomes with closed chromosomes and plasmids, plus methylation detection. The multiplexed approach enables processing up to 384 libraries per SMRT Cell at a cost of <$50 USD per sample [60].
Full-Length 16S/ITS Amplicon Sequencing for Microbiome Profiling
Protocol 2: Full-Length 16S Sequencing with PacBio
This protocol enables species- and strain-level taxonomic resolution of microbial communities by sequencing the entire 16S rRNA gene [61] [60].
Materials:
- PCR primers targeting full-length 16S rRNA gene (27F-1492R)
- High-fidelity DNA polymerase
- PacBio Barcodes for multiplexing
- Kinnex 16S rRNA kit (optional for increased throughput)
Procedure:
PCR Amplification
- Amplify the full-length 16S rRNA gene (approximately 1.5 kb) using barcoded primers.
- Use minimal PCR cycles (15-20) to reduce chimera formation.
- Pool amplified products from multiple samples.
Library Preparation
- Repair amplicon ends and ligate SMRTbell adapters.
- Size-select for full-length amplicons (approximately 1.5-1.6 kb).
- If using Kinnex kit, follow the concatenation protocol to increase throughput.
Sequencing
- Bind polymerase to SMRTbell templates.
- Sequence on PacBio system using appropriate sequencing chemistry.
- For Kinnex libraries: Generate reads containing multiple concatenated amplicons.
Data Analysis
- Demultiplex reads by barcode.
- If using Kinnex, bioinformatically break concatenated reads into individual amplicons.
- Cluster sequences into operational taxonomic units (OTUs) or amplicon sequence variants (ASVs).
- Classify taxa using reference databases (Silva, Greengenes).
Expected Outcomes: Species- and strain-level taxonomic profiling of microbial communities. With Kinnex technology, up to 1,536-plex sequencing can be achieved at <$5 per sample with >30,000 average reads per sample [60].
Workflow Visualization
Diagram 1: Microbial genome assembly and gene prediction workflow comparing sequencing approaches. Hybrid approaches leverage the complementary strengths of both technologies for improved variant detection [62].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential research reagents and solutions for microbial genomics studies
| Reagent/Solution | Function | Example Applications |
|---|---|---|
| PacBio HiFi plex prep kit 96 | Library preparation for highly accurate long-read sequencing | Microbial whole genome sequencing, metagenomic studies [60] |
| Oxford Nanopore Ligation Sequencing Kits | Library preparation for Nanopore sequencing | Real-time sequencing applications, field sequencing [58] |
| 10x Genomics Chromium Single Cell 3' Kits | Single-cell RNA sequencing library preparation | Single-cell gene expression profiling in microbial communities [63] |
| MAS-ISO-seq for 10x Genomics Kit | Full-length transcript sequencing from single cells | Single-cell isoform resolution in microbial eukaryotes [63] |
| Kinnex 16S rRNA Kit | High-throughput full-length 16S sequencing | Microbiome profiling at species and strain level [60] |
| Solid-phase reversible immobilization (SPRI) beads | DNA size selection and cleanup | Library preparation across all platforms [63] |
| High-fidelity DNA polymerase | Accurate PCR amplification | Amplicon sequencing, library amplification [61] |
The comparative analysis presented in this Application Note demonstrates that both short-read and long-read sequencing technologies offer distinct advantages for microbial genomics research. Short-read approaches remain valuable for applications requiring high base-level accuracy and low cost, while long-read technologies excel in resolving complex genomic regions, assembling complete genomes, and enabling more accurate gene prediction. The emerging paradigm of hybrid approaches, leveraging both technologies' complementary strengths, shows particular promise for maximizing variant detection accuracy while potentially reducing overall costs. For researchers focused on gene prediction in microbial genomes, long-read technologies provide superior contiguity and context that directly translates to more complete and accurate gene models, enabling deeper biological insights into microbial function, evolution, and ecological roles.
Linking Genetic Evidence to Clinical Success Rates
Table 1: Impact of Genetic Evidence on Clinical Trial Outcomes
| Trial Outcome | Genetic Support (Odds Ratio) | Key Implication |
|---|---|---|
| Overall Trial Progression | Increased Likelihood [64] | Genetically supported targets are more likely to advance through clinical stages. |
| Trials Stopped for Lack of Efficacy | OR = 0.61 (Significant Depletion) [64] | Absence of genetic support is a major risk factor for efficacy failure. |
| Trials Stopped for Safety Reasons | Depletion in genetic support; higher risk with constrained, broadly expressed targets [64] | Target properties (expression, constraint) influence safety profile. |
| FDA Drug Approvals (2021) | ~66% supported by human genetics [64] | Genetic evidence is a cornerstone of modern successful drug development. |
The pursuit of new therapeutic targets is a high-risk endeavor, with a majority of clinical trials failing due to a lack of efficacy or safety concerns. A powerful strategy to de-risk this pipeline is the integration of human genetic evidence during target selection. Retrospective analyses demonstrate that drug targets with genetic support are more than twice as likely to achieve regulatory approval [65]. This application note details protocols for leveraging genetic evidence and long-read genome sequencing to systematically identify and validate high-confidence microbial targets, thereby increasing the probability of clinical success.
Genetic Evidence Framework for Target Prioritization
The foundational step involves a rigorous genetic analysis to establish a causal link between a target and a disease, moving beyond mere association.
Analytical Protocol: Human Genetic Analysis
- Objective: To identify and prioritize microbial gene targets with human genetic evidence supporting their role in host disease.
- Methodology:
- Genome-Wide Association Studies (GWAS) Integration: Interrogate large-scale GWAS data for microbial taxa or gene families that are significantly associated with human disease states. A recent cross-trait analysis exemplifies this, identifying 42 pleiotropic genetic variants shared between lung and gastrointestinal diseases by integrating GWAS data from multiple populations [66].
- Colocalization Analysis: Determine if the same genetic variant influences both microbial abundance and disease risk using statistical methods (e.g., COLOC). Variants with a posterior probability of hypothesis H4 (PPH4) ≥ 0.5 are considered high-confidence candidate pleiotropic variants [66].
- Mendelian Randomization (MR): Apply MR techniques to infer causal relationships. For instance, a causal link between the gut microbiome genus Parasutterella, gastro-oesophageal reflux disease (GORD), and asthma was established using this method [66].
- Gene-Level Constraint Assessment: Evaluate the tolerance of the human ortholog of the microbial target to functional genetic variation. Targets that are highly constrained in human populations (e.g., pLI > 0.9) are associated with a greater likelihood of trial stoppage due to safety concerns, particularly in oncology [64].
Visualization: Genetic Evidence to Target Validation Pathway
The following diagram illustrates the logical workflow from genetic discovery to target validation.
Experimental Protocol: Long-Read Microbial Genome Assembly and Gene Prediction
Once a microbial target is genetically prioritized, its functional characterization requires a high-quality genome assembly. Long-read sequencing is critical for overcoming the challenges of repetitive regions and achieving complete, contiguous genomes.
Workflow: Microbial Genome Analysis
This protocol is adapted from the MIRRI ERIC bioinformatics platform for long-read data [9] [10].
Step 1: DNA Sequencing & Quality Control
- Technology: Pacific Biosciences (PacBio) HiFi or Oxford Nanopore Technologies (ONT) sequencing.
- Quality Control: Assess read quality and length using tools like NanoPlot (for ONT) or HiFi-specific QC metrics.
Step 2: Genome Assembly
Step 3: Assembly Evaluation
Step 4: Gene Prediction & Functional Annotation
Step 5: Multi-Omics Integration for Functional Validation
- Metatranscriptomics: Sequence community RNA to characterize context-specific biomolecular activity and identify actively transcribed pathogenic or protective genes [16].
- Metabolomics: Correlate microbial genetic potential with shifts in the metabolome (e.g., short-chain fatty acids, bile acids) to illuminate underlying mechanisms [67].
Visualization: Microbial Genome Analysis Pipeline
The following workflow diagram outlines the key steps in the microbial genome analysis protocol.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents and Platforms
| Item / Platform | Function / Application |
|---|---|
| MIRRI ERIC Platform | A user-friendly, web-accessible bioinformatics service for end-to-end long-read microbial genome analysis, from assembly to functional annotation [9] [10]. |
| Open Targets Platform | Integrates public resources to prioritize drug targets by aggregating genetic, genomic, and chemical data [64] [65]. |
| Prokka | A command-line software tool for the rapid annotation of prokaryotic genomes [9] [10]. |
| BRAKER3 | A pipeline for fully automated, evidence-based gene prediction in eukaryotic genomes [9] [10]. |
| InterProScan | A functional annotation tool that classifies proteins into families and predicts domains and important sites [9] [10]. |
| BUSCO | A tool to assess genome assembly and annotation completeness based on evolutionarily informed expectations of gene content [9] [10]. |
| Fecal Microbiota Transplantation (FMT) | A therapeutic intervention to test causal effects of microbiota; efficacy depends on stable donor strain engraftment, measurable via metagenomics [67]. |
| Experimental Evolution | A method to study microbial adaptation (e.g., antibiotic resistance) in controlled laboratory settings, informing on evolutionary trajectories and resistance mechanisms [68]. |
Application Notes
Advancing Microbial Genome Analysis Through Integrated Bioinformatics Platforms
The comprehensive analysis of microbial genomes is crucial for uncovering their ecological roles, evolutionary history, and potential applications in health, biotechnology, and environmental science [9]. Long-read sequencing technologies from PacBio and Oxford Nanopore Technologies (ONT) have revolutionized this field by generating significantly longer DNA fragments, enabling more complete genome assemblies and access to previously challenging genomic regions [1] [10]. However, transforming raw sequencing data into biological insights requires sophisticated computational resources and expertise, creating barriers for many researchers [9].
To address these challenges, integrated bioinformatics platforms have been developed that provide complete workflows from genome assembly to functional annotation. The MIRRI-IT platform exemplifies this approach by combining state-of-the-art tools (Canu, Flye, BRAKER3, Prokka, InterProScan) within a reproducible, scalable workflow built on Common Workflow Language and accelerated through high-performance computing infrastructure [9] [10]. This service provides a user-friendly web interface alongside advanced computational capabilities, making powerful genomic analysis accessible to non-specialists while maintaining reproducibility and quality assessment through standardized metrics [10].
Key innovations include the combination of multiple assemblers to enhance assembly quality and completeness, integrated gene prediction tailored for both prokaryotic and eukaryotic organisms, and functional protein annotation through connection to multiple external databases [9]. The platform has been validated through case studies involving clinically and environmentally significant microorganisms including Scedosporium dehoogii, Klebsiella pneumoniae, and Candida auris, demonstrating its ability to produce reliable, biologically meaningful results [10].
Lineage-Specific Protein Prediction Expands Functional Understanding of Microbial Communities
Microbial ecosystems utilize diverse genetic codes and gene structures that are often overlooked in standard metagenomic analyses, leading to spurious protein predictions and limiting functional understanding [22]. Lineage-specific gene prediction approaches that incorporate correct genetic codes based on taxonomic assignment of genetic fragments can dramatically improve protein discovery and functional annotation.
Applied to 9,634 human gut metagenomes and 3,594 genomes, this lineage-specific approach increased the landscape of captured expressed microbial proteins by 78.9% compared to conventional methods [22]. The optimized pipeline specifically enhanced prediction of small proteins, capturing 3,772,658 small protein clusters that formed an improved microbial protein catalogue of the human gut (MiProGut) [22]. This expansion enabled discovery of previously hidden functional groups and provided a more comprehensive resource for studying protein ecology—the ecological distribution of proteins or functions as the unit of study [22].
The practical utility of this approach was demonstrated through the development of InvestiGUT, a tool that integrates protein sequences with sample metadata to identify associations between protein prevalence and host parameters [22]. This enables direct study of protein ecology, moving beyond taxonomy-based inference to directly investigate how proteins and their functions distribute across microbial ecosystems and interact with host physiology [22].
Long-Read Metagenomic Sequencing Unlocks Soil Microbial Dark Matter for Drug Discovery
Soil represents one of the most complex and diverse microbial ecosystems on Earth, yet its vast genetic potential has remained largely untapped due to technical challenges in DNA extraction and sequencing [69]. Recent advances in long-read sequencing, coupled with optimized DNA extraction methods, have enabled unprecedented access to this microbial "dark matter."
A terabase-scale long-read sequencing study of forest soil utilizing optimized nanopore sequencing generated read lengths with N50 > 30 kbp—200 times longer than conventional 150-bp short-read technology [69]. This yielded megabase-sized assemblies, including 563 complete or near-complete circular metagenomic genomes from a single soil sample [69]. In comparison, previous Illumina-based soil metagenomic studies typically produced assemblies with N50 of approximately 1.6 kbp, even with >3 Tbp of sequencing data [69].
The large contiguous assemblies enabled direct identification of complete biosynthetic gene clusters (BGCs) encoding natural product pathways [69]. Through a synthetic bioinformatic natural product (synBNP) approach that couples bioinformatics prediction with chemical synthesis, researchers converted unearthed nonribosomal peptide BGCs directly into bioactive molecules [69]. This led to the discovery of novel antibiotics with rare modes of action, including a potent cardiolipin-binding broad-spectrum antibiotic and a ClpX-targeting antibiotic, both showing activity against multidrug-resistant pathogens [69].
Table 1: Comparison of Sequencing Technologies for Metagenomic Applications
| Platform | Average Read Length | Throughput per Run | Error Rate | Key Metagenomic Applications |
|---|---|---|---|---|
| Illumina NovaSeq | 150-250 bp | Up to 6 Tb | ~0.1% | Marker gene studies, shallow metagenomic profiling |
| PacBio Sequel II/Revio | 15-20 kb | 25-90 Gb | ~0.1% | High-quality metagenome-assembled genomes, complete BGC assembly |
| ONT PromethION | 20+ kb | 100-120 Gb | 1-3% | Large-scale metagenomic assembly, direct RNA sequencing |
Table 2: Impact of Long-Read Sequencing on Soil Metagenome Assembly Quality
| Assembly Metric | Short-Read Illumina | PacBio Long-Read | Optimized Nanopore |
|---|---|---|---|
| Assembly N50 | ~1.6 kbp | ~36 kbp | ~262 kbp |
| Contigs >1 Mbp | Rare | Limited | 3,200+ |
| Complete circular genomes from single soil | Not achievable | Dozens | 206 |
| BGC characterization | Fragmented, incomplete | Improved | Complete pathways |
Experimental Protocols
Protocol for Microbial Genome Assembly and Annotation from Long-Read Data
Experimental Workflow
Step-by-Step Methodology
Phase 1: DNA Extraction and Library Preparation for Long-Read Sequencing
Soil Microbial Cell Separation: Resuspend 1 g of soil sample in 10 mL of phosphate buffer. Layer onto a nycodenz gradient solution and centrifuge at 10,000 × g for 30 minutes at 4°C. Recover the bacterial cell band from the gradient interface [69].
DNA Purification: Process the bacterial suspension through a skim-milk wash to remove impurities. Extract high-molecular-weight DNA using Monarch HMW DNA extraction kit, following manufacturer's instructions with extended incubation times for complete cell lysis [69].
Size Selection and Quality Control: Use Oxford Nanopore's small fragment eliminator kit to retain DNA fragments >10 kbp. Assess DNA quality and fragment size distribution using pulsed-field gel electrophoresis or Fragment Analyzer systems. DNA should show predominant fragments >20 kbp for optimal results [69].
Phase 2: Genome Assembly and Quality Assessment
Multi-Assembler Approach: Process quality-filtered reads through at least two assemblers simultaneously. Recommended tools and parameters:
- Flye:
--nano-hqfor Nanopore data or--pacbio-hififor PacBio data, with--metaflag for metagenomic samples [10] - Canu:
-pacbioor-nanoporeparameters with correctedErrorRate=0.035 for metagenomic mode [9] - metaFlye: Use for complex metagenomic samples, particularly with
--metaand--carefulparameters [69]
- Flye:
Assembly Evaluation: Assess assembly quality using multiple metrics:
Phase 3: Gene Prediction and Functional Annotation
Repeat Masking: Identify and mask repetitive elements using RepeatMasker with RepBase libraries and species-specific repeats constructed by RepeatModeler [40]. This prevents spurious gene predictions in repetitive regions.
Lineage-Specific Gene Prediction: Implement taxonomic assignment of contigs using Kraken 2 or similar tools. Apply optimized gene prediction tools based on taxonomy:
Functional Annotation: Annotate predicted proteins using InterProScan for domain architecture and Gene Ontology terms. Cross-reference with UniProtKB/Swiss-Prot for validated functional assignments [9] [40].
Phase 4: Experimental Validation and Functional Exploration
Transcriptomic Validation: Isolate RNA from source material and prepare RNA-seq libraries. Map sequencing reads to predicted genes using Kallisto or STAR aligner. Confirm expression of predicted genes, with particular attention to small proteins and lineage-specific predictions [22] [40].
Gene Expansion Analysis: Use CAFE5 software to identify significantly expanded gene families compared to related organisms. Validate expansions through manual curation in Apollo genome browser [40].
Functional Characterization: For biosynthetic gene clusters, perform heterologous expression or chemical synthesis of predicted natural products. Test bioactivity against relevant pathogen panels [69].
Protocol for Metagenomic Biosynthetic Gene Cluster Discovery and Characterization
BGC Discovery Workflow
Step-by-Step Methodology
Phase 1: BGC Identification and Prioritization from Metagenomic Assemblies
BGC Prediction: Screen assembled contigs using antiSMASH with comprehensive analysis settings (
--clusterblast --subclusterblast --knownclusterblast --smcogs --tigrfam). For nonribosomal peptide and polyketide clusters, use additional specialized tools like PRISM for structural prediction [69].Cluster Boundary Definition: Define precise BGC boundaries by identifying flanking core biosynthetic genes and evaluating GC content shifts. For complete circular assemblies, verify cluster completeness by confirming presence of all essential biosynthetic modules and regulatory elements [69].
Novelty Assessment: Compare predicted BGCs against known cluster databases (MIBiG) using clusterblast algorithm. Prioritize clusters with <70% similarity to known BGCs for further investigation. Focus on clusters from underrepresented taxonomic groups identified through phylogenetic analysis [69].
Phase 2: Bioinformatic Product Prediction and Synthesis Planning
Chemical Structure Prediction: For nonribosomal peptide synthetase (NRPS) clusters, predict amino acid sequence from adenylation domain specificities using NRPSpredictor2. For polyketide synthases (PKS), predict carbon chain backbone from ketosynthase domain analysis [69].
Product Modification Prediction: Identify genes encoding tailoring enzymes (methyltransferases, oxidases, glycosyltransferases) within cluster boundaries and predict their impact on final product structure [69].
Synthesis Feasibility Assessment: Evaluate predicted structures for chemical synthesis feasibility considering complexity, stereocenters, and unusual building blocks. Prioritize structurally novel compounds with predicted bioactivity based on similarity to known bioactive scaffolds [69].
Phase 3: Chemical Synthesis and Bioactivity Testing
Abiotic Synthesis: Perform total chemical synthesis of predicted natural products using solid-phase peptide synthesis for NRPS-derived compounds or traditional organic synthesis for other scaffold types [69].
Structure Validation: Confirm synthesized compound structure using LC-MS/MS and NMR spectroscopy. Compare experimental data with predicted spectroscopic properties to validate bioinformatic predictions [69].
Bioactivity Screening: Test synthesized compounds against panels of multidrug-resistant bacterial pathogens including MRSA, VRE, and carbapenem-resistant Enterobacteriaceae. Perform secondary assays to identify mode of action for active compounds, including membrane permeability assays, protein binding studies, and genetic approaches [69].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Category | Tool/Reagent | Specific Function | Application Notes |
|---|---|---|---|
| Sequencing Platforms | PacBio Revio | HiFi long-read sequencing (15-18 kb average) | High accuracy (>99.5%), ideal for complete genome assembly [1] |
| ONT PromethION | Nanopore long-read sequencing (20+ kb average) | Lower cost, direct RNA/DNA modification detection [1] | |
| Assembly Tools | Flye | Scalable long-read assembler | Specialized for noisy reads, metagenomic mode available [9] [10] |
| Canu | Adaptive long-read assembler | Corrects errors, trims reads, produces assembled contigs [9] | |
| metaFlye | Metagenome-specific assembler | Handles uneven coverage, species diversity [69] | |
| Gene Prediction | BRAKER3 | Eukaryotic gene prediction | Uses RNA-seq and protein evidence, integrates AUGUSTUS [9] |
| Prokka | Prokaryotic gene annotation | Rapid annotation, integrates multiple tools [9] | |
| Pyrodigal | Metagenomic gene finding | Optimized for fragmented assemblies [22] | |
| Functional Analysis | InterProScan | Protein domain annotation | Integrates multiple databases, provides GO terms [9] |
| antiSMASH | BGC identification | Predicts secondary metabolite clusters [69] | |
| BUSCO | Assembly completeness | Measures gene content completeness against lineage datasets [40] | |
| Experimental Validation | CETSA | Cellular target engagement | Confirms drug-target interaction in intact cells [70] |
| Kallisto | RNA-seq quantification | Fast transcript abundance estimation [40] | |
| MO:BOT platform | Automated 3D cell culture | Standardized organoid screening for compound testing [71] |
Conclusion
The integration of long-read sequencing with advanced gene prediction methodologies represents a transformative advancement in microbial genomics. By leveraging comprehensive bioinformatics platforms, lineage-specific approaches, and robust validation frameworks, researchers can achieve unprecedented accuracy in genome annotation. These developments are already enhancing our understanding of microbial diversity, antibiotic resistance mechanisms, and functional ecology. Future directions will likely involve greater incorporation of artificial intelligence for prediction accuracy, standardized validation protocols across platforms, and expanded applications in personalized medicine and environmental monitoring. The continued refinement of these tools promises to unlock new therapeutic targets and deepen our comprehension of microbial contributions to human health and disease.
References
- 1. Unveiling microbial diversity: harnessing long-read ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11955098/]
- 2. The application of long-read sequencing in clinical settings [https://pmc.ncbi.nlm.nih.gov/articles/PMC10410870/]
- 3. mining for Genome discovery: progress at the front end - PMC drug [https://pmc.ncbi.nlm.nih.gov/articles/PMC8788784/]
- 4. The applications and advantages of nanopore sequencing ... [https://www.nature.com/articles/s44259-025-00157-5]
- 5. Long-read sequencing vs short-read sequencing [https://frontlinegenomics.com/long-read-sequencing-vs-short-read-sequencing/]
- 6. Long-read sequencing myths: debunked. Part 1- HiFi ... [https://www.pacb.com/blog/long-read-sequencing-myths-debunked-part-1-hifi-sequencing/]
- 7. and long-read whole-genome sequencing for microbial ... [https://pubmed.ncbi.nlm.nih.gov/41222256/]
- 8. Genome-resolved long-read sequencing expands known ... [https://www.nature.com/articles/s41564-025-02062-z]
- 9. Long-read microbial genome assembly, gene prediction ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1632189/full]
- 10. Long-read microbial genome assembly, gene prediction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12256462/]
- 11. Ten steps to get started in Genome Assembly and Annotation [https://pmc.ncbi.nlm.nih.gov/articles/PMC5850084/]
- 12. A Hitchhiker's Guide to long-read genomic analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12047252/]
- 13. Microbial dark matter sequences verification in amplicon ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2023.1247119/full]
- 14. Genome-resolved long-read sequencing expands known ... [https://pubmed.ncbi.nlm.nih.gov/40707831/]
- 15. Software and bioinformatics powering long-read sequencing [https://www.pacb.com/blog/sequencing-101-software-and-bioinformatics-powering-long-read-sequencing/]
- 16. Experimental design and quantitative analysis of microbial ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1359-z]
- 17. Quantitatively Partitioning Microbial Genomic Traits among ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6714891/]
- 18. Statistical Analysis Tool [https://img.jgi.doe.gov/docs/statsTool/]
- 19. Evaluation of strategies for the assembly of diverse bacterial ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-5381-7]
- 20. Assess the quality of your genome assembly | How-to Guides [https://australianbiocommons.github.io/how-to-guides/genome_assembly/assembly_qc]
- 21. A fast and flexible quality control and signal summarization ... [https://www.sciencedirect.com/science/article/pii/S2001037025000182]
- 22. Lineage-specific microbial protein prediction enables large ... [https://www.nature.com/articles/s41467-025-58442-w]
- 23. Prokka: rapid prokaryotic genome annotation [https://github.com/tseemann/prokka]
- 24. Gaius-Augustus/BRAKER [https://github.com/Gaius-Augustus/BRAKER]
- 25. Comparison of two annotation tools - Helixer and Braker3 [https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/comparison-braker-helixer-annotation/tutorial.html]
- 26. Comparison of genomic assembly and annotation based ... [https://www.sciencedirect.com/science/article/pii/S0923250825000804]
- 27. Long-read de novo assembly of the spekboom ... [https://www.nature.com/articles/s41597-025-06019-z]
- 28. Functional annotation of protein sequences [https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/functional/tutorial.html]
- 29. Using InterProScan like a pro [https://medium.com/computer-says-no/using-interproscan-like-a-pro-ad18b8c3ccc0]
- 30. Using InterProScan like a pro [https://lotus.au.dk/blog/bioinformatics/2016/11/28/interproscan.html]
- 31. InterProScan Plugin [https://www.geneious.com/plugins/interpro-scan]
- 32. 4.7. Annotation Methods and Functional Enrichment [https://bio-protocol.org/exchange/minidetail?id=7857555&type=30]
- 33. a user-friendly, comprehensive functional annotation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7789693/]
- 34. AMRomics: a scalable workflow to analyze large microbial ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10620-8]
- 35. AMRViz enables seamless genomics analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11100100/]
- 36. AMRomics: a scalable workflow to analyze large microbial ... [https://www.ora.ox.ac.uk/objects/uuid:7a150ca0-cdf6-4093-bb13-5ebe9efdf616]
- 37. amromics/amrviz: Visualization of antimicrobial resistance ... [https://github.com/amromics/amrviz]
- 38. Genetic code flexibility in microorganisms: novel mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4712924/]
- 39. Quantitative measures for the management and comparison of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2653490/]
- 40. Protocol for gene annotation, prediction, and validation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9494284/]
- 41. HPC & Cloud Pipelines for Genomic Data [https://hypersense-software.com/blog/2025/05/07/hpc-cloud-bioinformatics-genomic-data/]
- 42. What is high performance computing (HPC) [https://cloud.google.com/discover/what-is-high-performance-computing]
- 43. HPC Resource Management Strategies for Cost ... [https://www.simops.com/post/hpc-resource-management-strategies-for-cost-optimization-and-performance-maximization]
- 44. The Complexity of Eukaryotic Genomes - The Cell - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK9846/]
- 45. Understanding the causes of errors in eukaryotic protein ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03855-1]
- 46. Sequencing 101: long-read sequencing [https://www.pacb.com/blog/long-read-sequencing/]
- 47. Genomic reproducibility in the bioinformatics era - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11312195/]
- 48. common-workflow-language/cwltool [https://github.com/common-workflow-language/cwltool]
- 49. Workflow management software for pipeline development ... [https://www.biostars.org/p/254257/]
- 50. Installation and configuration — soma-workflow version 3.2 ... [https://brainvisa.info/soma-workflow-3.2/sphinx/install_config.html]
- 51. Programming APIs - Daniel's Blog [https://gridengine.eu/index.php/programming-apis]
- 52. Impact of microbial genome completeness on metagenomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9935889/]
- 53. N50, L50, and related statistics [https://en.wikipedia.org/wiki/N50,_L50,_and_related_statistics]
- 54. N50 statistics - Metagenomics wiki [https://www.metagenomics.wiki/tools/assembly/n50]
- 55. How To Assess The Quality Of An Assembly? (Is There No ... [https://www.biostars.org/p/61843/]
- 56. Update on the proposed minimal standards for the use of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10963913/]
- 57. Short-read sequencing — Knowledge Hub [https://www.genomicseducation.hee.nhs.uk/genotes/knowledge-hub/short-read-sequencing/]
- 58. Error Rate of PacBio vs Nanopore: How Accurate Are Long ... [https://www.cd-genomics.com/blog/pacbio-nanopore-error-rate-correction-strategies/]
- 59. Leveraging long-read sequencing technologies for ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1435416/full]
- 60. Microbial genomics [https://www.pacb.com/microbial-genomics/]
- 61. Finding the right fit: evaluation of short-read and long ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9176275/]
- 62. Joint processing of long- and short-read sequencing data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12296420/]
- 63. Comparison of single-cell long-read and short ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12231600/]
- 64. Genetic factors associated with reasons for clinical trial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11387188/]
- 65. List of AD Loci and Genes with Genetic Evidence Compiled ... [https://adsp.niagads.org/gvc-top-hits-list/]
- 66. A genome-wide cross-trait analysis characterizes the ... [https://www.nature.com/articles/s41467-025-58248-w]
- 67. Gut microbiome metagenomics in clinical practice [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562794/]
- 68. Microbial Experimental Evolution – a proving ground for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6680118/]
- 69. Bioactive molecules unearthed by terabase-scale long- ... [https://www.nature.com/articles/s41587-025-02810-w]
- 70. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 71. Inside ELRIG's Drug Discovery 2025 [https://www.drugtargetreview.com/article/190237/inside-elrigs-drug-discovery-2025/]