CASP Experiment: The Gold Standard in Protein Structure Prediction
This article provides a comprehensive overview of the Critical Assessment of protein Structure Prediction (CASP), the community-wide experiment that has driven progress in computational biology for three decades.
CASP Experiment: The Gold Standard in Protein Structure Prediction
Abstract
This article provides a comprehensive overview of the Critical Assessment of protein Structure Prediction (CASP), the community-wide experiment that has driven progress in computational biology for three decades. Aimed at researchers and drug development professionals, we explore CASP's foundational principles, its evolution in methodology from homology modeling to deep learning, and its role in validating groundbreaking tools like AlphaFold2. The article further details how CASP continues to tackle unsolved challenges in predicting protein complexes, RNA structures, and ligand interactions, while highlighting the real-world application of CASP-validated models in accelerating structural biology and therapeutic discovery.
CASP Unfolded: Understanding the Benchmark for Protein Folding
The Critical Assessment of Structure Prediction (CASP) is a community-wide, worldwide experiment that aims to advance methods of computing three-dimensional protein structure from amino acid sequence. Operating on a two-year cycle since 1994, CASP provides a rigorous framework for the blind testing of structure prediction methods, delivering an independent assessment of the state of the art to the research community and software users. The experiment was established in response to the fundamental challenge in molecular biology known as the "protein folding problem"—predicting a protein's native three-dimensional structure from its one-dimensional amino acid sequence. For decades, this problem stood as a grand challenge in science. CASP's primary goal has been to catalyze progress in solving this problem by objectively testing methods, identifying advances, and highlighting areas for future focus. The organization has become a cornerstone of structural bioinformatics, with more than 100 research groups regularly participating in what many view as the "world championship" of protein structure prediction [1] [2].
Historical Foundation and Key Objectives
The Mission of CASP
The mission of the Protein Structure Prediction Center, which organizes CASP, is to "help advance the methods of identifying protein structure from sequence." The Center facilitates the objective testing of these methods through the process of blind prediction [3]. The core components of this mission are the rigorous blind testing of computational methods and the independent evaluation of the results by assessors who are not participants in the predictions. By establishing the current state of the art, CASP helps identify what progress has been made and where future efforts may be most productively focused [3] [2].
A Brief History of CASP
CASP has been conducted every two years since its inception in 1994 [1]. The following table chronicles key developments over its thirty-year history.
Table 1: Historical Timeline of CASP Experiments
| CASP Round | Year | Key Milestones and Developments |
|---|---|---|
| CASP1 | 1994 | First experiment conducted [1]. |
| CASP4 | 2000 | First reasonable accuracy ab initio model built; residue-residue contact prediction introduced as a category [3] [1]. |
| CASP5 | 2002 | Secondary structure prediction dropped; disordered regions prediction introduced [1]. |
| CASP7 | 2006 | Introduction of model quality assessment and model refinement categories; redefinition of structure prediction categories to Template-Based Modeling (TBM) and Free Modeling (FM) [1]. |
| CASP11 | 2014 | First time a larger new fold protein (256 residues) was built with unprecedented accuracy; data-assisted modeling category included [3] [2]. |
| CASP12 | 2016 | Assembly modeling (complexes) assessed; notable progress from using predicted contacts [3]. |
| CASP13 | 2018 | Substantial improvement in template-free models using deep learning and distance prediction; won by DeepMind's AlphaFold [3] [1]. |
| CASP14 | 2020 | Extraordinary increase in accuracy with AlphaFold2; models competitive with experimental structures for ~2/3 of targets [3] [2] [1]. |
| CASP15 | 2022 | Enormous progress in modeling multimolecular protein complexes; accuracy of oligomeric models almost doubled [3]. |
| CASP16 | 2024 | Planned start in May 2024; includes special interest groups (SIGs) for continuous community engagement [3] [4]. |
In 2023, to foster continuous dialogue between the biennial experiments, CASP established three Special Interest Groups (SIGs): CASP-AI (focusing on artificial intelligence methods), CASP-NA (focusing on nucleic acid structure prediction), and CASP-Ensemble (focusing on conformational ensembles of biomolecules) [4]. These groups hold regular online meetings to discuss recent developments, helping to bridge gaps for newer members and between disciplines [4].
The CASP Experimental Protocol
The CASP experiment is designed as a rigorous double-blind test to ensure a fair assessment. Neither the predictors nor the organizers know the structures of the target proteins at the time predictions are made [1].
Target Selection and Release
Targets for structure prediction are proteins whose experimental structures (solved by X-ray crystallography, cryo-electron microscopy, or NMR spectroscopy) are soon-to-be made public or are currently on hold by the Protein Data Bank [1] [2]. In a typical CASP round (e.g., CASP14), structures of 50-70 proteins and complexes are received from the experimental community and released as prediction targets. For CASP14, these were divided into 68 tertiary structure targets and later organized into 96 evaluation units [2].
Prediction Categories and Evaluation Metrics
Predictors submit their computed structures within a strict timeframe (typically 3 weeks for human groups and 72 hours for automatic servers). The submitted models are then evaluated by independent assessors using a variety of metrics [2] [1].
Table 2: CASP Prediction Categories and Evaluation Methods
| Category | Description | Primary Evaluation Metrics |
|---|---|---|
| Tertiary Structure | Prediction of a single protein chain's 3D structure. | GDT_TS (Global Distance Test - Total Score), LDDT (Local Distance Difference Test) [2] [1]. |
| Template-Based Modeling (TBM) | Modeling using evolutionary-related structures (templates). | GDT_TS, with targets classified as TBM-Easy or TBM-Hard based on difficulty [2] [1]. |
| Free Modeling (FM) | Modeling with no detectable homology to known structures (ab initio). | GDT_TS, with visual assessment for loose resemblances in difficult cases [1] [3]. |
| Assembly Modeling | Prediction of multimolecular protein complexes (quaternary structure). | Interface Contact Score (ICS/F1), LDDT of the interface (LDDTo) [3]. |
| Model Refinement | Improving the accuracy of a starting model. | Change in GDT_TS from the starting model [3]. |
| Contact/Distance Prediction | Predicting spatial proximity of residue pairs. | Precision of top-ranked predictions [3]. |
| Model Quality Assessment | Estimating the accuracy of a protein model. | Correlation between predicted and observed accuracy [1]. |
The GDTTS score is the primary metric for evaluating the backbone accuracy of tertiary structure models. It represents the percentage of well-modeled residues in the model compared to the experimental target structure, with a higher score indicating greater accuracy. A GDTTS above 50 generally indicates the correct fold, while scores above 90 are considered competitive with experimental accuracy [2] [1].
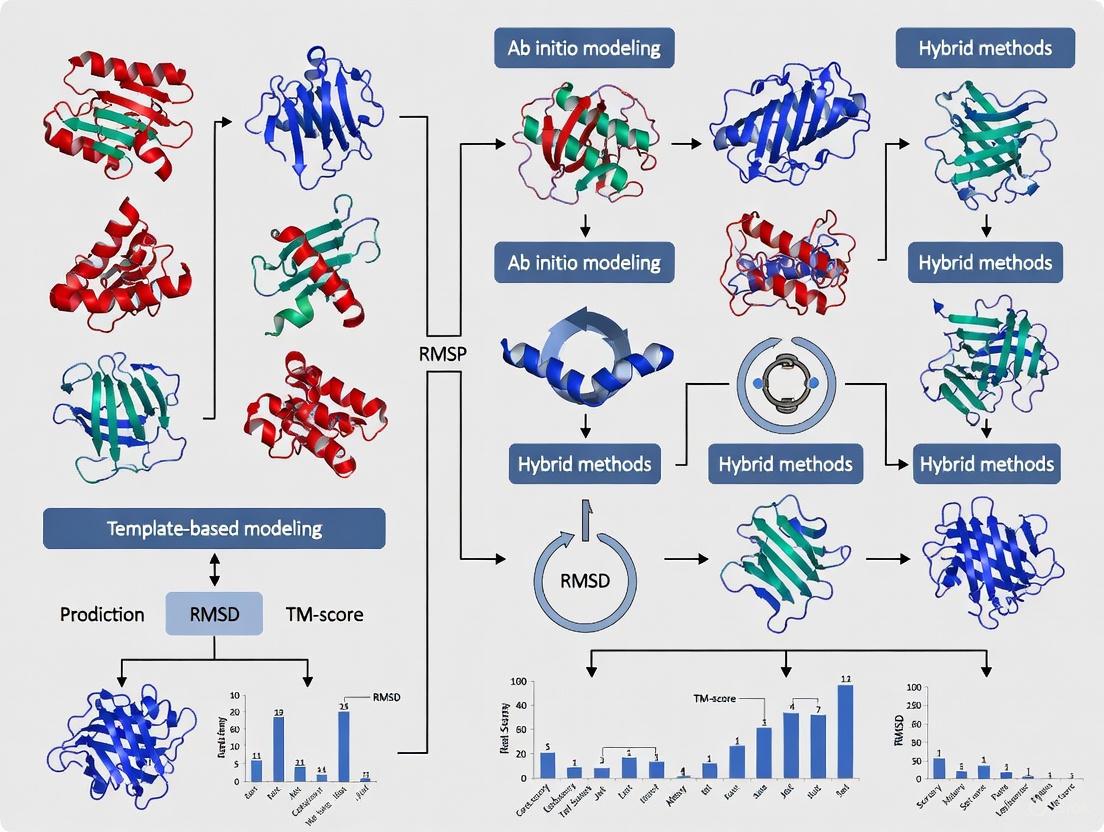
The following diagram illustrates the end-to-end workflow of a CASP experiment.
Quantitative Assessment of Progress
CASP's rigorous evaluation has provided clear, quantitative evidence of the remarkable progress in protein structure prediction, particularly in recent years.
The Leap in Accuracy in CASP14
CASP14 (2020) marked a watershed moment. The advanced deep learning method AlphaFold2, developed by DeepMind, produced models competitive with experimental accuracy for approximately two-thirds of the targets [2]. The trend line for CASP14 starts at a GDT_TS of about 95 for the easiest targets and finishes at about 85 for the most difficult targets. This represents a dramatic improvement over previous years, where accuracy fell off sharply for targets with less available evolutionary information [2].
Table 3: Historical Progress in CASP Backbone Accuracy (GDT_TS)
| CASP Round | Year | Approx. Average GDT_TS for Easy Targets | Approx. Average GDT_TS for Difficult Targets |
|---|---|---|---|
| CASP7 | 2006 | ~75 [3] | Significantly lower |
| CASP12 | 2016 | Information not available in sources | ~81 for a specific small domain (T0866-D1) [3] |
| CASP13 | 2018 | ~80 | ~65 [2] |
| CASP14 | 2020 | ~95 | ~85 [2] |
This leap in performance was not limited to a single group. The performance of the best servers in CASP14 was similar to the best performance of all groups in CASP13, indicating a rapid dissemination of advanced methods through the community [2].
Expansion into Complexes and Ensembles
Following the success in single-chain prediction, CASP has expanded its focus. CASP15 (2022) showed "enormous progress in modeling multimolecular protein complexes," with the accuracy of oligomeric models almost doubling in terms of the Interface Contact Score (ICS) compared to CASP14 [3]. Furthermore, the newly formed CASP-Ensemble SIG is exploring the assessment of conformational ensembles, recognizing that biomolecules adopt dynamic, multi-state structures rather than single static conformations [4].
Essential Research Reagents and Computational Tools
The CASP experiment relies on a suite of computational tools and resources. The following table details key resources that form the foundation of modern structure prediction, as utilized by participants.
Table 4: Key Research Reagent Solutions in Protein Structure Prediction
| Resource / Tool | Type | Primary Function in CASP |
|---|---|---|
| Protein Data Bank (PDB) | Database | Repository of experimentally solved protein structures used as templates and for method training [1]. |
| Multiple Sequence Alignment (MSA) | Data | Collection of evolutionarily related sequences; provides information for deep learning methods on residue co-evolution and constraints [2] [4]. |
| AlphaFold2 & OpenFold | Software | End-to-end deep learning systems that predict protein 3D structure from amino acid sequence and MSA; set new standards in accuracy [2] [4]. |
| Molecular Dynamics (MD) | Software | Computational simulations of physical movements of atoms and molecules; used for model refinement and studying dynamics [4]. |
| Rosetta | Software | A comprehensive software suite for de novo protein structure prediction and design, often used for template-free modeling and refinement [1]. |
| CASP Assessment Metrics (GDT_TS, LDDT) | Algorithm | Standardized metrics for objectively comparing the accuracy of predicted models against experimental structures [1] [2]. |
Over three decades, the Critical Assessment of Structure Prediction has evolved from a small-scale challenge into a large, global community experiment that has fundamentally shaped the field. CASP has provided the objective framework necessary to measure progress, from the early days of comparative modeling to the recent revolution driven by deep learning. The mission to solve the protein folding problem has been largely achieved for single proteins, a conclusion starkly evidenced by the quantitative results from CASP14. The experiment now looks toward new frontiers, including the accurate prediction of multimolecular complexes, conformational ensembles, and the integration of computational models with experimental data to solve ever more challenging biological problems. Through its rigorous, blind assessment protocol and its engaged community, CASP continues to drive innovation, ensuring that computational structure prediction remains a powerful tool for researchers and drug development professionals worldwide.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, biennial experiment that has been the cornerstone of protein structure prediction research since 1994 [3] [1]. Its primary mission is to establish the state of the art in modeling protein structure from amino acid sequence through objective, blind testing of methods [5]. The integrity and scientific value of this massive undertaking—involving over 100 research groups submitting tens of thousands of predictions—rests upon a foundational principle: the double-blind protocol [5] [1]. This rigorous framework ensures that assessments are unbiased, progress is measured authentically, and the results faithfully guide the field's future direction. This paper deconstructs the double-blind methodology that empowers CASP to deliver authoritative evaluations of computational protein structure prediction.
The Mechanics of the Double-Blind Protocol
The double-blind protocol in CASP is a carefully orchestrated process designed to eliminate any possibility of subjective bias or unfair advantage. The "double-blind" nature means that two key parties in the experiment are kept ignorant of critical information until after predictions are submitted.
Target Selection and Anonymity
The process begins with the selection of "target" proteins whose structures have been recently determined experimentally but are not yet publicly available. These targets are typically structures soon-to-be solved by X-ray crystallography, NMR spectroscopy, or cryo-electron microscopy, and are often held on hold by the Protein Data Bank [1]. The critical point is that the amino acid sequences of these targets are provided to predictors without any accompanying structural information [3].
The Two Key "Blinds"
- Predictors are blind to experimental structures: Participating research groups have no access to the experimental structures of the target proteins at the time they are making their predictions [1]. This prevents them from tailoring their methods to a known answer and forces algorithms to rely solely on the amino acid sequence and their underlying principles.
- Assessors are blind to predictor identity: The independent scientists who evaluate the accuracy of the submitted models do so without knowing which research group or method produced any given model [5]. This prevents any conscious or unconscious bias based on a group's reputation or past performance.
The entire workflow, from target release to final assessment, is summarized below.
Quantifying Success: CASP Evaluation Metrics
The objectivity of the double-blind protocol is complemented by rigorous, quantitative evaluation. The primary metric for assessing the backbone accuracy of a predicted model is the Global Distance Test Total Score (GDTTS) [1]. The GDTTS score, measured on a scale of 0 to 100, calculates the percentage of well-modeled residues in a model by measuring the Cα atom positions against the experimental structure [5] [1]. As a rule of thumb, models with a GDT_TS above 50 generally have the correct overall topology, while those above 75 contain many correct atomic-level details [5]. The dramatic progress in CASP, particularly with the advent of deep learning, is unmistakable when viewed through this objective lens.
Table 1: Key Evaluation Metrics in the CASP Experiment
| Metric/Aspect | Description | Significance |
|---|---|---|
| GDT_TS | Global Distance Test Total Score; measures Cα atom positions [1]. | Primary score for backbone accuracy; >50 indicates correct fold, >75 high atomic-level detail [5]. |
| Template-Based Modeling (TBM) | Category for targets with identifiable structural templates [1]. | Assesses ability to leverage evolutionary information from known structures. |
| Free Modeling (FM) | Category for targets with no detectable templates (most challenging) [1]. | Tests true de novo structure prediction capabilities. |
| Interface Contact Score (ICS/F1) | Measures accuracy of interfaces in multimeric complexes [3]. | Critical for evaluating the prediction of protein-protein interactions. |
The Scientist's Toolkit: Essential Research Reagents in CASP
The CASP experiment relies on a suite of "research reagents"—both data and software—that form the essential toolkit for participants and assessors alike.
Table 2: Essential Research Reagents & Resources in CASP
| Resource | Type | Function in the Experiment |
|---|---|---|
| Target Sequences | Data | The fundamental input for predictors; amino acid sequences of soon-to-be-published structures [1]. |
| Protein Data Bank (PDB) | Database | Source of "on-hold" target structures and repository of known structures used for template-based modeling [1]. |
| CASP Prediction Center | Web Infrastructure | Central platform for distributing target sequences and collecting blinded model submissions [3]. |
| GDT_TS Algorithm | Software Tool | The standardized algorithm for quantifying model accuracy, ensuring consistent and comparable evaluation [1]. |
| Multiple Sequence Alignments | Data | Evolutionary information derived from protein families; a critical input for modern deep learning methods [5]. |
The Fruit of Rigor: Key Outcomes Enabled by the Protocol
The strict adherence to the double-blind protocol has allowed CASP to authoritatively document the field's most groundbreaking achievements. The most profound of these was the confirmation in the CASP14 experiment that DeepMind's AlphaFold2 had produced models competitive with experimental accuracy for roughly two-thirds of the targets [2]. This milestone, validated through an unbiased process, represented a solution to the classical protein folding problem for single proteins [2]. The protocol has also reliably captured progress in other complex areas, such as multimeric protein complex prediction (CASP15 showed a near-doubling of accuracy in interface prediction) [3] and the utility of models for aiding experimentalists in solving structures via molecular replacement [3].
Table 3: Documented Progress in CASP Through Blind Assessment
| CASP Edition | Key Documented Advance | Quantified Improvement |
|---|---|---|
| CASP13 (2018) | Emergence of deep learning for contact/distance prediction [5]. | Best model accuracy (GDT_TS) on difficult targets sustained at >60 [5]. |
| CASP14 (2020) | AlphaFold2 demonstrates atomic-level accuracy [2]. | ~2/3 of targets had models competitive with experiment (GDT_TS >90) [2]. |
| CASP15 (2022) | Major progress in modeling multimolecular complexes [3]. | Interface prediction accuracy (ICS) almost doubled compared to CASP14 [3]. |
The double-blind protocol is the engine of credibility for the CASP experiment. By rigorously enforcing anonymity for both predictors and assessors, CASP generates an unbiased, quantitative record of the state of the art in protein structure prediction. This framework has proven its worth by reliably validating every major breakthrough in the field, from the early successes of statistical methods to the recent revolution driven by deep learning. As the field continues to tackle ever more complex challenges—such as the prediction of large multi-protein complexes and the conformational changes underling protein function—the double-blind protocol of CASP will remain the gold standard for objective assessment, ensuring that future progress is measured with the same unwavering rigor.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind experiment held every two years to objectively determine the state of the art in computing three-dimensional protein structures from amino acid sequences [1]. The primary goal of CASP is to advance computational methods by providing rigorous blind testing and independent evaluation [6] [7]. Since its inception in 1994, CASP has served as the gold-standard assessment, creating a unique framework where participants worldwide predict protein structures for sequences whose experimental structures are unknown but soon-to-be-solved [8] [1]. The experiment has witnessed dramatic progress, particularly with the introduction of deep learning methods like AlphaFold, which in recent rounds have demonstrated accuracy competitive with experimental structures for single proteins and have spurred enormous advances in modeling protein complexes [3] [9] [10]. This technical guide delineates the complete lifecycle of a CASP target protein, from its selection as an unsolved biological puzzle to its final role in assessing cutting-edge prediction methodologies.
The CASP Lifecycle: A Stage-by-Stage Breakdown
The lifecycle of a CASP target is a meticulously orchestrated process involving collaboration between experimentalists, organizers, predictors, and assessors. The diagram below illustrates the core workflow and logical relationships between these stages.
Figure 1: The end-to-end workflow of a CASP target protein, from identification by experimentalists to final assessment and publication, highlighting the key stages and responsible parties.
Stage 1: Target Identification and Submission
The lifecycle begins when structural biologists submit prospective targets to the CASP organizers. Target providers are typically X-ray crystallographers, NMR spectroscopists, or cryo-EM scientists who have determined or expect to determine a protein structure whose coordinates are not yet publicly available [7] [1]. The preferred method is direct submission via the Prediction Center web interface, though email submission and designation during PDB submission are also available [9]. The critical requirement is that the experimental data must not be publicly available until after computed structures have been collected to maintain the blind nature of the experiment [9]. For CASP16, the deadline for target submission was July 1, 2024 [7].
Stage 2: Target Release and Predictor Registration
CASP organizers release approved targets through the official CASP website during the "modeling season". For CASP16, this ran from May 1 to July 31, 2024 [7]. Participation is open to all, and research groups must register with the Prediction Center [7]. The targets are announced with their amino acid sequences and sometimes additional information, such as subunit stoichiometry for complexes, which may be released in stages to test methods under different information conditions [7]. The experiment is double-blinded: predictors cannot access the experimental structures, and assessors do not know the identity of those making submissions during evaluation [8].
Stage 3: Sequence Analysis and Feature Generation
Upon receiving a target sequence, predictors conduct in-depth bioinformatic analyses. A crucial first step is the construction of a Multiple Sequence Alignment (MSA) by gathering homologous sequences from genomic databases [8] [10]. For modern deep learning methods, the next step involves generating evolutionary coupling statistics and pairwise features that may indicate which residue pairs are likely to be in spatial proximity [11] [10]. Advanced methods like AlphaFold's Evoformer block then process these MSAs and residue-pair representations through repeated layers of a novel neural network architecture to create an information-rich foundation for structure prediction [10].
Stage 4: 3D Structure Prediction
This core stage involves translating the processed sequence information into atomic coordinates. The following table summarizes the primary methodologies employed for different prediction categories in CASP.
Table 1: Key Protein Structure Prediction Methodologies Assessed in CASP
| Method Category | Core Principle | Typical Applications | Key Innovations (Examples) |
|---|---|---|---|
| Template-Based Modeling (TBM) | Identifies structural templates (homologous proteins of known structure) and builds models through sequence alignment and comparative modeling [3] [1]. | Proteins with detectable sequence or structural similarity to known folds. | More accurate alignment; combining multiple templates; improved regions not covered by templates [3]. |
| Free Modeling (FM) / Ab Initio | Predicts structure without detectable homologous templates, using physical principles or statistical patterns [3] [1]. | Proteins with novel folds or no detectable homology. | Accurate 3D contact prediction using co-evolutionary analysis and deep learning [3] [11]. |
| Deep Learning (e.g., AlphaFold) | Uses neural networks trained on known structures and sequences to directly predict atomic coordinates from MSAs and pairwise features [8] [10]. | All target types, with particularly high accuracy for single domains [6] [10]. | Evoformer architecture; end-to-end differentible learning; iterative refinement ("recycling") [10]. |
Stage 5: Model Submission
Predictors submit their final 3D structure models in a specified format through the Prediction Submission form or by email [7]. Each model contains the predicted 3D coordinates of all or most atoms for the target protein. For CASP16, approximately 100 research groups submitted more than 80,000 models for over 100 modeling entities, illustrating the massive scale of the experiment [7]. Server predictions are made publicly available shortly after the prediction window for a specific target closes, fostering a collaborative and transparent environment [7].
Stage 6: Experimental Structure Determination
In parallel with the prediction season, the target providers finalize their experimental structures. CASP requires the experimental data by August 15 for assessment, though the data can remain confidential until after the evaluation period [9]. These experimentally determined structures, solved through techniques like X-ray crystallography, NMR, or cryo-EM, serve as the ground truth or "gold standard" against which all computational models are rigorously evaluated [6] [1].
Stage 7: Blind Assessment and Evaluation
Independent assessors, who are expert scientists not involved in the predictions, compare the submitted models with the experimental structures. The assessment employs quantitative metrics and qualitative analysis, with the specific criteria varying by prediction category.
Table 2: Key Quantitative Metrics for Evaluating CASP Predictions
| Evaluation Metric | What It Measures | Interpretation | Primary Application |
|---|---|---|---|
| GDT_TS (Global Distance Test Total Score) | The average percentage of Cα atoms in the model that can be superimposed on the native structure under multiple distance thresholds (1, 2, 4, and 8 Å) [8]. | 0-100 scale; higher scores indicate better overall fold accuracy. A score >~90 is considered competitive with experimental accuracy [6] [3]. | Single protein and domain structures [3]. |
| GDT_HA (High Accuracy) | Similar to GDT_TS but uses more stringent distance thresholds (0.5, 1, 2, and 4 Å) [1]. | Measures high-quality structural agreement, particularly for well-predicted regions. | High-accuracy template-based models [1]. |
| lDDT (local Distance Difference Test) | A local, superposition-free score that evaluates the local consistency of distances in the model compared to the native structure [10]. | More robust to domain movements than global scores. Reported as pLDDT (predicted lDDT) by AlphaFold as an internal confidence measure [10]. | Local model quality and accuracy estimation. |
| ICS (Interface Contact Score) / F1 | For complexes, measures the accuracy of residue-residue contacts at the subunit interface [3]. | 0-1 scale; higher scores indicate more accurate protein-protein interaction interfaces. | Protein complexes and assemblies [3]. |
| RMSD (Root Mean Square Deviation) | The average distance between equivalent atoms (e.g., Cα atoms) after optimal superposition [10]. | Measured in Ångströms (Å); lower values indicate better atomic-level accuracy. | Overall and local atomic accuracy. |
Stage 8: Results Publication and Knowledge Integration
The CASP lifecycle concludes with the public dissemination of results. All predictions and numerical evaluations are made available through the Prediction Center website [7]. A conference is held to discuss the results (for CASP16, tentatively scheduled for December 1-4, 2024) [7]. Finally, the proceedings, including detailed assessments, methods descriptions, and analyses of progress, are published in a special issue of the journal PROTEINS: Structure, Function, and Bioinformatics [6] [7]. This completes the cycle, transforming a single target protein from a private sequence into a public benchmark that advances the entire field.
Successful navigation of the CASP lifecycle relies on a suite of computational and data resources.
Table 3: Essential Research Reagents and Resources for CASP
| Resource/Solution | Type | Primary Function in CASP |
|---|---|---|
| Protein Data Bank (PDB) | Data Repository | The single worldwide archive of structural data of biological macromolecules; provides the foundational training data for knowledge-based methods and stores the final experimental targets [8]. |
| Multiple Sequence Alignment (MSA) Tools | Computational Tool | Generates alignments of homologous sequences from genomic databases; essential for extracting evolutionary constraints and co-evolutionary signals for contact prediction [8] [10]. |
| AlphaFold & Related DL Models | Software/Algorithm | Deep learning systems that directly predict 3D atomic coordinates from amino acid sequences and MSAs; represent the current state-of-the-art in accuracy [6] [10]. |
| Molecular Dynamics Software | Computational Tool | Uses physics-based simulations for model refinement; can slightly improve initial models by sampling conformational space near the starting structure [11] [8]. |
| CASP Prediction Center | Web Infrastructure | The central hub for the experiment: distributes target sequences, collects submitted models, provides evaluation tools, and disseminates results [3] [7]. |
The lifecycle of a CASP target protein embodies a unique and powerful collaborative model in scientific research. From its genesis in an experimental lab to its role as a blind test for computational methods and its final contribution to published literature, each target plays a crucial part in driving the field forward. The rigorous, community-wide assessment provided by CASP has been instrumental in benchmarking progress, most notably catalyzing the revolutionary advances brought by deep learning. As the experiment continues to evolve, incorporating new challenges like protein-ligand complexes, RNA structures, and conformational ensembles, the structured lifecycle of a CASP target will remain fundamental to transforming amino acid sequences into biologically meaningful three-dimensional structures.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind experiment conducted every two years since 1994 to objectively assess the state of the art in computing protein three-dimensional structure from amino acid sequence [1]. This rigorous experiment provides a framework for testing protein structure prediction methods through blind testing, where predictors calculate structures for proteins whose experimental configurations are not yet public [3] [12]. A fundamental requirement of this assessment is objective, quantitative metrics to evaluate the accuracy of predicted models against experimentally determined reference structures. The Global Distance Test Total Score (GDT_TS) has emerged as the primary metric for this evaluation, serving as the gold standard for comparing predicted and experimental structures in CASP and beyond [13].
Understanding GDT_TS: Calculation and Interpretation
Fundamental Principles and Calculation Methodology
The Global Distance Test (GDT) was developed to provide a more robust measure of protein structure similarity than Root-Mean-Square Deviation (RMSD), which is sensitive to outlier regions caused by poor modeling of individual loops in an otherwise accurate structure [13]. The conventional GDT_TS score is computed over the alpha carbon atoms and is reported as a percentage ranging from 0 to 100, with higher values indicating closer approximation to the reference structure [13].
The GDT algorithm calculates the largest set of amino acid residues' alpha carbon atoms in the model structure that fall within a defined distance cutoff of their position in the experimental structure after iteratively superimposing the two structures [13]. The algorithm was originally designed to calculate scores across 20 consecutive distance cutoffs from 0.5 Å to 10.0 Å [13]. However, the standard GDT_TS used in CASP assessment is the average of the maximum percentage of residues that can be superimposed under four specific distance thresholds: 1, 2, 4, and 8 Ångströms [13].
Table 1: GDT_TS Distance Cutoffs and Their Implications
| Distance Cutoff (Å) | Structural Interpretation | Typical Accuracy Level |
|---|---|---|
| 1 Å | Very high atomic-level accuracy | Near-experimental quality |
| 2 Å | High backbone accuracy | Competitive with experiment |
| 4 Å | Correct fold determination | Structurally useful model |
| 8 Å | Overall topological similarity | Basic fold recognition |
Variations and Extended GDT Metrics
Over successive CASP experiments, the GDT framework has evolved to include specialized variants addressing specific assessment needs:
- GDT_HA (High Accuracy): Uses smaller cutoff distances (typically 0.5, 1, 2, and 4 Å) to more heavily penalize larger deviations, emphasizing high-precision modeling [13]. This metric was introduced for the high-accuracy category in CASP7 [13].
- GDC_SC (Global Distance Calculation for Sidechains): Extends the assessment to side chain atoms using predefined "characteristic atoms" near the end of each residue [13].
- GDC_ALL (All-Atom GDC): Incorporates full-model information, evaluating the accuracy of all atoms rather than just the protein backbone [13].
GDT_TS in Practice: CASP Assessment and Performance Benchmarks
The CASP Assessment Framework
In CASP experiments, protein structures soon to be solved by X-ray crystallography, NMR spectroscopy, or cryo-electron microscopy are selected as targets [1]. Predictors submit their models based solely on amino acid sequences, and these predictions are subsequently compared to the experimental structures when they become publicly available [14]. The evaluation occurs across multiple categories, with tertiary structure prediction being a core component throughout all CASP experiments [1].
Target structures are classified into difficulty categories based on their similarity to known structures: Template-Based Modeling Easy (TBM-Easy), TBM-Hard, Free Modeling/TBM (FM/TBM), and Free Modeling (FM) for the most challenging targets with no detectable homology [2]. Historically, model accuracy strongly correlated with these categories, but recent advances have substantially reduced this dependence [2].
Performance Benchmarks and Historical Progress
GDTTS has been instrumental in quantifying the remarkable progress in protein structure prediction, particularly the breakthroughs demonstrated in recent CASP experiments. According to CASP assessments, a GDTTS score of approximately 90 is informally considered competitive with experimental methods [14].
Table 2: CASP Performance Benchmarks and GDT_TS Interpretation
| GDT_TS Score Range | Interpretation | CASP Benchmark |
|---|---|---|
| 90-100 | Competitive with experimental accuracy | AlphaFold2 CASP14 median: 92.4 GDT_TS [14] |
| 80-90 | High accuracy | CASP14 best models for difficult targets [2] |
| 60-80 | Correct fold with structural utility | CASP13 best performance for difficult targets [2] |
| <50 | Incorrect or largely inaccurate fold | Pre-deep learning era for difficult targets [2] |
The CASP14 experiment in 2020 marked a paradigm shift, with AlphaFold2 achieving a median GDTTS of 92.4 overall across all targets, with an average error of approximately 1.6 Ångströms [10] [14]. This performance was competitive with experimental structures for about two-thirds of the targets [2]. Surprisingly, the best model trend line in CASP14 started at a GDTTS of about 95 and finished at about 85 for the most difficult targets, demonstrating only a minor fall-off in accuracy despite decreasing evolutionary information [2].
Methodological Protocols: Calculating and Applying GDT_TS
Standard GDT_TS Calculation Protocol
The technical implementation of GDT_TS calculation follows a specific methodology:
- Input Preparation: The predicted model and experimental reference structure are prepared with identical amino acid sequences and residue numbering.
- Residue Correspondence Establishment: The algorithm establishes residue correspondence, typically assuming identical sequences for CASP targets.
- Iterative Superposition: The structures undergo iterative superposition to identify optimal alignment.
- Distance Calculation: For each residue, the Cα distance between the model and reference is calculated after superposition.
- Residue Counting at Cutoffs: The algorithm identifies the largest set of residues that can be superimposed under each distance cutoff (1, 2, 4, and 8 Å).
- Averaging: The final GDT_TS is computed as the average of these four percentages.
The following workflow visualizes this calculation process:
Accounting for Uncertainty in GDT_TS Measurements
Protein structures are not static entities but exist as ensembles of conformational states, introducing uncertainty in atomic positions that affects GDTTS measurements [15]. Research has demonstrated that the uncertainty of GDTTS scores, quantified by their standard deviations, increases for lower scores, with maximum standard deviations of 0.3 for X-ray structures and 1.23 for NMR structures [15]. This uncertainty arises from:
- Structural flexibility inherent in protein molecules
- Experimental limitations in structure determination methods
- Dynamic properties of target proteins
For high-accuracy models (GDT_TS > 70), the uncertainty is relatively small, but becomes more significant for lower-quality models [15]. Time-averaged refinement techniques for X-ray structures and ensemble approaches for NMR structures help quantify this uncertainty [15].
Table 3: Key Research Resources for Protein Structure Prediction and Validation
| Resource/Reagent | Type | Function and Application |
|---|---|---|
| LGA (Local-Global Alignment) | Software Algorithm | Primary tool for GDT_TS calculation and structure comparison [13] |
| Protein Data Bank (PDB) | Database | Repository of experimental protein structures used for template-based modeling and method training [10] |
| AlphaFold | Prediction Method | Deep learning system that demonstrated GDT_TS scores competitive with experiment in CASP14 [10] [14] |
| CASP Assessment Data | Benchmark Dataset | Curated targets and predictions from past experiments for method development and validation [3] |
| Multiple Sequence Alignments (MSAs) | Bioinformatics Data | Evolutionary information used as input for modern deep learning prediction methods [10] |
The GDTTS metric has proven indispensable for quantifying progress in protein structure prediction, particularly through the CASP experiments. As the field advances with deep learning methods like AlphaFold2 routinely producing models with GDTTS scores above 90 [10] [14], the role of GDTTS is evolving. While it remains crucial for assessing backbone accuracy, the focus is expanding to include all-atom accuracy, complex assembly prediction, and accuracy estimation [12] [7]. The continued development and refinement of assessment metrics like GDTTS and its variants will ensure rigorous evaluation of the next generation of structure prediction methods, furthering their application in drug discovery and basic biological research [16].
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind experiment established in 1994 to advance methods for computing three-dimensional protein structure from amino acid sequence [17] [3]. CASP operates as a rigorous testing ground where research groups worldwide predict protein structures that have been experimentally determined but not yet publicly released [6] [17]. By evaluating predictions against the experimental benchmarks, independent assessors establish the current state of the art, identify progress, and highlight areas for future focus [6] [3]. This experiment is foundational to structural biology because protein function is dictated by its 3D structure, and accurate prediction is crucial for understanding biological processes and accelerating drug development [17].
The Evolution of CASP Assessment Categories
The CASP experiment has systematically evolved its assessment categories to track progress across the diverse challenges in protein structure modeling. The core categories have matured in response to methodological breakthroughs.
Template-Based Modeling (TBM)
Template-Based Modeling assesses methods that build protein models using structures of related proteins as templates [3]. For over a decade, progress in this category was incremental, but CASP12 (2016) marked a significant acceleration in accuracy due to improved sequence-template alignment, multiple template combination, and better model refinement [3]. The emergence of deep learning in CASP14 created another step-change, with models achieving near-experimental accuracy (GDT_TS>90) for approximately two-thirds of targets [3].
Free Modeling (FM) /Ab InitioModeling
Free Modeling (originally called ab initio modeling) represents the most challenging task: predicting structures without identifiable templates from existing databases [3]. Early progress was limited to small proteins (~120 residues). CASP11 and CASP12 showed substantial improvements through the successful use of predicted contacts as constraints [3]. CASP13 registered another leap forward through deep learning techniques predicting inter-residue distances [3]. By CASP14, methods like AlphaFold2 produced models with backbone accuracy competitive with experiments for many targets, effectively solving aspects of the classical protein-folding problem for single domains [6] [17] [3].
Quaternary Structure (Assembly) Modeling
Assembly Modeling (assessment of multimolecular protein complexes) was introduced in CASP12 [3]. CASP15 (2022) demonstrated enormous progress, with accuracy nearly doubling in terms of Interface Contact Score (ICS) compared to CASP14 [3]. Deep learning methods originally developed for monomeric proteins were successfully extended to model oligomeric complexes, significantly outperforming earlier methods [6] [3].
Refinement
The Refinement category tests the ability of methods to improve model accuracy by correcting structural deviations from experimental reference structures [3]. CASP assessments have identified two methodological trends: molecular dynamics methods that provide consistent but modest improvements, and more aggressive methods that can achieve substantial refinement but with less consistency [3].
Contact Prediction
Contact Prediction evaluates the accuracy of predicting spatially proximate residue pairs in the native structure [3]. This category witnessed sustained improvement from CASP11 to CASP13, where precision jumped from 27% to 70% for the best-performing methods [3]. These advances directly contributed to improved accuracy in free modeling by providing strong constraints for 3D model construction [3].
Data-Assisted Modeling
Data-Assisted Modeling involves predicting structures using low-resolution experimental data (NMR, cross-linking, cryo-EM, etc.) combined with computational methods [3]. This hybrid approach has shown promise in improving model accuracy, as demonstrated in CASP12 where cross-linking assisted models showed significant improvement over non-assisted predictions [3].
Table: Key CASP Assessment Categories and Their Evolution
| Category | Primary Focus | Key Evolutionary Milestones |
|---|---|---|
| Template-Based Modeling (TBM) | Building models using known protein structures as templates | CASP12 (2016): Significant accuracy improvements through better alignment and template combination [3]CASP14 (2020): Deep learning methods (e.g., AlphaFold2) achieved near-experimental accuracy [3] |
| Free Modeling (FM) | Predicting structures without homologous templates (ab initio) | CASP11-12: Improved accuracy using predicted contacts as constraints [3]CASP13: Major leap from deep learning and distance prediction [3]CASP14: AlphaFold2 produced models competitive with experiment [6] [3] |
| Quaternary Structure (Assembly) | Modeling multimolecular protein complexes | CASP12: Category introduced [3]CASP15: Accuracy dramatically improved through extended deep learning methods [3] |
| Refinement | Improving model accuracy towards experimental structures | CASP10-14: Identification of consistent molecular dynamics methods and powerful but less consistent aggressive methods [3] |
| Contact Prediction | Predicting spatially proximate residue pairs | CASP11-13: Precision nearly tripled from 27% to 70% [3] |
| Data-Assisted Modeling | Combining computational methods with experimental data | CASP11-13: Demonstrated significant accuracy improvements when integrating experimental constraints [3] |
Quantitative Tracking of Progress in CASP
CASP employs rigorous quantitative metrics to evaluate prediction accuracy, allowing objective tracking of methodological progress across experiments. The Global Distance Test (GDT_TS) is a primary metric measuring the average percentage of Cα atoms in a model that fall within a threshold distance of their correct positions in the experimental structure after optimal superposition [3]. For assembly prediction, the Interface Contact Score (ICS or F1) measures accuracy in modeling residue-residue contacts across protein interfaces [3].
Table: Quantitative Progress Across CASP Experiments (2006-2022)
| CASP Experiment | Template-Based Modeling (Avg. GDT_TS) | Free Modeling (Avg. GDT_TS) | Contact Prediction (Top Precision %) | Notable Methodological Advances |
|---|---|---|---|---|
| CASP7 (2006) | ~70-80 (est.) | ~40-50 (est.) | <10% (est.) | First reasonable ab initio models for small proteins [3] |
| CASP11 (2014) | ~75-85 (est.) | ~50-60 (est.) | 27% | Baker team ranked first; deep learning introduced for structure prediction [17] [3] |
| CASP12 (2016) | Significant improvement over CASP11 | Improved via predicted contacts | 47% | Burst of progress in TBM; contact prediction precision nearly doubled [3] |
| CASP13 (2018) | Continued improvement | 65.7 (from 52.9 in CASP12) | 70% | AlphaFold1 debut; substantial improvement in FM via deep learning and distance prediction [17] [3] |
| CASP14 (2020) | ~92 (average) | ~85 for difficult targets | No significant increase | AlphaFold2; models competitive with experiment for ~2/3 of targets [6] [3] |
| CASP15 (2022) | High accuracy maintained | High accuracy maintained | Data not shown | Assembly modeling accuracy nearly doubled (ICS) [3] |
Methodological Breakthroughs and Experimental Protocols
The extraordinary progress tracked by CASP has been driven by fundamental methodological breakthroughs, particularly the integration of deep learning and evolutionary information.
The Deep Learning Revolution
The transformation in protein structure prediction is exemplified by the evolution of AlphaFold. AlphaFold1 (CASP13) used convolutional neural networks (CNNs) to analyze 2D maps of distances between amino acids, predicting inter-residue distances and optimizing structures using gradient descent [17]. AlphaFold2 (CASP14) implemented a radically different architecture that moved beyond predetermined distance constraints to directly process sequence information including multiple sequence alignments (MSA) and pair representations [17]. Its core innovation was the Evoformer module—a modified Transformer algorithm that uses attention mechanisms to learn complex relationships directly from amino acid sequences [17].
AlphaFold2's High-Level Workflow
Experimental Protocol in CASP
The CASP experimental protocol follows a rigorous blind assessment paradigm:
- Target Selection: Organizers provide amino acid sequences of proteins whose structures have recently been experimentally determined but not published [17].
- Prediction Phase: Participant groups submit their three-dimensional structure predictions using only sequence information [3].
- Assessment: Independent assessors evaluate predictions against experimental structures using standardized metrics like GDT_TS and ICS, without knowledge of which groups generated which models [6] [3].
- Results Publication: Comprehensive analysis is published in special issues of Proteins: Structure, Function, and Bioinformatics [6] [3].
The Scientist's Toolkit: Essential Research Reagents
Table: Key Research Reagents and Resources in Protein Structure Prediction
| Resource/Reagent | Type | Function in Protein Structure Prediction |
|---|---|---|
| Protein Data Bank (PDB) | Database | Primary repository of experimentally determined protein structures used for method training and validation [17] |
| Multiple Sequence Alignments (MSA) | Data | Collections of evolutionarily related sequences used to infer structural constraints and co-evolutionary patterns [17] |
| AlphaFold2 | Software | End-to-end deep learning system that predicts 3D structures from amino acid sequences with high accuracy [17] |
| Evoformer | Algorithm | Transformer-based architecture that processes MSA and pair representations to learn structural relationships [17] |
| CASP Targets | Dataset | Blind test cases with experimentally solved structures but unreleased coordinates, used for objective assessment [3] |
| Molecular Dynamics Software | Software | Simulates physical movements of atoms and molecules, used for structure refinement [3] |
CASP's evolving assessment categories have systematically tracked the field's transformation from modest template-based modeling to the accurate ab initio prediction of single proteins and complex multimolecular assemblies. The quantitative progress documented in CASP demonstrates that AI methods, particularly deep learning, have fundamentally changed structural biology [6] [17]. These advances have immediate practical applications, with CASP14 models already being used to solve problematic crystal structures and correct experimental errors [6] [3]. For drug development professionals, these breakthroughs enable rapid structural characterization of therapeutic targets, potentially accelerating drug discovery pipelines. As CASP continues to evolve its assessment categories to address more complex challenges like protein design and functional prediction, it will continue to serve as the essential benchmark for tracking progress in computational structural biology.
From Templates to AI: The Methodological Revolution in CASP
The Critical Assessment of protein Structure Prediction (CASP) is a biennial, community-wide blind experiment established in 1994 to objectively assess the state of the art in predicting protein three-dimensional structure from amino acid sequence [1]. It functions as a rigorous testing ground where predictors worldwide submit models for proteins whose structures have been experimentally determined but are not yet public. Independent assessors then evaluate these submissions, providing an unbiased overview of methodological capabilities and progress [5]. Within this framework, Template-Based Modeling (TBM) has historically been the most reliable method for predicting protein structures when a related protein of known structure (a "template") can be identified [1] [3]. TBM leverages the evolutionary principle that structural homology is more conserved than sequence homology, allowing for the construction of accurate models even with low sequence similarity. This guide details the core methodologies, experimental validation, and practical applications of TBM within the context of CASP, providing researchers and drug development professionals with a technical overview of this foundational approach.
The CASP Experimental Framework for TBM
The Double-Blind Protocol
A cornerstone of the CASP experiment is its double-blind protocol, which ensures an objective assessment. Predictors receive only the amino acid sequences of the target proteins and have no access to the experimental structures during the prediction phase. Simultaneously, the assessors evaluate the submitted models without knowing the identity of the predictors [1] [5]. This eliminates bias and guarantees that the assessment purely reflects the predictive power of the computational methods.
Target Difficulty Categorization
CASP classifies targets based on their similarity to known structures in the Protein Data Bank (PDB), which directly dictates the applicability of TBM. The official CASP classification is as follows [2]:
- TBM-Easy: Targets for which straightforward homology modeling is possible, typically with clear and easily identifiable templates.
- TBM-Hard: Targets where homology modeling is still the primary approach, but the structural homology is more remote or difficult to detect, often requiring advanced sequence alignment or threading techniques [1]. It is critical to note that the distinction between TBM and the more challenging "Free Modeling" (FM) category has become increasingly blurred with the advent of deep learning methods like AlphaFold, which integrate principles from both categories to achieve remarkable accuracy [2].
Core Methodology of Template-Based Modeling
The TBM workflow is a multi-stage process that transforms a target sequence and a template structure into a refined 3D model. The following diagram illustrates the key steps and their logical relationships.
Step 1: Template Identification and Selection
The initial and crucial step is to identify one or more experimentally solved protein structures (templates) that are homologous to the target sequence.
- Experimental Protocols:
- Sequence-Based Search: Tools like BLAST or HHsearch are used to search the PDB for sequences with significant similarity to the target. A high sequence identity often indicates a suitable template [1].
- Protein Threading: For distantly related homologs where sequence identity is low, protein threading methods can be more effective. These methods score the compatibility of the target sequence with the structural folds of potential templates in the library, even in the absence of clear sequence similarity [1].
- Selection Criteria: The ideal template is selected based on factors including sequence identity, the quality and resolution of the experimental template structure, and coverage of the target sequence.
Step 2: Target-Template Alignment
A precise alignment between the target amino acid sequence and the sequence (and structure) of the template is generated. This alignment defines how the coordinates of the template will be transferred to the target.
- Experimental Protocols: This step can use the same tools as template identification (e.g., HHsearch). Advanced methods may employ iterative algorithms and multiple sequence alignment information to improve the accuracy of the alignment, especially in low-identity scenarios.
Step 3: Model Building
The core framework of the model is constructed by transferring the backbone coordinates from the template to the target based on the sequence alignment.
- Experimental Protocols:
- Copying Conserved Regions: Amino acids that are identical between the target and template can have their coordinates directly copied.
- Handling Variations: For aligned but non-identical residues, side-chain conformations are modeled. In regions where the target has an insertion not present in the template (a "gap" in the alignment), the structure must be built from scratch.
Step 4: Loop Modeling
Regions corresponding to gaps in the target-template alignment, typically loops, are the most variable and difficult to model. Specialized methods are required for this step.
- Experimental Protocols:
- Knowledge-Based Methods: Searching a database of protein loops for fragments that fit the geometric constraints of the "stem" regions.
- De Novo Methods: Using conformational sampling algorithms, such as those implemented in Rosetta, to generate and score possible loop conformations [1].
Step 5: Side-Chain Refinement
The conformations of side chains, even in well-aligned regions, are optimized to remove steric clashes and find energetically favorable rotamers.
- Experimental Protocols: This involves using rotamer libraries—statistical distributions of side-chain dihedral angles observed in high-resolution structures—and employing energy minimization algorithms to select the most stable conformations.
Step 6: Model Validation and Quality Assessment
The final model must be checked for structural integrity and reliability.
- Experimental Protocols:
- Geometric Checks: Tools like MolProbity assess stereochemical quality, including bond lengths, angles, and Ramachandran plot outliers.
- Physical Plausibility: Checking for atomic clashes, solvation energy, and other physicochemical properties.
- Model Quality Estimation: Methods like ModFOLD or ProQ2 estimate the local and global accuracy of the model, which is a dedicated assessment category in CASP [1].
Quantitative Assessment in CASP
Primary Metrics for Model Accuracy
The CASP assessment uses robust metrics to quantitatively compare predicted models against the experimental target structure. The primary metric for the backbone is the Global Distance Test (GDT). The most common variant is the GDTTS (Total Score), which represents the average of four values: GDT1, GDT2, GDT4, and GDT8. These correspond to the percentage of Cα atoms in the model that can be superimposed on the corresponding atoms in the experimental structure under different distance thresholds (1, 2, 4, and 8 Ångströms) [1] [2]. A higher GDTTS indicates a more accurate model.
Table 1: Key Metrics for Evaluating TBM Models in CASP
| Metric | Definition | Interpretation |
|---|---|---|
| GDT_TS | Average percentage of Cα atoms within 1, 2, 4, and 8 Å of their correct positions after optimal superposition. | Primary measure of overall backbone accuracy. >90: Competitive with experiment. >80: High accuracy. >50: Generally correct fold [3] [2]. |
| GDT_HA | Same as GDT_TS but uses tighter distance thresholds (0.5, 1, 2, and 4 Å). | Measures "High-Accuracy" details, assessing atomic-level precision. |
| RMSD | Root Mean Square Deviation of atomic positions (typically Cα atoms) between model and target. | Measures average deviation. Sensitive to local errors; less informative for global fold. |
| MolProbity Score | Comprehensive evaluation of stereochemistry, clashes, and rotamer outliers. | Validates the geometric and physical plausibility of the model. |
Documented Performance and Historical Progress
TBM has shown consistent and dramatic improvements over the history of CASP, driven by better algorithms, template libraries, and the integration of deep learning.
Table 2: Historical Progress of TBM Accuracy in CASP (Data compiled from CASP reports)
| CASP Round | Key Trends and Average Performance Highlights |
|---|---|
| CASP10 (2012) | Baseline for a decade of progress. Models were accurate but with room for improvement. |
| CASP12 (2016) | A "burst of progress": backbone accuracy improved more in 2014-2016 than in the preceding 10 years [3]. |
| CASP13 (2018) | Significant improvement driven by the integration of deep learning for contact/distance prediction, even for TBM targets [5]. |
| CASP14 (2020) | "Extraordinary increase" in accuracy. AlphaFold2 models for TBM targets reached an average GDT_TS of ~92, significantly surpassing models from simple template transcription [3]. |
The data shows that modern TBM methods, particularly those enhanced by deep learning, have moved beyond simple template transcription. They now produce models that are significantly more accurate than the best available templates, achieving near-experimental accuracy for the majority of targets [3] [2].
The Scientist's Toolkit: Essential Reagents for TBM
Table 3: Key Resources for Template-Based Modeling
| Resource / Tool | Type | Primary Function in TBM |
|---|---|---|
| PDB (Protein Data Bank) | Database | The central repository of experimentally determined protein structures, serving as the source for all potential templates [1]. |
| BLAST | Software | Performs rapid sequence similarity searches to identify potential homologous templates in the PDB [1]. |
| HHsearch/HHblits | Software | Employs hidden Markov models (HMMs) for sensitive profile-based sequence searches and alignments, crucial for finding distant homologs [1] [5]. |
| Rosetta | Software Suite | Provides powerful algorithms for de novo loop modeling, side-chain packing, and overall structural refinement, especially when template coverage is incomplete [1]. |
| Modeller | Software | A widely used package for comparative (homology) model building, which spatial restraints derived from the template to construct the target model. |
| MolProbity | Software | A structure-validation tool that checks the stereochemical quality of the built model, identifying clashes, rotamer outliers, and Ramachandran deviations. |
| AlphaFold2 | Software | A deep learning system whose architecture and training have revolutionized the field. While a full Free Modeling tool, its principles are now integrated into modern TBM pipelines, and its public models can serve as highly accurate starting templates [18]. |
Template-Based Modeling, rigorously tested and refined within the CASP experiment, remains a cornerstone of computational structural biology. The methodology has evolved from simple homology modeling to a sophisticated process that, when augmented by modern deep learning techniques, can produce models of near-experimental quality. The quantitative assessments from CASP unequivocally demonstrate this dramatic progress, with GDT_TS scores for TBM targets now consistently exceeding 90 for a majority of targets [3] [2]. For researchers in drug discovery, this level of accuracy makes TBM an indispensable tool for tasks ranging from understanding protein function and elucidating mechanisms of disease to structure-based drug design and virtual screening. The continued integration of experimental data (e.g., from cryo-EM, NMR, or cross-linking mass spectrometry) into the modeling process promises to further enhance the reliability and scope of TBM, solidifying its role as a critical technology for advancing human health.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, biennial experiment that serves as the gold standard for objectively testing computational methods that predict protein three-dimensional structure from amino acid sequence [1]. Since its inception in 1994, CASP has categorized the protein folding problem into distinct challenges, one of the most difficult being Template-Free Modeling (FM), also known as ab initio or de novo prediction [19] [1]. This category specifically addresses the prediction of protein structures that possess novel folds—those with no detectable structural homology to any known template in the Protein Data Bank (PDB) [20] [2]. For researchers and drug development professionals, the ability to accurately model these novel folds is paramount for understanding the structure and function of proteins unique to pathogens or disease processes, where no prior structural information exists. The evolution of FM methodologies within the CASP framework, from early physical models to the recent revolution in deep learning, represents one of the most significant frontiers in computational structural biology.
The Theoretical Divide: Template-Based vs. Template-Free Modeling
Computational protein structure prediction methods are broadly classified into two categories:
- Template-Based Modeling (TBM): This approach leverages known protein structures (templates) that share evolutionary or structural relationships with the target sequence. It includes Homology Modeling, Comparative Modeling, and Threading (fold-recognition) [19]. TBM is highly reliable when sequence identity with a template is above 30%, but its major limitation is its inability to predict new protein folds not represented in existing databases [21] [19].
- Template-Free Modeling (FM): FM methods predict structure without relying on identifiable structural templates. They address the core of the "protein folding problem" by attempting to compute the native conformation—the one at the lowest Gibbs free energy level—from physical principles and knowledge-based statistics derived from known protein structures [21] [19].
The following diagram illustrates the logical decision process and the position of FM within a generalized protein structure prediction workflow, as formalized by CASP:
Evolution of Methodologies in Template-Free Modeling
Early and Hybrid Methodologies
The immense conformational space available to even a small protein made exhaustive search impossible. Early FM strategies focused on reducing search space and designing effective energy functions to guide the search toward native-like states [21].
- Fragment Assembly: Pioneered by tools like ROSETTA, this method breaks the target sequence into short fragments (typically 1-20 residues) that are frequently retrieved from unrelated protein structures based on sequence similarity and local structure propensity [19] [22]. Full-length models are then assembled from these fragments using stochastic search algorithms like Monte Carlo simulations, guided by knowledge-based force fields that favor hydrophobic burial and specific steric interactions [22].
- Lattice Models: Approaches like TOUCHSTONE II represented protein conformations on discrete lattices, significantly simplifying the conformational search but potentially losing accuracy due to the discrete representation [21].
- Continuous Space Sampling: Methods like the one in RAPTOR++ aimed to sample protein conformations in a continuous space using directional statistics and Conditional Random Fields (CRFs) to model backbone angle distributions, thus avoiding the discretization artifacts of lattice models [21].
- Hybrid Approaches: As the field evolved, the line between TBM and FM blurred. Many successful "FM" predictors in CASP began using very remote templates or server predictions as starting points for further refinement [20]. Tools like Bhageerath and QUARK exemplify hybrid protocols that combine knowledge-based potentials from fragments with physics-based approaches [19].
The Deep Learning Revolution
A paradigm shift occurred with the integration of deep learning, particularly in CASP13 and CASP14.
- CASP13 (2018): Saw a substantial improvement in FM accuracy, primarily driven by the use of deep learning to predict inter-residue contacts and distances at various thresholds. This provided powerful spatial constraints that guided the model assembly process more effectively than previous methods [3] [2].
- CASP14 (2020): Marked an extraordinary breakthrough with AlphaFold2. It employed an end-to-end deep learning architecture based on an Evoformer neural network that jointly processed multiple sequence alignments (MSAs) and pairwise features [2]. Rather than predicting contacts, it directly output accurate 3D coordinates. AlphaFold2's performance was so high that for about two-thirds of the targets, its models were considered competitive with experimental accuracy in backbone measurement (GDTTS >90) [3] [2]. The trend line for CASP14 started at a GDTTS of about 95 for easy targets and finished at about 85 for the most difficult FM targets, a dramatic reduction in the accuracy gap between TBM and FM [2].
The following workflow summarizes the key methodological evolution in FM:
Quantitative Assessment in CASP
Key Metrics for Evaluation
CASP employs rigorous, superposition-dependent and independent metrics to evaluate model quality. The primary measures for FM include:
- GDTTS (Global Distance Test Total Score): The primary metric in CASP, expressing the average percentage of Cα atoms in a model that can be superimposed on the corresponding atoms in the experimental structure within multiple distance thresholds (1, 2, 4, and 8 Å). A higher GDTTS (0-100 scale) indicates a better model, with scores above 50 generally indicating a correct fold [1] [2].
- TM-Score (Template Modeling Score): A protein size-independent metric that measures the structural similarity between a model and the native structure. A TM-score >0.5 indicates the same fold, while a score <0.17 indicates a random similarity [22].
- QCS (Quality Control Score) and Other Scores: CASP10 introduced additional superposition-independent scores to provide a more comprehensive assessment, especially for clustering and selecting the best models from a large pool of predictions [20].
Performance Evolution in CASP FM
The table below summarizes the quantitative progress in FM as observed through recent CASP experiments, highlighting the dramatic leap in performance.
Table 1: Evolution of Template-Free Modeling Performance in CASP
| CASP Experiment | Key Methodological Advance | Representative Performance on FM Targets (GDT_TS) | Noteworthy Tools/Servers |
|---|---|---|---|
| CASP7 (2006) | Early fragment assembly and knowledge-based potentials | ~75 for small protein domains (e.g., T0283-D1) [3] | ROSETTA, RAPTOR++ [21] [3] |
| CASP9 & 10 | Hybrid approaches using remote templates; sustained progress for small proteins (<150 residues) [19] | Improved accuracy for targets up to 256 residues [3] | QUARK, Zhang-server (leading servers in CASP10) [20] |
| CASP12 (2016) | Use of predicted contacts as constraints for modeling | ~81 for specific targets (e.g., T0866-D1) [3] | – |
| CASP13 (2018) | Major improvement from deep learning-based contact/distance prediction | Average GDT_TS increased from 52.9 (CASP12) to 65.7 [3] | AlphaFold (v1), other DL methods [3] [2] |
| CASP14 (2020) | End-to-end deep learning (direct coordinate prediction) | Trend line: ~85 (difficult FM) to ~95 (easier FM); 2/3 of all targets had GDT_TS >90 [2] | AlphaFold2 [2] |
Table 2: Essential Research Reagents and Computational Tools for FM
| Tool / Resource | Type | Primary Function in FM | Relevance to Drug Development |
|---|---|---|---|
| QUARK | Software Suite / Server | Ab initio structure prediction by replica-exchange Monte Carlo simulations guided by a knowledge-based force field and fragment assembly [22]. | Model novel drug targets for structure-based drug design when no templates exist. |
| ROSETTA | Software Suite | Comprehensive suite for macromolecular modeling; its ab initio protocol uses fragment assembly and a sophisticated energy function [19]. | Protein engineering, enzyme design, and protein-protein interaction prediction. |
| MODELER | Software Suite | Comparative modeling, but often used in conjunction with FM methods for loop modeling or final model refinement [21]. | Generate complete models where parts of a structure are novel and other parts are template-based. |
| PSI-BLAST | Algorithm / Database | Generates Position-Specific Iterated (PSI) multiple sequence alignments (MSAs) to derive evolutionary profiles [21]. | Provides crucial evolutionary constraints for both traditional and modern DL-based FM methods. |
| PSIPRED | Algorithm | Predicts protein secondary structure from amino acid sequence [21]. | Offers structural constraints to guide the conformational search in knowledge-based FM. |
| AlphaFold2 | Deep Learning System | End-to-end deep network that directly predicts 3D atomic coordinates from sequence and MSA data [2]. | Generate highly accurate structural models for entire proteomes, revolutionizing target identification. |
| CASP Data Archive | Database | Repository of all CASP targets, predictions, and evaluation results for benchmarking new methods [3] [1]. | Benchmark in-house prediction pipelines and assess the expected accuracy for a given target class. |
The journey of Template-Free Modeling within the CASP experiment has evolved from a formidable challenge to a domain where computational methods, particularly deep learning, have demonstrated unprecedented accuracy. The field has transitioned from relying on physical principles and fragment assembly to leveraging deep learning-predicted constraints and, finally, to the end-to-end structure prediction embodied by AlphaFold2. This progress has effectively blurred the lines between FM and TBM, as the latest methods seem to rely less on explicit homologous templates and more on evolutionary information embedded in multiple sequence alignments [2].
For researchers and drug development professionals, the implications are profound. The ability to rapidly generate accurate structural models for proteins with novel folds opens new avenues for understanding disease mechanisms, exploring previously "undruggable" targets, and accelerating structure-based drug discovery. While challenges remain—particularly for large multi-domain proteins, dynamic ensembles, and membrane proteins—the advances showcased in CASP have irrevocably transformed the role of computational prediction in structural biology, making it an indispensable tool in the scientist's toolkit.
The Critical Assessment of protein Structure Prediction (CASP) stands as the global benchmark for evaluating protein folding methodologies. For decades, this biannual experiment quantified incremental progress but fell short of achieving the ultimate goal: computational prediction competitive with experimental structures. The 2020 CASP14 assessment marked a historic inflection point, characterized by the performance of AlphaFold2, an artificial intelligence system developed by DeepMind. This whitepaper provides an in-depth technical analysis of how AlphaFold2's novel architecture redefined the possible in structural biology. We detail its core methodological breakthroughs, quantify its performance against experimental data and other methods, and summarize the subsequent ecosystem of AI tools it inspired. Furthermore, we contextualize its impact within the CASP framework and outline the new frontiers of research it has opened, providing researchers and drug development professionals with a comprehensive guide to the current and future landscape of protein structure prediction.
The CASP Experiment: The Benchmark for Protein Folding Research
Since 1994, the Critical Assessment of protein Structure Prediction (CASP) has served as a community-wide, blind experiment to objectively assess the state of the art in predicting protein 3D structure from amino acid sequence [1]. Its primary goal is to advance methods by providing rigorous, independent evaluation. During each CASP round, organizers release amino acid sequences for proteins whose structures have been experimentally determined but are not yet public. Predictors worldwide submit their computed models, which are then compared against the ground-truth experimental structures [1] [2].
A key feature of CASP is its double-blind protocol; neither predictors nor organizers know the target structures during the prediction window, ensuring an unbiased assessment [1]. The evaluation is rigorous, relying on metrics like the Global Distance Test (GDT_TS), a score from 0-100 that measures the percentage of Cα atoms in a model positioned within a threshold distance of their correct location in the experimental structure [1] [2]. Historically, CASP targets have been categorized by difficulty, from Template-Based Modeling (TBM), where evolutionary related structures can guide prediction, to the most challenging Free Modeling (FM) category, which involves proteins with no recognizable structural homologs [2].
For over two decades, CASP documented steady but slow progress. However, as one overview noted, "accurate computational approaches are needed to address this gap" between the billions of known protein sequences and the small fraction with experimentally solved structures [10]. This longstanding challenge set the stage for a transformative breakthrough.
Before the Breakthrough: The Protein Folding Problem
The "protein folding problem" has been a grand challenge in biology for over 50 years. A protein's specific 3D structure, or native conformation, is essential to its function. Christian Anfinsen's pioneering work posited that this native structure is intrinsically determined by the protein's amino acid sequence [23]. Predicting this structure computationally from sequence alone proved immensely difficult.
Prior to the deep learning revolution, computational methods fell into two main categories [10] [23]:
- Template-Based Modeling (TBM): These methods relied on identifying evolutionarily related proteins of known structure (templates) in the Protein Data Bank (PDB) through sequence alignment. Models were built by copying and adapting the template structure. While often accurate when good templates existed, these methods failed for proteins without clear homologs.
- De Novo (or Free) Modeling: For proteins without templates, methods attempted to predict structure from physical principles and energy minimization. These approaches were computationally expensive and notoriously unreliable, especially for larger proteins.
The limitations of these approaches were starkly evident in pre-CASP14 results. While performance on TBM targets was strong, accuracy on FM targets was low, leaving a significant portion of the proteome inaccessible to reliable prediction [2] [3].
The AlphaFold2 Architecture: A Technical Deconstruction
AlphaFold2's performance at CASP14 was not an incremental improvement but a paradigm shift. Its novel end-to-end deep learning architecture moved beyond the previous paradigm of predicting inter-residue distances and assembling structures.
Input Representation and Feature Embedding
The system begins by generating a rich set of input features from the amino acid sequence [10]:
- Multiple Sequence Alignments (MSAs): The input sequence is searched against genetic databases to find homologous sequences. The resulting MSA captures evolutionary constraints and co-evolutionary signals, providing critical information about residue pairs that mutate in a correlated fashion.
- Templates: Structures of evolutionarily related proteins (if available) are incorporated as positional priors.
- Primary Sequence: The raw amino acid sequence and positional information.
These inputs are embedded into two primary representations that the network processes: an Nseq × Nres MSA representation and an Nres × Nres pair representation [10].
The Evoformer: The Core Innovation
The Evoformer is a novel neural network block that forms the trunk of the AlphaFold2 architecture. Its design treats structure prediction as a graph inference problem, where residues are nodes and their spatial relationships are edges [10]. The Evoformer's key innovation is structured, iterative information exchange between the MSA and pair representations using attention mechanisms [10].
- MSA → Pair Representation: Information flows from the MSA to the pair representation via an outer product, updating the pairwise relationships using evolutionary information.
- Pair → MSA Representation: The pair representation biases the attention operations within the MSA, allowing spatial reasoning to influence the processing of sequence information.
- Triangular Self-Attention and Multiplicative Updates: Within the pair representation, two operations enforce geometric consistency. Triangular self-attention and the triangle multiplicative update operate on sets of three residues, implicitly learning to satisfy triangle inequality constraints essential for real 3D structures [10]. This allows the network to reason about the protein fold as a coherent geometric whole.
The Structure Module: From Representations to 3D Coordinates
The structure module translates the refined representations from the Evoformer into an atomic-level 3D structure. Unlike previous methods that predicted distributions over distances or angles, AlphaFold2 directly predicts the 3D coordinates of all heavy atoms [10]. Key features include:
- Explicit Frame Representation: Each residue is represented by a local rigid body frame (rotation and translation).
- Equivariant Transformers: These networks respect the geometric symmetries of 3D space, ensuring that rotating the input sequence representation results in a corresponding rotation of the output structure.
- Iterative Refinement (Recycling): The entire network processes its own outputs recursively, allowing the structure to be refined over multiple iterations, which significantly improves accuracy [10].
Table 1: Core Components of the AlphaFold2 Architecture and Their Functions
| Component | Primary Function | Key Innovation |
|---|---|---|
| Input Embedding | Encodes MSAs, templates, and primary sequence into numerical representations. | Creates a rich, information-dense starting point for the network. |
| Evoformer Block | Processes MSA and pair representations to evolve a structural hypothesis. | Structured, iterative information exchange using triangular attention. |
| Structure Module | Generates 3D atomic coordinates from the processed representations. | Direct, end-to-end prediction of coordinates using equivariant transformers. |
| Recycling | The network processes its own output multiple times. | Enables iterative refinement of the structure, boosting accuracy. |
AlphaFold2's End-to-End Deep Learning Architecture
Quantifying the Breakthrough: CASP14 and Beyond
The CASP14 results demonstrated that AlphaFold2 was not merely better than its predecessors; it was in a class of its own.
Performance at CASP14
The official assessment concluded that AlphaFold2 produced models competitive with experimental structures in about two-thirds of cases [2]. The median backbone accuracy (Cα RMSD95) for AlphaFold2 was 0.96 Å, a resolution comparable to the width of a carbon atom. The next best method had a median accuracy of 2.8 Å [10]. In a landmark statement, CASP organizers declared that the results "represent a solution to the classical protein folding problem, at least for single proteins" [2].
Table 2: Key Quantitative Results from AlphaFold2 at CASP14 [10] [2]
| Metric | AlphaFold2 Performance | Next Best Method Performance | Implication |
|---|---|---|---|
| Backbone Accuracy (Median Cα RMSD95) | 0.96 Å | 2.8 Å | Atomic-level accuracy; competitive with some experimental methods. |
| All-Atom Accuracy (RMSD95) | 1.5 Å | 3.5 Å | Accurate placement of side chains, critical for functional sites. |
| Trend Line GDT_TS (Difficult Targets) | ~85 | ~60 (in CASP13) | Correct fold for nearly all targets, including the most difficult. |
| Targets with GDT_TS > 90 | ~2/3 of targets | Rare | Models are competitive with experimental structures. |
The accuracy extended beyond the backbone to all-heavy atoms, meaning side chains were positioned with high precision, a critical factor for understanding protein function and drug binding [10]. Furthermore, AlphaFold2's self-estimated accuracy metric, predicted Local Distance Difference Test (pLDDT), provided a reliable per-residue confidence score that strongly correlated with the true model quality, allowing researchers to identify less reliable regions [10] [24].
Comparison with Predecessors and Contemporaries
The leap from CASP13 to CASP14 was unprecedented. Figure 1 from the CASP14 overview shows the trend line for the best models starting at a GDT_TS of ~95 for easy targets and finishing at ~85 for the most difficult targets, a dramatic rise from the CASP13 trend line which finished below 65 for difficult targets [2]. This performance was vastly superior to other groups in CASP14, and notably, the standard server performance in CASP14 (which did not include AlphaFold2) matched the best human-group performance from CASP13, underscoring the scale of the discontinuity that AlphaFold2 represented [2].
The New Ecosystem: Tools and Extensions Inspired by AlphaFold2
The open-source release of AlphaFold2 catalyzed the development of a new ecosystem of computational tools, making high-accuracy structure prediction accessible and extending its capabilities.
Table 3: Essential Research Tools in the Post-AlphaFold2 Ecosystem
| Tool / Resource | Type | Primary Function | Reference |
|---|---|---|---|
| AlphaFold DB | Database | Provides pre-computed AlphaFold2 models for over 200 million proteins, covering nearly the entire UniProt proteome. | [24] |
| ColabFold | Server / Local | A faster, more accessible implementation combining MMseqs2 for rapid MSA generation with AlphaFold2 or RoseTTAFold. | [25] [24] |
| AlphaFold-Multimer | Algorithm | A version of AlphaFold2 specifically fine-tuned for predicting structures of protein complexes and multimers. | [24] |
| RoseTTAFold | Algorithm | A contemporaneous deep learning method from Baker lab that also uses a three-track network (sequence, distance, 3D). | [24] |
| ESMFold | Algorithm | A model based on a protein language model that can perform predictions from a single sequence, enabling ultra-fast screening. | [24] |
| MULTICOM | Tool | An example of an advanced system that refines AlphaFold2 predictions through better MSA sampling, model ranking, and refinement. | [25] |
Beyond Single Chains: Current Frontiers and Limitations
While AlphaFold2 solved the core folding problem for single chains, research has rapidly moved toward more complex challenges, many of which were incorporated as new categories in CASP15 [26].
- Protein Complexes and Assemblies: Predicting the structures of multi-chain protein complexes is a primary focus. CASP15 reported "enormous progress in modeling multimolecular protein complexes," with accuracy nearly doubling from CASP14 due to methods like AlphaFold-Multimer [3].
- Protein-Ligand Interactions: Accurately predicting how small molecules (e.g., drugs) bind to proteins is crucial for drug discovery. This was introduced as a pilot category in CASP15 [26].
- Conformational Ensembles and Dynamics: Proteins are dynamic, often sampling multiple conformations. CASP15 explored predicting ensembles of structures, such as those arising from allostery or local dynamics, often using data from cryo-EM maps or NMR [26].
- RNA and Protein-Nucleic Acid Complexes: Predicting RNA structures and their complexes with proteins represents a significant and challenging frontier, also piloted in CASP15 [26].
- Intrinsically Disordered Regions (IDRs): AlphaFold2's low pLDDT scores often correspond to biologically important IDRs that lack a fixed structure, presenting a limitation for the current model [23].
Current Research Frontiers Extending Beyond AlphaFold2's Core Breakthrough
The CASP experiment provided the rigorous, blind testing ground that for decades charted the arduous path toward solving the protein folding problem. AlphaFold2's performance at CASP14 stands as a watershed moment, demonstrating that a deep learning approach could achieve accuracy rivaling experimental methods for single protein chains. Its architectural innovations—particularly the Evoformer's information exchange and the end-to-end coordinate generation—were fundamental to this success. This breakthrough has not only provided a powerful tool for life science research and drug development but has also redefined the field's ambitions. The focus has now shifted from single-chain prediction to the more complex challenges of modeling the interactome, conformational dynamics, and the full machinery of life, setting the agenda for the next decade of computational structural biology.
The Critical Assessment of Structure Prediction (CASP) is a community-wide, biannual experiment that has served as the gold standard for objectively testing protein structure prediction methods since 1994 [1]. Initially focused on the classical "protein folding problem"—predicting a protein's three-dimensional structure from its amino acid sequence—CASP has dramatically evolved beyond its original scope. The extraordinary success of deep learning methods, particularly AlphaFold2 in CASP14, which demonstrated accuracy competitive with experimental structures, effectively provided a solution to the single-chain protein folding problem for many targets [2] [10]. This breakthrough has shifted the field's focus toward more complex challenges, including predicting protein complexes, RNA structures, and ligand interactions—frontiers essential for advancing structural biology and drug discovery.
This expansion reflects the growing understanding that biological function arises not from isolated proteins but from intricate macromolecular interactions. The accurate prediction of these complexes provides deeper insights into cellular mechanisms and creates new opportunities for therapeutic intervention, particularly for RNA-targeting drugs [27]. CASP has responded by introducing dedicated assessment categories for these challenges, fostering innovation in computational methods that integrate physical, evolutionary, and AI-driven approaches. This article examines the methodologies, assessments, and future directions of CASP's expanded horizons in predicting biological complexes.
Methodological Foundations: From Proteins to Complex Assemblies
The Core CASP Framework and Its Expansion
CASP operates on a rigorous blind testing principle. Organizers provide participants with amino acid sequences (and later, RNA sequences) of proteins and complexes whose structures have been recently solved but not yet publicly released [1]. Predictors submit their models within a strict timeframe, and independent assessors evaluate them by comparing them to the experimental reference structures. The primary metrics for evaluation include the Global Distance Test (GDT_TS), which measures the percentage of well-modeled residues, and root-mean-square deviation (RMSD) for atomic positions [1] [2].
The CASP14 experiment in 2020 marked a watershed moment. AlphaFold2 introduced a novel end-to-end deep learning architecture that incorporated evolutionary, physical, and geometric constraints of protein structures [10]. Its neural network jointly embedded multiple sequence alignments (MSAs) and pairwise features through a novel "Evoformer" module, then explicitly predicted 3D coordinates of all heavy atoms through a "structure module" that employed iterative refinement [10]. This approach demonstrated that computational models could achieve atomic accuracy competitive with experimental methods for many single-chain proteins.
CASP's Expanded Assessment Categories
With the single-chain protein problem largely solved, CASP has systematically expanded its assessment categories to address more complex biological questions:
- Protein Complexes: Assessment of protein-protein and protein-subunit interactions, often in collaboration with CAPRI (Critical Assessment of Predicted Interactions) [2] [7].
- RNA Structures: Introduced in CASP15, this category evaluates methods for predicting RNA three-dimensional structures, including single RNA molecules and RNA-protein complexes [28].
- Nucleic Acid-Ligand Complexes: CASP16 includes categories for predicting structures of nucleic acids (RNA and DNA) with proteins and organic ligands, reflecting growing therapeutic interest [7].
- Macromolecular Conformational Ensembles: Assessment of methods for predicting multiple conformational states, recognizing that biomolecules are dynamic entities [7].
- Integrative Modeling: Evaluation of approaches that combine computational modeling with sparse experimental data (e.g., SAXS, chemical crosslinking) [7].
Table 1: Key Expanded Assessment Categories in Recent CASP Experiments
| Category | First CASP | Primary Focus | Key Assessment Metrics |
|---|---|---|---|
| Protein Complexes | CASP2 (collab. with CAPRI) | Protein-protein interactions, subunit assembly | Interface Contact Score (ICS), DockQ [1] [2] |
| RNA Structures | CASP15 | 3D structure of RNA molecules and RNA-protein complexes | RMSD, Deformation Index (DI), INF [28] |
| Nucleic Acid-Ligand Complexes | CASP16 | Small molecule binding to RNA/DNA | Ligand RMSD, interaction network fidelity [7] |
| Model Accuracy Estimation | CASP7 | Self-assessment of model reliability | pLDDT, confidence scores [1] [10] |
| Refinement | CASP7 | Improving near-native models | RMSD improvement, GDT_TS improvement [11] |
Predicting Protein Complexes and Assemblies
Methodologies for Complex Prediction
Predicting the structures of protein complexes presents challenges beyond single-chain prediction, including identifying binding interfaces, modeling conformational changes upon binding, and accurately positioning subunits. Methods for complex prediction have evolved along several trajectories:
- Template-Based Modeling: Leveraging known complex structures from the PDB as templates when homologous complexes exist.
- Free Docking Approaches: Physically assembling individual subunit structures without template information, often using scoring functions to evaluate potential binding modes.
- Integrated Deep Learning: More recent methods adapt deep learning architectures similar to AlphaFold2 for complexes, incorporating interface predictions and co-evolutionary signals from multiple sequence alignments of interacting partners.
The assessment of complexes in CASP employs specialized metrics that focus on interface accuracy rather than global structure. The Interface Contact Score (ICS) measures the fraction of correct residue-residue contacts across subunit interfaces, while DockQ provides a composite score evaluating interface quality [2].
Key Findings and Challenges
CASP experiments have revealed both progress and persistent challenges in complex prediction:
- Accurate for High-Similarity Templates: Prediction accuracy remains high when close homologous templates are available, but drops significantly for novel complexes [2].
- Domain-Swapping and Flexibility: Complexes involving large conformational changes or domain swapping remain particularly challenging [2].
- Recent Advances: CASP15 saw "enormous progress" in protein complex prediction, though accuracy remained lower than for single proteins, indicating substantial room for advancement [7].
Table 2: CASP Assessment Metrics for Different Structure Types
| Structure Type | Primary Metrics | Key Challenges | Typical Performance Range |
|---|---|---|---|
| Single Proteins | GDT_TS (0-100), RMSD (Å) | Difficult targets with few homologs | GDT_TS: 85+ for easy targets, 60+ for hard targets [2] |
| Protein Complexes | Interface Contact Score, DockQ (0-1) | Interface prediction, conformational changes | Varies widely with template availability [2] |
| RNA Structures | RMSD (Å), Deformation Index, INF | Non-canonical pairs, flexible regions | RMSD: 2-15Å depending on size and complexity [28] |
| Ligand Complexes | Ligand RMSD, Interaction Fidelity | Binding site prediction, flexibility | Highly variable; emerging category [7] |
The New Frontier: RNA Structure Prediction
The CASP15 RNA Assessment
CASP15 (2022) marked the first formal assessment of RNA structure prediction, representing a significant expansion beyond proteins. This initiative emerged from the earlier RNA-Puzzles experiment, which had established preliminary benchmarks for RNA modeling [28]. Twelve RNA-containing targets were released for prediction, ranging from single RNA molecules to RNA-protein complexes. Forty-two research groups submitted models, which were evaluated using both traditional protein-inspired metrics and RNA-specific measures.
The assessment employed a dual approach: the CASP-recruited team used a Z-score ranking system (ZRNA) based on multiple metrics, while the RNA-Puzzles team employed RNA-specific measures including the Deformation Index (DI) and Interaction Network Fidelity (INF) [28]. Despite differences in methodology, both assessments independently identified the same top-performing groups: AIchemy_RNA2 (first), Chen (second), with RNAPolis and GeneSilco tied for third.
RNA-Specific Evaluation Metrics
RNA structure prediction requires specialized metrics that account for its unique structural features:
- Deformation Index (DI): Combines RMSD with Interaction Network Fidelity (DI = RMSD/INF), providing a composite measure that penalizes both geometric deviation and errors in interaction networks [28].
- Interaction Network Fidelity (INF): Evaluates the accuracy of predicted base-base interactions using the Matthews Correlation Coefficient, calculated separately for Watson-Crick pairs (INFWC), non-Watson-Crick pairs (INFNWC), and stacking interactions (INF_STACK) [28].
- Stereochemical Quality: Assessed using MolProbity's clash score, which counts the number of steric clashes per thousand residues [28].
Interestingly, the top-performing groups in CASP15 RNA did not use deep learning approaches, which performed significantly worse than more traditional methods—the opposite trend observed in protein structure prediction [28]. This suggests that RNA structure prediction remains a distinct challenge where evolutionary and physical constraints may not be as easily captured by current deep learning architectures.
Diagram 1: CASP15 RNA structure prediction assessment workflow showing the independent evaluations that reached identical conclusions about method performance. The top groups did not use deep learning approaches.
Ligand Interaction Prediction: Emerging Approaches
The Growing Importance of RNA-Ligand Targeting
Accurate prediction of RNA-ligand interactions has gained significant attention for its therapeutic potential. Historically, RNA was considered challenging to target with small molecules, but recent advances have catalyzed interest in RNA-targeted drug discovery for antiviral, anticancer, and metabolic applications [27]. CASP16 introduced categories specifically for predicting protein-organic ligand complexes and nucleic acid-ligand interactions, reflecting this growing importance.
Traditional computational methods for RNA-ligand interaction prediction include molecular docking (predicting binding orientations) and molecular dynamics simulations (modeling dynamic behavior) [27]. However, these methods are computationally demanding and often struggle to capture the complexity and flexibility of RNA-ligand interactions.
AI-Driven Advances and Challenges
Artificial intelligence, particularly deep learning, is revolutionizing RNA-ligand interaction prediction:
- Binding Site Identification: AI-driven tools like RNAsite and RNACavityMiner use machine learning ensemble algorithms and deep neural networks to predict binding pockets on RNA structures [27].
- Structure Modeling: AI methods are being developed to predict RNA-ligand complex structures, though they currently lag behind protein-ligand prediction in accuracy [27].
- Binding Affinity Prediction: Machine learning models are improving virtual screening by predicting RNA-ligand binding affinities, accelerating drug discovery pipelines [27].
Key challenges persist, including the limited availability of experimentally validated RNA-ligand complex structures for training models, and the intrinsic flexibility of RNA structures which adopt multiple conformational states [27]. Future progress will likely depend on integrating AI with expanded experimental datasets and incorporating physics-based modeling approaches.
Experimental Protocols and Methodologies
CASP Evaluation Workflow
The CASP experiment follows a rigorous, standardized protocol to ensure fair and objective assessment:
- Target Identification and Release: Experimental groups provide protein/RNA sequences whose structures are soon to be solved. CASP organizers release these sequences to predictors during the "prediction season" [1].
- Model Submission: Participants have approximately three weeks to submit their predictions (shorter deadlines for servers). Models must follow specific format guidelines [7].
- Independent Assessment: Assessors evaluate models using standardized metrics without knowledge of the methods used. Evaluation includes both global and local accuracy measures [28] [2].
- Results Publication: All predictions and evaluations are made publicly available. Detailed analyses are published in special issues of the journal PROTEINS [1].
RNA Structure Assessment Methodology
The pioneering CASP15 RNA assessment employed these specific methodological approaches:
RNA-Puzzles Style Assessment:
- Structures were superposed using the McLachlan algorithm as implemented in Profit.
- Base pairs were annotated using MC-Annotate, classifying them as Watson-Crick or non-Watson-Crick.
- Interaction Network Fidelity (INF) was calculated as the geometric mean of positive predictive value and sensitivity.
- Deformation Index (DI) was computed as RMSD divided by INF [28].
CASP-Style Assessment:
- Implemented ZRNA, a weighted Z-score average of multiple metrics.
- Used US-align for TM-score computation and Local-Global Alignment for GDT_TS.
- Calculated INF scores using ClaRNA for base pair assignment robustness.
- Employed OpenStructure for lDDT computation and PHENIX for clashscores [28].
Diagram 2: Standard CASP experimental workflow showing the double-blind evaluation process that ensures objective assessment of prediction methods.
Table 3: Essential Research Tools for CASP-Style Structure Prediction
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| AlphaFold2 | Deep Learning Network | End-to-end protein structure prediction | Single-chain proteins, now adapted for complexes [10] |
| Evoformer | Neural Network Module | Jointly embeds MSAs and pairwise features | Core innovation in AlphaFold2 architecture [10] |
| US-align | Evaluation Tool | Structural alignment for TM-score computation | Protein and RNA structure comparison [28] |
| MC-Annotate | Analysis Tool | RNA base-pair annotation and classification | RNA-specific structure assessment [28] |
| MolProbity | Validation Suite | Stereochemical quality analysis | Clashscores, RNA backbone validation [28] |
| ClaRNA | Analysis Tool | RNA contact classification | Base pair assignment in low-resolution models [28] |
| ZRNA | Assessment Pipeline | Comprehensive RNA model evaluation | CASP15 RNA assessment workflow [28] |
| PDB | Database | Repository of experimental structures | Template source, training data for AI methods [10] |
The expansion of CASP into complexes, RNA, and ligand interactions represents the evolving frontier of computational structural biology. While extraordinary progress has been made in single-protein structure prediction, these more complex challenges remain only partially solved. Key future directions include:
- Integration of Multi-scale Data: Combining computational predictions with sparse experimental data (NMR, cryo-EM, crosslinking) to solve larger and more complex assemblies [11].
- Dynamic Ensembles: Moving beyond static structures to model conformational ensembles and dynamics, as reflected in the new CASP16 category for macromolecular ensembles [7].
- Hybrid AI-Physics Approaches: Integrating deep learning with physics-based simulations to improve accuracy, particularly for RNA and ligand interactions where data scarcity limits pure AI approaches [27].
- Accessible Tools: Developing publicly available servers and tools that bring state-of-the-art prediction methods to the broader scientific community [2].
As CASP continues to evolve, it will likely further expand into predicting the effects of mutations, designing functional proteins and RNAs, and modeling increasingly complex cellular assemblies. These advances will continue to transform structural biology and drug discovery, providing unprecedented insights into the molecular machinery of life.
The Critical Assessment of Structure Prediction (CASP) is a community-wide, worldwide experiment that has taken place every two years since 1994 to objectively test methods for predicting protein three-dimensional structure from amino acid sequence [1]. This experiment serves as a rigorous blind testing ground where predictors compute structures for proteins whose experimental structures are soon-to-be solved but not yet public, ensuring an unbiased assessment of methodology [1] [2]. The primary evaluation metric in CASP is the Global Distance Test - Total Score (GDT_TS), which calculates the percentage of well-modeled residues in the predicted structure compared to the experimental target [1]. CASP has historically categorized targets by difficulty: Template-Based Modeling (TBM) for targets with detectable homology to known structures, and Free Modeling (FM) for the most difficult targets with no detectable homology [2]. For over two decades, CASP has documented incremental progress in the field, but recent experiments have witnessed revolutionary advances that have fundamentally transformed the relationship between computational prediction and experimental structural biology.
The CASP14 Revolution: AlphaFold2 and the Accuracy Breakthrough
The CASP14 round in 2020 marked an extraordinary turning point in protein structure prediction. Deep-learning methods from DeepMind's AlphaFold2 consistently delivered computed structures rivaling experimental accuracy, with the system scoring around 90 on the 100-point GDTTS scale for moderately difficult protein targets [1] [2]. The trend curve for CASP14 started at a GDTTS of approximately 95 and finished at about 85 for the most difficult targets, representing a dramatic improvement over previous CASPs [2]. Historically, the most accurate models were obtained using information about experimentally determined homologous structures (template-based modeling), but CASP14 demonstrated that AlphaFold2 models were only marginally more accurate when such information was available, indicating the method's remarkable ability to predict structures de novo [2].
Table 1: CASP14 Results Summary for AlphaFold2
| Metric | Performance | Context |
|---|---|---|
| Average GDT_TS | >90 for ~2/3 of targets | Competitive with experimental accuracy [3] |
| Minimum GDT_TS | ~85 for most difficult targets | Far exceeds previous CASP performance [2] |
| Key Advancement | Accuracy improvement 2018-2020 > 2004-2014 | Represents accelerated progress [3] |
| Experimental Applications | Four structures solved using AlphaFold2 models | Direct practical utility demonstrated [3] |
The implications of this accuracy breakthrough are profound. For approximately two-thirds of targets in CASP14, the computed models reached GDT_TS values greater than 90, making them competitive with experimental structures in backbone accuracy [2] [3]. When models achieve this level of accuracy, they transition from theoretical predictions to practical tools that can directly assist experimental structural biology and drug discovery efforts.
Molecular Replacement Using CASP Models: Methodologies and Protocols
The Molecular Replacement Technique
Molecular replacement is a common method in X-ray crystallography for determining the phase problem by using a known homologous structure as a search model [3]. Before the CASP14 breakthrough, molecular replacement typically required experimentally-solved structures with significant sequence similarity to the target protein. The extraordinary accuracy of AlphaFold2 models has fundamentally changed this paradigm, enabling computational models to successfully solve crystal structures even for targets with limited or no homology information [3].
Experimental Protocol for Molecular Replacement with CASP Models
The following workflow outlines the standardized methodology for utilizing CASP models in molecular replacement experiments:
Case Studies and Success Stories
In CASP14, four structures were solved with the aid of AlphaFold2 models, demonstrating the practical utility of these predictions for experimental structural biology [3]. A post-CASP analysis showed that models from other groups would also have been effective in some cases, indicating that the methodology was becoming more widespread [3]. These were all challenging targets with limited or no homology information available for at least some domains, highlighting the power of the new methods across all classes of modeling difficulty [3].
One notable pre-CASP example includes the crystal structure of Sla2 ANTH domain of Chaetomium thermophilum (CASP11 target T0839), which was determined by molecular replacement using CASP models with a GDT_TS of 62.8 [3]. While such successes were exceptional in earlier CASPs, they have become increasingly common with the improved accuracy of deep learning-based methods.
Table 2: Molecular Replacement Success Cases Using CASP Models
| CASP Target | Model Used | GDT_TS | Application Outcome |
|---|---|---|---|
| T0839 (CASP11) | TS184_1 | 62.8 | Structure solved by molecular replacement [3] |
| Multiple targets (CASP14) | AlphaFold2 | >90 | Four structures solved using models [3] |
| T1064 (SARS-CoV-2 ORF8) | AlphaFold2 | 87 | Impressive atomic-level agreement [2] |
Applications in Rational Drug Design
From Structure to Drug Discovery
The high accuracy of CASP models, particularly in side-chain positioning, enables their direct use in structure-based drug design [2]. The atomic-level agreement between AlphaFold2 models and experimental structures for main chain and side chain atoms (as demonstrated with SARS-CoV-2 ORF8) provides confidence for virtual screening and binding site identification [2]. The reliability of these models eliminates previous uncertainties in computational drug design where inaccuracies in predicted binding sites could lead to failed experimental validation.
Workflow for Drug Design Using CASP Models
The integration of CASP models into rational drug design follows a systematic process:
COVID-19 Response: A Case Study in Rapid Response
The CASP community demonstrated the practical utility of these methods during the COVID-19 pandemic by working together to compute and evaluate models for ten of the most challenging SARS-CoV-2 proteins of unknown structure [2]. This represented the most extensive community modeling experiment in CASP history and produced immediately useful results for the global research effort. The accurate model of SARS-CoV-2 ORF8 (CASP target T1064) with a GDT_TS of 87 provided researchers with a reliable structural framework for investigating the protein's function and potential as a drug target [2].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for CASP-Based Structural Biology
| Reagent/Resource | Function/Application | Examples/Specifications |
|---|---|---|
| AlphaFold2 | Protein structure prediction | Deep learning system; achieves GDT_TS >85 for most targets [2] |
| GDT_TS | Model accuracy assessment | Percentage of well-modeled residues; >90 indicates experimental quality [1] [2] |
| Molecular Replacement Software | Phase problem solution | PHASER, MolRep; use CASP models as search models [3] |
| CASP Models | Experimental structure solution | Four structures solved in CASP14 using AlphaFold2 models [3] |
| Confidence Metrics | Model quality estimation | pLDDT; identifies low-confidence regions for removal before MR [3] |
The CASP experiments have documented the remarkable progress in protein structure prediction, culminating in methods that now produce models competitive with experimental structures in accuracy. This transformation has moved protein structure prediction from a theoretical exercise to a practical tool that directly assists experimental structural biology and drug discovery. The demonstrated success of CASP models in molecular replacement for solving crystal structures and their application in rational drug design represents a paradigm shift in structural biology.
As the field progresses, future CASP experiments will likely focus on increasingly challenging targets, including membrane proteins, large multi-protein complexes, and protein-nucleic acid interactions. The integration of these accurate computational models with experimental structural biology will continue to accelerate, potentially reducing the time and cost associated with traditional structure determination methods. For drug discovery professionals, these advances provide immediate access to reliable protein structures for targets that were previously intractable to experimental determination, opening new avenues for therapeutic development.
Pushing the Boundaries: CASP's Role in Identifying Current Challenges
The Critical Assessment of Structure Prediction (CASP) is a community-wide, worldwide experiment that has taken place every two years since 1994 to objectively test protein structure prediction methods through rigorous blind testing [1]. This experiment provides an independent assessment of the state of the art in protein structure modeling to the research community and software users, with more than 100 research groups routinely participating [1]. For decades, CASP has served as the definitive benchmark for progress in solving the fundamental "protein folding problem" – predicting a protein's three-dimensional structure from its amino acid sequence alone. The CASP experiment operates as a double-blind test where predictors are given amino acid sequences of proteins whose structures have been experimentally determined but not yet made public, ensuring no group has prior structural information [1].
A revolutionary shift occurred during CASP14 (2020) when DeepMind's AlphaFold2 demonstrated unprecedented accuracy in predicting single protein chains, with models competitive with experimental structures for approximately two-thirds of targets [2]. According to CASP co-founders, this achievement represented a solution to the classical single-chain protein folding problem [2]. However, this breakthrough has illuminated a more formidable challenge: the accurate prediction of protein complex assemblies. In living organisms, proteins typically perform their functions by interacting to form complexes [29]. Determining these multi-chain structures is crucial for understanding and manipulating biological functions, yet accurately capturing inter-chain interaction signals remains a formidable challenge [29]. This whitepaper examines the persistent technical hurdles in protein complex assembly prediction within the framework of CASP experiments, addressing the critical gap between single-chain success and multi-chain challenges.
The Evolving CASP Framework for Assessing Complex Assembly
Historical Development of Assembly Assessment in CASP
Protein assembly prediction has been progressively integrated into the CASP framework through several developmental phases. Early attempts at evaluating oligomeric predictions began in CASP9 as part of template-based assessment, but participation and performance were limited [30]. A more substantial collaboration with CAPRI (Critical Assessment of PRedicted Interactions) occurred in CASP11, where despite low participation, several groups submitted accurate models by CAPRI standards [30]. The first independent assessment category dedicated solely to protein assembly was established in CASP12, marking a significant milestone in recognizing the importance of quaternary structure prediction [30].
In CASP12, assembly prediction was evaluated based on a three-level difficulty scale: "Easy" targets had templates with detectable sequence similarity and the same assembly; "Medium" targets had templates sharing partial subsets of chains in the same association mode; and "Hard" targets had no available oligomeric templates [30]. This classification helped distinguish template-based from template-free assembly predictions, revealing substantial challenges in the latter category. The evaluation metrics introduced in CASP12, including Interface Contact Similarity (ICS) and Interface Patch Similarity (IPS), provided standardized measures for assessing interface accuracy [30].
Contemporary CASP Assessment Categories
The remarkable success of deep learning methods in CASP14 prompted a significant reorganization of CASP categories to reflect new challenges and priorities. CASP15 and the upcoming CASP16 feature revised assessment frameworks that acknowledge the transformed landscape of protein structure prediction [7] [26]. The current categories relevant to complex assembly include:
- Assembly Modeling: Assesses the ability to correctly model domain-domain, subunit-subunit, and protein-protein interactions, continuing collaboration with CAPRI [26].
- Protein Complexes: Evaluates modeling of subunit-subunit and protein-protein interactions, with new options for predicting stoichiometry [7].
- Nucleic Acid Structures and Complexes: Includes RNA and DNA single structures and complexes with proteins [7].
- Protein-Organic Ligand Complexes: Focuses on drug-like compounds from pharmaceutical discovery projects [7] [31].
- Macromolecular Conformational Ensembles: Assesses methods for predicting structure ensembles, ranging from disordered regions to conformations involved in allosteric transitions [7].
Table 1: CASP Assembly Assessment Evolution
| CASP Edition | Assembly Assessment Features | Key Metrics | Notable Limitations |
|---|---|---|---|
| CASP9 (2010) | Preliminary attempt in template-based category | Limited participation | Only six groups submitted oligomeric models for most targets |
| CASP11 (2014) | Collaboration with CAPRI | CAPRI evaluation standards | Only five groups submitted models for most oligomeric targets |
| CASP12 (2016) | First independent assembly category | ICS, IPS scores | No successful residue contact predictions for hard targets |
| CASP14 (2020) | Joint CASP/CAPRI assessment | Interface accuracy measures | Limited to 22 quaternary structure targets |
| CASP15 (2022) | Realigned categories post-AlphaFold2 | TM-score, LDDTo | Accuracy not yet as high as for single proteins |
| CASP16 (2024) | Stoichiometry prediction option | LDDT-PLI for ligands | Ongoing challenges with flexible complexes |
Technical Hurdles in Protein Complex Prediction
Fundamental Limitations in Current Methodologies
Despite substantial progress, current protein complex prediction methods face several fundamental limitations that distinguish them from the largely solved single-chain problem. The accurate modeling of both intra-chain and inter-chain residue-residue interactions among multiple protein chains presents significantly greater complexity than tertiary structure prediction [29]. This challenge manifests in several critical areas:
Multiple Sequence Alignment (MSA) Pairing Limitations: For protein complexes, monomeric MSAs derived from individual chains must be systematically paired across different chains to generate comprehensive paired MSAs that capture inter-chain co-evolutionary signals [29]. However, popular sequence search tools such as HHblits, Jackhammer, and MMseqs are primarily designed for constructing monomeric MSAs and cannot be directly applied to paired MSA construction [29]. This limitation particularly compromises prediction accuracy for tightly intertwined complexes or highly flexible interactions, such as antibody-antigen systems, where identifying orthologs between interacting proteins is challenging due to the absence of species overlap [29].
The Soft Disorder Problem: Statistical evidence demonstrates that "soft disorder" – regions characterized by high flexibility, amorphous structure, or missing residues in experimental structures – plays a crucial role in complex assembly [32]. Analysis of the entire set of X-ray crystallographic structures in the PDB revealed that new interfaces tend to form at residues characterized as softly disordered in preceding complexes in assembly hierarchies [32]. This soft disorder modulates assembly pathways, with the location of disordered regions changing as the number of partners increases. This inherent flexibility presents a fundamental challenge for static structure prediction methods, as accurately forecasting these disorder-mediated assembly paths requires understanding conformational dynamics rather than single static structures.
Scoring Function Limitations: The modest correlation between predicted and experimental binding affinities (maximum Kendall's τ = 0.42 in CASP16, well below the theoretical maximum of ~0.73) highlights persistent challenges in scoring function development [31]. Notably, providing experimental structures in the second stage of CASP16's affinity challenge did not improve predictions, suggesting that scoring functions themselves represent a key limiting factor rather than structural accuracy alone [31].
Specific Challenging Systems
Certain protein complex types present particularly formidable challenges for current prediction methods:
Antibody-Antigen Complexes: These systems often lack clear inter-chain co-evolutionary signals, making established MSA pairing strategies ineffective [29]. The DeepSCFold study noted that virus-host and antibody-antigen systems typically don't exhibit inter-chain co-evolution, creating fundamental limitations for methods relying solely on sequence-level co-evolutionary information [29].
Immune-Related and Viral-Host Complexes: CASP16 specifically identified immune-related complexes and viral-host complexes as particularly challenging targets that remain informative for method development [7]. The transient nature and exceptional flexibility of these complexes contribute to their prediction difficulty.
Large Multimeric Assemblies: As complex size increases, the cumulative effect of small interface errors can lead to significant deviations in overall topology. The statistical evidence linking soft disorder migration with increasing complex size further complicates prediction of large assemblies [32].
Table 2: Quantitative Performance Gaps in Protein Complex Prediction
| Assessment Metric | Single Chain Performance | Complex Performance | Performance Gap |
|---|---|---|---|
| Backbone Accuracy (GDT_TS) | ~95 (Easy) to ~85 (Hard) in CASP14 [2] | Significantly lower than single proteins [7] | 10-30 GDT_TS points |
| Interface Contact Prediction | Not applicable | 0% success for hard targets in CASP12 [30] | Fundamental method limitation |
| Antibody-Antigen Interface Prediction | Not applicable | 24.7% improvement needed over AlphaFold-Multimer [29] | Substantial room for improvement |
| Affinity Prediction (Kendall's τ) | Not applicable | 0.42 (max) in CASP16 vs 0.73 theoretical max [31] | ~42% of potential unmet |
| Ligand Pose Prediction (LDDT-PLI) | Not applicable | 0.69 (best groups) vs 0.8 (AlphaFold3) in CASP16 [31] | Automated methods outperform human groups |
Experimental Protocols for Assembly Assessment
CASP Target Selection and Evaluation Workflow
The CASP experiment follows a rigorous protocol to ensure unbiased assessment of protein complex prediction methods. The process begins with target identification and proceeds through structured evaluation phases:
Target Selection and Release: CASP organizers collaborate with structural biologists to identify protein structures soon to be solved or recently solved but not yet publicly available [1]. For complexes, the oligomeric state is carefully determined through collaboration with experimentalists using tools like Evolutionary Protein-Protein Interface Classifier (EPPIC) and PISA, supplemented by experimental evidence such as size exclusion chromatography data [30]. Targets are released with sequence information and stoichiometry, though CASP16 introduced the option to initially release targets without stoichiometry information to test ab initio complex formation prediction [7].
Model Submission and Collection: Predictors typically have a three-week window to submit their models, while automated servers must return models within 72 hours [2]. Each predictor can submit up to five different models per target assembly but must designate their best model as number one [30]. For CASP16, nearly 100 research groups submitted more than 80,000 models across 300 targets in five prediction categories [7].
Evaluation Metrics and Assessment: Independent assessors evaluate models using standardized metrics. For complexes, key evaluation measures include:
- Interface Contact Similarity (ICS): Combines precision and recall of interface contact predictions using the F1-measure [30]. ICS = 2·P·R/(P+R), where P is contact precision and R is contact recall.
- Interface Patch Similarity (IPS): Measures similarity between interface patches of model and target using Jaccard coefficient [30]. IPS = |IM ∩ IT|/|IM ∪ IT|, where IM and IT are interface residues of model and target respectively.
- LDDT-PLI: A version of Local Distance Difference Test adapted for protein-ligand interactions [31].
- TM-score: Template Modeling score used for global structure similarity assessment [29].
The following workflow diagram illustrates the complete CASP experimental process for protein complexes:
Advanced Methodological Approaches
Recent methodological advances address specific challenges in complex assembly prediction:
DeepSCFold Protocol: This recently developed pipeline uses sequence-based deep learning models to predict protein-protein structural similarity (pSS-score) and interaction probability (pIA-score) purely from sequence information [29]. The method constructs paired MSAs by integrating structural similarity assessments between monomeric query sequences and their homologs with interaction pattern identification across distinct monomeric MSAs. DeepSCFold demonstrated significant improvements over state-of-the-art methods, achieving 11.6% and 10.3% TM-score improvements compared to AlphaFold-Multimer and AlphaFold3 respectively on CASP15 targets [29].
Soft Disorder Integration: Advanced methods now incorporate soft disorder predictions to identify potential interface regions. The correlation between AlphaFold2's low confidence residues (pLDDT) and regions of soft disorder provides a pathway for using confidence metrics as interface predictors [32]. This approach acknowledges that new interfaces tend to settle into the floppy parts of a protein, mediating assembly order.
Multi-Source Biological Integration: Leading methods incorporate multi-source biological information including species annotations, UniProt accession numbers, and experimentally determined complexes from the PDB to construct paired MSAs with enhanced biological relevance [29]. This integration helps compensate for absent co-evolutionary signals in challenging cases.
Research Reagent Solutions for Complex Assembly Prediction
Table 3: Essential Computational Tools for Protein Complex Prediction
| Tool/Resource | Type | Primary Function | Application in Complex Prediction |
|---|---|---|---|
| AlphaFold-Multimer | Deep Learning Model | Protein complex structure prediction | Baseline method for multimer prediction, extending AlphaFold2 architecture [29] |
| DeepSCFold | Computational Pipeline | Sequence-derived structure complementarity | Predicts structural similarity and interaction probability from sequence [29] |
| ESMPair | MSA Construction Tool | Paired MSA generation | Ranks monomeric MSAs using ESM-MSA-1b and integrates species information [29] |
| DiffPALM | MSA Construction Tool | Protein sequence pairing | Employs MSA transformer to estimate amino acid probabilities for pairing [29] |
| PISA | Interface Analysis Tool | Biological assembly assignment | Helps distinguish biological interfaces from crystal contacts [30] |
| EPPIC | Interface Classification | Evolutionary interface classification | Analyzes protein-protein interfaces in crystal lattices [30] |
| DeepUMQA-X | Quality Assessment | Complex model quality estimation | In-house method for selecting top models in DeepSCFold pipeline [29] |
| AlphaFold3 | Deep Learning Model | Protein-ligand complex prediction | Automated baseline method for ligand pose prediction [31] |
The accurate prediction of protein complex assembly remains a formidable challenge despite the revolutionary progress in single-chain protein structure prediction. The CASP experiments have systematically documented both the persistent hurdles and encouraging advances in this critical domain. Current limitations in MSA pairing, soft disorder handling, and scoring function development continue to separate complex prediction from the accuracy achieved for single chains.
Future progress will likely depend on improved integration of conformational dynamics, better handling of transient and flexible interactions, and more sophisticated approaches to capturing interaction patterns beyond sequence-level co-evolution. The recent development of methods like DeepSCFold that leverage structural complementarity information represents a promising direction. Furthermore, the systematic assessment framework provided by CASP ensures that advances will be rigorously validated through blind testing, maintaining the scientific integrity of this rapidly evolving field.
For researchers and drug development professionals, current protein complex prediction tools provide valuable structural hypotheses that require experimental validation. As methods continue to mature, the integration of computational predictions with experimental structural biology will likely accelerate the understanding of cellular function and facilitate drug discovery targeting protein-protein interactions.
{ abstract: While computational structure prediction has been revolutionized by deep learning, significant gaps remain in the reliability of accuracy estimation, particularly for macromolecular interfaces and nucleic acid-containing complexes. Within the framework of the Critical Assessment of protein Structure Prediction (CASP) experiments, this review quantifies these limitations, detailing how current methods struggle with functionally critical regions like protein-protein interfaces, ligand-binding sites, and non-canonical RNA structures. We present standardized assessment data from recent CASP rounds, describe the experimental protocols used for evaluation, and provide a toolkit to guide researchers in applying and developing these methods. }
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind experiment conducted every two years to objectively assess the state of the art in protein structure modeling [1]. Its core principle is the rigorous, blinded testing of methods by inviting research groups to predict structures of proteins whose sequences are known but whose experimental structures are not yet public [2] [33]. Since its inception in 1994, CASP has been the definitive "gold standard" for tracking progress in the field, with the primary evaluation metric being the Global Distance Test Total Score (GDT_TS), which measures the percentage of α-carbons in a predicted model within a threshold distance of the experimental structure [1] [34]. The experiment has catalyzed major advances, most notably the extraordinary leap in accuracy demonstrated by AlphaFold2 in CASP14, which delivered models competitive with experimental structures for approximately two-thirds of the targets [2] [35]. CASP has since expanded its scope beyond single-domain proteins to include critical assessment categories such as protein complexes, structure refinement, model accuracy estimation (EMA), and, most recently, nucleic acid structures and their complexes [3] [7].
The Critical Role and Current State of Accuracy Estimation
Estimation of Model Accuracy (EMA), also known as Quality Assessment (QA), is a fundamental sub-task in computational structural biology. Its goal is to predict the quality of a computational model without knowledge of the true native structure, enabling researchers to select the best models from a pool of decoys and to understand which regions of a model can be trusted for downstream biological applications [36] [37]. EMA methods are broadly classified into two categories:
- Single-model methods evaluate the intrinsic properties of one structure, using features such as statistical potentials, stereochemical checks, and compatibility with predicted secondary structure or solvent accessibility [36] [37].
- Multi-model (consensus) methods leverage the structural similarity between a model and other independently generated models for the same target, operating on the principle that structurally similar regions are more likely to be correct [36] [37].
The advent of deep learning has transformed EMA. Modern methods now integrate traditional features with inter-residue distance and contact predictions derived from deep multiple sequence alignments, using deep residual networks and other architectures to achieve state-of-the-art performance [37]. However, despite these advances, the reliability of accuracy estimation is not uniform across all types of structural elements. As this review will detail, significant estimation gaps persist at complex interfaces and for nucleic acids, posing a challenge for applications in functional analysis and drug design.
Quantifying the Reliability Gap at Interfaces
The accuracy of predicted models for protein complexes saw "enormous progress" in CASP15, with the average quality of multimeric models increasing dramatically [3] [7]. Despite this, assessing the reliability of these models, particularly at the interfaces where subunits interact, remains a substantial challenge. The performance of EMA methods for complexes has not kept pace with the ability to generate the models themselves.
Assessment Metrics and Quantitative Performance
CASP evaluation of complexes and their interfaces employs specific metrics distinct from those used for single chains. The Interface Contact Score (ICS), also known as the F1 score, is a key measure that evaluates the precision and recall of residue-residue contacts across a subunit interface [3]. The overall fold similarity is measured by LDDT (Local Distance Difference Test), including a specific variant for interfaces [3]. The quantitative performance from recent CASP experiments is summarized in Table 1.
Table 1: Quantitative Assessment of Model and EMA Performance in CASP
| Category | Key Metric | CASP14 Performance (2020) | CASP15 Performance (2022) | Current Challenges |
|---|---|---|---|---|
| Single Domain Proteins | Average GDT_TS (Best Models) | ~2/3 of targets >90 (competitive with experiment) [2] | Not quantified in results | Near-experimental accuracy for many targets. |
| Protein Complexes (Assembly Modeling) | Interface Contact Score (ICS/F1) | Outperformed by large margin [3] | Almost doubled since CASP14 [3] | Accuracy remains lower than for single proteins [7]. |
| Model Accuracy Estimation (EMA) | Loss in GDT_TS (Lower is better) | Top multi-model methods: 0.073 - 0.081 [37] | No longer a standalone category for single proteins [7] | Server-predicted accuracy (pLDDT) is now highly reliable for single proteins [7]. |
| Nucleic Acid Structures | Functional Region Accuracy | Category introduced in CASP15 [7] | Predictions "often lack accuracy in the regions of highest functional importance" [38] | Poor modeling of non-canonical interactions and functional interfaces [38]. |
Experimental Protocols for Interface Assessment
The protocol for assessing the accuracy of protein complexes in CASP is as follows [3] [2]:
- Target Selection and Release: Experimental groups provide structures of soon-to-be-published protein complexes. Targets are divided into evaluation units, often focusing on the key interacting subunits.
- Blind Prediction: Modeling groups submit predicted three-dimensional structures of the complexes within a set timeframe.
- Evaluation Against Experimental Structure: The submitted models are compared to the solved experimental structure using the LGA (Local-Global Alignment) software to calculate ICS and LDDT scores.
- Independent Assessment: An independent team of assessors analyzes the results to determine the state of the art, identify progress, and highlight remaining weaknesses, particularly at subunit interfaces.
The following workflow diagram illustrates the CASP experiment's structure for assessing interfaces:
Specific Challenges in Nucleic Acid Structure Assessment
The introduction of a dedicated nucleic acid (NA) structure category in CASP15 and its continuation in CASP16 highlights a growing recognition of their biological importance and the distinct challenges they present. The 2025 evaluation of CASP16 NA targets reveals that while blind prediction can achieve reasonable global folds for some complex RNAs, the accuracy plummets in the most functionally critical regions [38].
Key Failure Modes and Functional Consequences
The primary failure modes of nucleic acid structure prediction, as identified by the experimental providers of CASP targets, are [38]:
- Inaccuracy in Non-Canonical Regions: Predictions consistently show "inaccuracies in non-canonical regions where, for example, the nucleic-acid backbone bends, deviating from an A-form helix geometry, or a base forms a non-standard hydrogen bond." These regions are not random errors but are "integral to forming functionally important regions such as RNA enzymatic active sites."
- Poor Modeling of Functional Interfaces: The assessment concludes that "the modeling of conserved and functional interfaces between nucleic acids and ligands, proteins, or other nucleic acids remains poor." This represents a critical gap for understanding the molecular basis of gene regulation and therapeutic intervention.
- Challenge of Structural Ensembles: For some targets, the single experimental structure used for assessment may not represent the only conformation the complex occupies in solution, posing a fundamental challenge for the prediction community to model functionally relevant dynamics [38].
The following diagram illustrates the specific structural challenges in nucleic acid modeling that lead to functional inaccuracies:
Experimental Protocols for Nucleic Acid Assessment
The protocol for the nucleic acid category in CASP involves close collaboration with the RNA-Puzzles community and is evaluated by experts in the field [38] [7]:
- Target Provision: Experimentalists provide RNA and DNA structures, including complexes with proteins, prior to public release.
- Structure Prediction: Groups submit three-dimensional models for these NA targets. Methods range from traditional physics-based and comparative modeling to newer deep learning approaches, which have so far been less dominant for NAs than for proteins [7].
- Evaluation by Structure Providers: A unique aspect of the NA assessment is the direct involvement of the experimentalists who solved the structure. They provide a qualitative and quantitative evaluation of the models, with a specific focus on the accuracy of functionally important motifs and interfaces [38].
- Analysis of Functional Relevance: The final assessment highlights the disconnect between reasonable global folds and inaccurate functional sites, pushing the field to move beyond global metrics and focus on biological utility [38].
The Scientist's Toolkit: Research Reagent Solutions
To engage with CASP-related research or to apply its methodologies, scientists rely on a suite of computational tools and resources. The following table details key components of the modern structural bioinformatician's toolkit.
Table 2: Essential Research Reagents and Resources in Protein & Nucleic Acid Structure Prediction
| Tool/Resource Name | Type | Primary Function | Relevance to Gaps |
|---|---|---|---|
| AlphaFold2/3 | Structure Prediction Server/Software | Predicts 3D structures of proteins and their complexes from sequence [2] [35]. | Baseline for high-accuracy single-chain protein prediction; limitations remain for some complexes and nucleic acids. |
| MESHI_consensus | Accuracy Estimation Method | Estimates model quality using a tree-based regressor and 982 structural and consensus features [36]. | Demonstrates integration of diverse feature types to improve EMA. |
| MULTICOM EMA Suite | Accuracy Estimation Method | A family of deep learning methods that integrate inter-residue distance predictions to estimate model quality [37]. | Showcases the value of distance-based features for EMA. |
| CASP Assessment Metrics (ICS, LDDT) | Evaluation Software | Algorithms for quantitatively comparing predicted and experimental structures [3] [1]. | Essential for objectively quantifying gaps at interfaces and for nucleic acids. |
| DeepDist | Inter-residue Distance Predictor | Predicts distances between residue pairs from a multiple sequence alignment [37]. | Provides features for EMA methods; critical for single-model quality assessment. |
| CASP/CAPRI Target Data | Data Resource | Publicly available datasets of targets, predictions, and evaluations from past experiments [3] [7]. | Essential benchmark data for training new methods and testing against state-of-the-art. |
The CASP experiments have systematically illuminated a path from the solved problem of single-domain protein structure prediction to the next frontier: achieving reliable modeling of complex biological assemblies and nucleic acids. The quantitative data and experimental protocols reviewed here demonstrate that while structure generation has advanced remarkably, the parallel challenge of accurately estimating the reliability of those models at functionally critical sites like interfaces and active loops remains only partially met. For drug development professionals, this underscores the necessity of carefully interpreting computational models, particularly when analyzing protein-protein interactions for biologic therapeutics or targeting RNA structures with small molecules. The future of the field, as guided by CASP's blind assessments, lies in developing integrated methods that leverage sparse experimental data, better model conformational dynamics, and, most importantly, prioritize the prediction of functional accuracy over mere global structural similarity.
Within the field of computational structural biology, the advent of deep learning has been revolutionary. The Critical Assessment of protein Structure Prediction (CASP) experiments have documented this progress, with methods like AlphaFold2 demonstrating accuracy competitive with experimental structures for many single-domain proteins. However, the narrative of deep learning's omnipotence is incomplete. This whitepaper, framed within the context of CASP findings, delineates the specific areas where classical computational methods maintain a competitive edge. Drawing on the most recent CASP assessments, we provide quantitative evidence that for challenges such as predicting RNA structures, modeling protein-ligand complexes for drug design, and simulating conformational ensembles, classical physics-based and knowledge-based approaches continue to outperform deep learning. This document serves as a technical guide for researchers and drug development professionals, offering a balanced perspective on selecting the right tool for the problem at hand.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind experiment conducted every two years since 1994 to objectively assess the state of the art in protein structure modeling [1]. In CASP, predictors are given amino acid sequences of proteins with soon-to-be-released structures and challenged to compute their three-dimensional forms. Independent assessors then rigorously evaluate the submitted models against the experimental structures. CASP has historically categorized predictions based on methodology and difficulty, including Template-Based Modeling (TBM) and Template-Free Modeling (FM), the latter also known as ab initio or de novo modeling [39] [1].
The CASP14 experiment in 2020 marked a paradigm shift, with AlphaFold2 (AF2) achieving accuracy "competitive with experiment" for about two-thirds of single protein targets [2]. This success established deep learning as the dominant force for predicting monomeric protein structures. However, subsequent CASP experiments have revealed the boundaries of this approach. CASP15 (2022) and the latest CASP16 have highlighted specific, critically important areas where deep learning models have not yet surpassed classical methods. These areas are characterized by complex molecular interactions, limited training data, and a strong dependence on physical laws that are not yet fully captured by data-driven pattern recognition [12] [40] [41]. This paper synthesizes these findings to provide a clear-eyed view of the current technological landscape.
Persistent Challenges for Deep Learning
Protein-Ligand Complex Prediction
The prediction of how small molecule ligands bind to proteins is paramount for rational drug design. While recent co-folding models like AlphaFold3 (AF3) and RoseTTAFold All-Atom (RFAA) have shown impressive initial results, critical studies question their understanding of fundamental physics.
Performance Gap with Classical Docking: As of CASP16, deep learning methods for organic ligand-protein structures, while substantially more successful than traditional ones on a relatively easy target set, often still fall short of experimental accuracy [40]. Benchmarking studies have shown that AF3 can achieve high accuracy in "blind docking" scenarios. However, a 2025 adversarial study revealed significant limitations. When binding site residues were mutated to unrealistic amino acids (e.g., all to glycine or phenylalanine), deep learning models continued to place the ligand in the original site, despite the loss of all favorable interactions and the introduction of steric clashes, indicating a potential overfitting to statistical correlations in the training data rather than a robust understanding of physical principles [41].
Methodological Shortcomings: Classical docking tools like AutoDock Vina and GOLD are built on physics-based force fields that explicitly calculate van der Waals forces, electrostatic interactions, and solvation effects. In contrast, deep learning models appear to rely heavily on pattern memorization. The same 2025 study found that these models largely memorize ligands from their training data and do not generalize effectively to unseen ligand structures [41]. Their performance seems more tied to pocket-finding ability than to resolving detailed atomic interactions.
Experimental Protocol for Validating Protein-Ligand Predictions: To assess the physical realism of a protein-ligand prediction method, researchers can employ a binding site mutagenesis challenge:
- Select a Target Complex: Choose a high-resolution structure of a protein with a bound ligand (e.g., ATP bound to CDK2).
- Generate Adversarial Mutations: Systematically mutate all binding site residues to residues that alter the site's chemistry and steric profile (e.g., all to Glycine to remove side-chains, or all to Phenylalanine to create a steric block).
- Run Predictions: Submit both the wild-type and mutated sequences to the deep learning and classical methods.
- Evaluate Physical Plausibility: Analyze the resulting models. A physically realistic method should predict the ligand is displaced or adopts a completely different pose when its binding site is destroyed. The persistence of the original binding mode in the face of disruptive mutations indicates a lack of genuine physical understanding [41].
RNA Structure Prediction
The first inclusion of RNA structure prediction in CASP15 revealed a striking divergence from the success seen with proteins.
Quantitative Performance Gap: In CASP15, the classical approaches produced better agreement with experiment than the new deep learning ones, and the overall accuracy was recognized as limited [12]. This performance gap has persisted, with CASP16 results noting that deep learning methods are "notably unsuccessful at present and are not superior to traditional approaches" [40]. Both classical and deep learning methods produce poor results in the absence of structural homology, but classical methods, often based on physics-based simulations and comparative analysis, maintain a lead.
Root Causes: The underlying reasons are twofold. First, the database of known RNA structures is orders of magnitude smaller than the Protein Data Bank (PDB), providing deep learning models with far less training data. Second, the physical forces stabilizing RNA structures, such as long-range electrostatic interactions and metal ion binding, are complex and not as easily inferred from sequence data alone as they are for proteins. This gives methods with explicit physical models an inherent advantage.
Modeling Macromolecular Ensembles and Flexibility
Proteins and RNA are dynamic, often adopting multiple conformations critical for their function. CASP has begun to assess methods for predicting these ensembles.
Limited Accuracy of Deep Learning: The assessment of macromolecular ensembles in CASP16 was limited by a small target set. However, the general conclusion was that in the absence of structural templates, results tend to be poor, and the detailed structures of alternative conformations are usually of relatively low accuracy [40]. Deep learning models, trained predominantly on static snapshots from crystallographic structures, often struggle to generate genuine, functionally relevant conformational diversity.
Strength of Classical Simulation Methods: Classical methods, particularly Molecular Dynamics (MD) simulations, excel in this domain. Methods like all-atom MD and structure-based models (Gō models) can simulate the physical pathway of conformational change [42]. For example, simulations of large proteins like serpins have provided critical insights into folding intermediates, misfolding, and oligomerization pathways that are poorly accessible to current deep learning approaches [42]. These methods explicitly compute the forces between atoms over time, allowing them to naturally model dynamics and transitions.
The following workflow outlines the general process of a CASP experiment and the points at which classical and deep-learning methods are typically applied, highlighting the categories where classical methods retain an advantage.
Comparative Performance Data from CASP
The following tables synthesize quantitative and qualitative findings from recent CASP experiments to provide a clear, data-driven comparison between classical and deep learning methodologies across different problem domains.
Table 1: Performance Comparison in Key Biomolecular Modeling Categories (CASP15 & CASP16)
| Modeling Category | Deep Learning Performance | Classical Method Performance | Key CASP Findings |
|---|---|---|---|
| Protein-Ligand Complexes | Promising but limited generalization; potential overfitting to training data [41]. Short of experimental accuracy on harder targets [40]. | Superior for challenging cases; more robust and physically realistic predictions [40]. | Deep learning models fail adversarial physical tests; classical physics-based docking (e.g., AutoDock Vina) remains more reliable for novel ligand interactions [41]. |
| RNA Structures | "Notably unsuccessful" and not superior to classical methods [40]. | Superior and produces better agreement with experiment [12] [40]. | Accuracy is limited for both approaches without structural homology, but classical methods maintain a lead [40]. |
| Macromolecular Ensembles | Results tend to be poor without structural templates; low accuracy for alternative conformations [40]. | Superior in sampling conformational diversity and pathways [40] [42]. | Molecular Dynamics (MD) and structure-based models can provide insights into folding intermediates and dynamics that deep learning cannot yet match [42]. |
| Single-Protein Monomers | Highly accurate, often competitive with experimental structures (GDT_TS >90 for many targets) [3] [2]. | Lower accuracy, especially without good templates. | AlphaFold2 and its derivatives largely solved the single-domain protein folding problem as classically defined [12] [2]. |
Table 2: Detailed Experimental Protocols for Key Assessment Categories
| Experiment Type | Core Objective | Classical Methodology | Deep Learning Methodology | Key Evaluation Metrics |
|---|---|---|---|---|
| Binding Site Mutagenesis [41] | Test physical understanding of protein-ligand interactions. | Physics-based docking (e.g., AutoDock Vina) with explicit force fields. | End-to-end co-folding (e.g., AlphaFold3, RFAA) with mutated sequence. | Ligand displacement upon mutation; absence of steric clashes; physical plausibility of pose. |
| RNA Structure Prediction [12] [40] | Predict 3D structure from nucleotide sequence. | Comparative modeling, fragment assembly, and physics-based simulations. | DL models trained on known RNA structures (architecture varies). | RMSD (Root Mean Square Deviation); percentage of correctly predicted base pairs. |
| Conformational Ensemble Modeling [40] [42] | Predict multiple native-state conformations. | All-atom MD simulations; structure-based models (Gō models). | Generation of multiple outputs from a single sequence (e.g., via sampling). | Agreement with experimental data for alternative states (NMR, cryo-EM); diversity and realism of generated ensemble. |
| Template-Free Modeling (FM) [3] [39] | Predict structure without homologous templates. | Fragment assembly (e.g., Rosetta, QUARK) with Monte Carlo/REMC sampling. | Deep learning based on co-evolution and attention mechanisms (e.g., AlphaFold2). | GDT_TS (Global Distance Test Total Score); Cα RMSD. |
This table details key computational tools and resources relevant to the fields discussed, highlighting their primary function and methodological basis.
Table 3: Key Research Reagent Solutions in Computational Structure Prediction
| Tool/Resource Name | Type/Function | Methodological Basis | Relevance to Classical/Deep Learning Divide |
|---|---|---|---|
| AutoDock Vina [41] | Protein-ligand docking software. | Classical (Physics-based scoring function, Monte Carlo search). | A benchmark classical method for ligand docking; used to test physical realism of deep learning predictions. |
| GROMACS/AMBER | Molecular Dynamics (MD) simulation packages. | Classical (Newtonian physics, empirical force fields). | Essential for simulating macromolecular dynamics, flexibility, and folding pathways—a key advantage over static DL models. |
| Rosetta [39] [43] | Suite for macromolecular modeling (structure prediction, design, docking). | Classical (Knowledge-based and physics-based energy functions, fragment assembly). | A versatile classical toolkit; used for ab initio folding, loop modeling, and protein design where DL may be less effective. |
| AlphaFold2/3 [3] [2] [41] | Protein and biomolecular complex structure prediction. | Deep Learning (Attention-based neural networks, co-evolutionary data). | The state-of-the-art for single proteins; performance in complexes and ligand binding is under active scrutiny. |
| RoseTTAFold All-Atom [41] | Biomolecular complex structure prediction. | Deep Learning (Three-track neural network). | Similar to AF3; aims to predict proteins, nucleic acids, and small molecules but shows similar physical limitations. |
| I-TASSER [39] | Hierarchical protein structure modeling. | Classical/Hybrid (Threading, fragment assembly, replica-exchange Monte Carlo). | A leading classical method in pre-AlphaFold2 CASP experiments; exemplifies template-based and ab initio refinement. |
| CASP Database [3] | Repository of targets, predictions, and results. | Community Experiment & Benchmarking Platform. | The primary source for objective, blind performance data on all methods, classical and deep learning. |
The evidence from successive CASP experiments makes it clear that the field of computational structural biology is not in a post-deep-learning era, but rather in a hybrid one. While deep learning has conclusively solved the structure prediction problem for a large class of single-domain proteins, its limitations are equally well-documented. Classical, physics-based methods continue to excel in domains where generalization, robust physical understanding, and the modeling of dynamics are paramount. These include predicting the structures of RNA, accurately modeling protein-ligand interactions for drug discovery on novel targets, and simulating the conformational ensembles of flexible macromolecules.
The most promising future direction lies not in a competition between the two paradigms, but in their intelligent integration. As noted in the CASP16 overview, two trends are particularly encouraging: "One is the combination of traditional physics-inspired methods and deep learning, and the other is the expected increase in training data, especially for ligand-protein complexes" [40]. For researchers and drug development professionals, this implies a continued need for a diverse toolkit. The choice of method must be problem-specific, leveraging the sheer predictive power of deep learning where it is proven to work, while relying on the tried-and-tested physical principles of classical methods to tackle more complex, dynamic, or data-sparse biological questions.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind assessment experiment established in 1994 that has long served as the gold standard for evaluating protein structure prediction methods [1]. This biennial experiment provides an independent mechanism for objectively testing structure prediction methods by challenging researchers to predict protein structures for sequences whose experimental structures are not yet public [7] [44]. For nearly three decades, CASP has catalyzed research, monitored progress, and established the state of the art in protein structure modeling [14].
The 2020 CASP14 assessment marked a transformational moment when DeepMind's AlphaFold2 demonstrated accuracy competitive with experimental structures in a majority of cases [10] [45]. With this breakthrough achieving a median Global Distance Test (GDT) score of 92.4 (where ~90 is considered competitive with experimental methods), the protein structure prediction landscape fundamentally changed [14]. Rather than rendering CASP obsolete, this success prompted a strategic realignment of the experiment's focus toward more complex biological challenges that remain unsolved [7] [46].
Strategic Shifts in CASP's Assessment Categories
Category Realignment in Response to AI Advances
Following the dramatic improvements in single protein chain prediction accuracy demonstrated by AlphaFold2 and subsequent AI methods, CASP has systematically reorganized its assessment categories to address frontiers where significant challenges remain. This restructuring, maintained from CASP15 into CASP16, redirects focus toward biologically relevant complexes and dynamics [7].
Table: Evolution of CASP Assessment Categories Pre- and Post-AlphaFold2
| CASP14 (2020) Categories | CASP16 (2024) Categories | Rationale for Change |
|---|---|---|
| High Accuracy Modeling | Single Proteins and Domains (refined emphasis) | Shift from establishing baseline accuracy to assessing fine-grained precision |
| Template-Based Modeling | - (discontinued as separate category) | Integration into single protein category as methods matured |
| Topology/Free Modeling | - (discontinued as separate category) | Dramatically reduced distinction between template-based and free modeling |
| Assembly | Protein Complexes | Increased focus on subunit interactions with option to predict stoichiometry |
| - | Protein-organic ligand complexes | New category addressing drug design applications |
| - | Nucleic acid structures and complexes | Expanded scope beyond proteins alone |
| - | Macromolecular conformational ensembles | New focus on structural dynamics beyond static structures |
| Data Assisted | Integrative modeling | Reintroduced category combining AI with sparse experimental data |
Quantitative Performance Improvements in CASP15
The strategic shift in categories follows demonstrated success in protein complex prediction in CASP15 (2022), which showed "enormous progress in modeling multimolecular protein complexes" [3]. The accuracy of models almost doubled in terms of the Interface Contact Score (ICS/F1) and increased by one-third in terms of the overall fold similarity score (LDDT₀) [3]. This progress established that deep learning methodology, which had revolutionized monomeric modeling in CASP14, could be successfully extended to multimeric modeling.
Table: CASP15 Performance Metrics Demonstrating Progress in Complex Prediction
| Metric | CASP14 Performance | CASP15 Performance | Improvement | Significance |
|---|---|---|---|---|
| Interface Contact Score (ICS/F1) | Baseline | Nearly doubled | ~100% | Measures accuracy of protein-protein interfaces |
| Fold Similarity Score (LDDT₀) | Baseline | Increased by 1/3 | ~33% | Assesses overall structural accuracy |
| Example Target T1113o | - | F1=92.2; LDDT₀=0.913 | - | Demonstrates near-experimental accuracy for complexes |
Methodological Framework: CASP's Experimental Protocol
Target Selection and Blind Assessment Protocol
CASP's experimental validity hinges on its rigorous blind assessment protocol. The experiment depends on the generous contribution of protein sequences from structural biologists who have recently determined or are in the process of determining structures but have not yet made them public [7] [44]. This ensures that predictors cannot have prior information about the protein's structure that would provide an unfair advantage [1].
The CASP16 timetable follows a standardized protocol [7]:
- Target Release (May 1 - July 31, 2024): Sequences of unknown structures are posted on the CASP website
- Model Submission (May - August 2024): Participants worldwide submit predicted structures
- Evaluation Phase (August - October 2024): Independent assessors compare models to newly released experimental structures
- Conference (December 2024): Results are presented and discussed
Assessment Methodologies and Metrics
CASP employs sophisticated evaluation metrics to assess different aspects of structural accuracy. The primary metric for single protein structures is the Global Distance Test (GDT), which measures the percentage of amino acid residues within a threshold distance from their correct positions [14] [1]. Additional metrics have been developed for specific categories:
- Protein Complexes: Interface Contact Score (ICS/F1) and interface LDDT [3]
- Accuracy Estimation: Predicted local distance difference test (pLDDT) at individual residue level [7]
- Ligand Binding: Atomic accuracy metrics for organic molecule interactions [7]
Independent assessors in each category are leading experts who apply these metrics consistently while retaining the flexibility to incorporate new evaluation methods as the field advances [7].
Frontier Challenges: CASP16 Focus Areas
Protein Complexes and Stoichiometry Prediction
While CASP15 demonstrated substantial progress in modeling multimolecular complexes, accuracy remains lower than for single proteins, creating significant opportunity for advancement [7]. CASP16 introduced the novel challenge of optionally predicting complex stoichiometry—the number and arrangement of subunits in protein assemblies [7]. For suitable targets, CASP16 initially releases sequences without stoichiometric information, collects models, then re-releases targets with stoichiometry data provided, enabling assessment of both scenarios.
Protein-Ligand Interactions and Drug Discovery Applications
The limited success of deep learning methods in predicting protein-organic ligand interactions revealed an important capability gap [7]. CASP16 includes specific target sets related to drug design, recognizing that accurate prediction of small molecule binding is crucial for pharmaceutical applications [7]. This category assesses whether AI methods can now compete with more traditional molecular docking approaches that have historically dominated this domain.
Nucleic Acids and Mixed Macromolecular Complexes
The underwhelming performance of deep learning methods for RNA structure prediction in CASP15 highlighted another frontier [7]. CASP16 has expanded this category to include both RNA and DNA single structures and complexes, as well as complexes of nucleic acids with proteins [7]. This reflects the biological importance of RNA-protein complexes and chromatin organization, while testing whether new architectures can overcome previous limitations.
Conformational Ensembles and Integrative Modeling
CASP16 maintains two categories that address protein dynamics and experimental integration [7]:
- Macromolecular Conformational Ensembles: Assesses methods for predicting multiple conformations and alternative states, moving beyond single static structures
- Integrative Modeling: Evaluates approaches that combine deep learning with sparse experimental data (SAXS, crosslinking) to determine structures of large complexes
Table: Key Research Reagents and Resources in CASP16
| Resource | Type | Function in CASP | Relevance to AlphaFold Era |
|---|---|---|---|
| Protein Data Bank (PDB) | Database | Source of experimental structures for training and validation | AlphaFold was trained on ~170,000 structures from PDB [10] |
| Multiple Sequence Alignments (MSAs) | Bioinformatics | Evolutionary information for co-evolution analysis | Critical input for AlphaFold's Evoformer architecture [10] |
| UniProt Database | Database | Comprehensive protein sequence repository | Source of ~180 million sequences for genomic-scale prediction [14] |
| AlphaFold Server | Software Tool | Provides free access to AlphaFold3 for non-commercial research | Enables broad community access to state-of-the-art prediction [45] |
| CASP Target Database | Database | Repository of all CASP targets, predictions, and results | Enables retrospective analysis and method development [3] |
| pLDDT Confidence Metric | Assessment | Per-residue estimate of prediction reliability | Allows users to identify trustworthy regions of models [10] |
| Cross-linking Mass Spectrometry Data | Experimental | Sparse distance constraints for integrative modeling | Enhances accuracy for large complexes where pure AI struggles [7] |
CASP has successfully navigated the transition from benchmarking basic folding accuracy to addressing more complex biological questions in the AlphaFold era. The experiment continues to catalyze method development by focusing on unsolved challenges: modeling flexible systems, predicting complex assemblies, and characterizing functional interactions. Current assessments probe whether new architectures, including large language models and diffusion-based approaches, can overcome remaining limitations [7].
The fundamental epistemological challenges highlighted by critics—including the limitations of static structures for representing dynamic biological reality and the environmental dependence of protein conformations—continue to shape CASP's evolution [47]. By focusing on conformational ensembles and integrative modeling, CASP acknowledges that the next frontiers involve predicting how structures change in response to cellular conditions, binding partners, and functional states.
For researchers and drug development professionals, CASP's ongoing assessment provides crucial guidance on which biological questions can reliably be addressed with current AI tools, and which still require complementary experimental approaches. As CASP co-founder John Moult noted, "In 2020, news headlines repeated John Moult's words... that artificial intelligence had 'solved' a long-standing grand challenge in biology" [46]. CASP's continued adaptation ensures it remains relevant not by denying this achievement, but by defining what comes next.
Beyond the Scoreboard: Validating CASP Models in the Real World
The Critical Assessment of protein Structure Prediction (CASP) experiments represent the gold standard for independent verification of computational protein structure modeling methods. This whitepaper examines the experimental frameworks and metrics CASP employs to objectively validate prediction accuracy against experimental data. We detail how methodologies from X-ray crystallography, cryo-electron microscopy (cryo-EM), and nuclear magnetic resonance (NMR) spectroscopy provide the ground truth for benchmarking predicted models, with focus on the breakthrough performance of AlphaFold2 in CASP14 and subsequent methodological advancements. The establishment of rigorous blind testing protocols has transformed protein structure prediction from a computational challenge to a practical tool for structural biologists and drug discovery researchers.
The CASP Experimental Framework
Principles of Blind Testing
CASP (Critical Assessment of Structure Prediction) operates as a community-wide experiment conducted every two years since 1994 to advance methods of computing three-dimensional protein structure from amino acid sequence [2] [1]. The core principle involves fully blinded testing where participants predict structures for proteins whose experimental structures are imminent but not yet public [33] [1]. This ensures objective assessment without prior knowledge bias. Targets are obtained through collaborations with experimental structural biologists who provide protein sequences for which structures are soon to be solved by X-ray crystallography, NMR, or cryo-EM but are temporarily withheld from public databases [2] [1].
The CASP infrastructure manages the distribution of target sequences to registered modeling groups worldwide, who then submit their predicted structures within strict timeframes (typically 3 weeks for human groups and 72 hours for automated servers) [2]. This process generates thousands of predictions that are systematically compared to the corresponding experimental structures once they become available. Independent assessment teams then evaluate the results using standardized metrics and methodologies [2] [33].
Target Categorization and Difficulty Assessment
CASP classifies targets based on modeling difficulty and the availability of structural templates:
- TBM (Template-Based Modeling): Targets with detectable sequence or structural similarity to proteins of known structure. These are subdivided into TBM-Easy and TBM-Hard categories based on the straightforwardness of template identification and alignment [2].
- FM (Free Modeling): Targets with no detectable homology to known structures, representing the most challenging prediction category [2] [1].
- FM/TBM: Targets with only remote structural homologies to known folds [2].
In recent CASP experiments, the distinction between these categories has become less pronounced with the advent of deep learning methods that achieve high accuracy even without obvious templates [2]. Additionally, CASP has expanded to include assessments of multimeric protein complexes, structure refinement, model quality estimation, and prediction of inter-residue contacts and distances [3] [2].
Quantitative Metrics for Model Validation
Primary Accuracy Measures
CASP evaluation employs rigorous quantitative metrics to compare predicted models against experimental reference structures:
- GDT_TS (Global Distance Test Total Score): The primary metric measuring the percentage of Cα atoms in the predicted model that fall within a defined distance threshold (typically 1-8 Å) of their correct positions in the experimental structure after optimal superposition. Higher scores indicate better accuracy, with scores above ~90 considered competitive with experimental methods [2] [1] [18].
- LDDT (Local Distance Difference Test): A local consistency measure that evaluates distance differences in a model compared to the native structure, less sensitive to domain movements than global measures [3].
- RMSD (Root Mean Square Deviation): Measures the average distance between equivalent atoms in superimposed structures, particularly useful for assessing local refinement improvements [11].
- Interface Contact Score (ICS/F1): Used specifically for assessing quaternary structure predictions of protein complexes, measuring accuracy at interfacial residues [3].
Table 1: Key Accuracy Metrics in CASP Validation
| Metric | Calculation Method | Interpretation | Application Context |
|---|---|---|---|
| GDT_TS | Percentage of Cα atoms within successive distance thresholds (1, 2, 4, 8 Å) after optimal superposition | 0-100 scale; >90 considered competitive with experimental methods | Overall model accuracy assessment |
| LDDT | Local distance differences between atoms in the model versus native structure | 0-1 scale; more local and less sensitive to domain movements | Model quality estimation, especially for multi-domain proteins |
| Cα RMSD | Root mean square deviation of Cα atomic positions after superposition | Lower values indicate better accuracy; sensitive to outliers | Local structure and refinement assessment |
| ICS/F1 | Precision and recall of interfacial residue contacts in complexes | 0-1 scale; measures interface prediction accuracy | Quaternary structure assessment |
Historical Progress in Prediction Accuracy
CASP data demonstrates remarkable progress in prediction accuracy over its 30-year history. The performance leap in CASP14 (2020) represented a paradigm shift, with AlphaFold2 achieving a median GDT_TS of 92.4 across all targets, rivaling experimental accuracy for approximately two-thirds of targets [2]. This contrasted with previous CASPs where accuracy declined sharply for more difficult targets with fewer available templates.
Table 2: Evolution of Model Accuracy Across CASP Experiments
| CASP Edition | Year | Best System GDT_TS (FM Targets) | Best System GDT_TS (TBM Targets) | Notable Methodological Advances |
|---|---|---|---|---|
| CASP7 | 2006 | ~75 (for small domains) | ~85 | First reasonable ab initio models for small proteins |
| CASP11 | 2014 | ~60 | ~85 | Improved contact prediction enabling first accurate large FM targets |
| CASP13 | 2018 | ~65 | ~88 | Deep learning revolution in contact/distance prediction |
| CASP14 | 2020 | ~85 | ~95 | AlphaFold2 end-to-end deep learning architecture |
| CASP15 | 2022 | ~80 (complexes) | ~90 (complexes) | Extension to multimeric assemblies |
The CASP14 trend line started at a GDT_TS of about 95 for the easiest targets and finished at about 85 for the most difficult targets, indicating only minor benefit from homologous structure information compared to previous experiments where accuracy fell sharply with decreasing evolutionary information [2]. This demonstrated that the new generation of methods could achieve high accuracy primarily from sequence information alone.
Experimental Methodologies for Ground Truth Validation
Experimental Structure Determination Methods
CASP relies on three principal experimental methods to provide the reference structures against which predictions are validated:
X-ray Crystallography: Provides high-resolution atomic structures (typically 1.5-3.0 Å resolution) for proteins that can form regular crystals. The majority of CASP targets (42 of 52 in CASP14) are determined using this method [2].
Cryo-Electron Microscopy (cryo-EM): Particularly valuable for large protein complexes that are difficult to crystallize. Seven CASP14 targets were determined using cryo-EM [2].
Nuclear Magnetic Resonance (NMR) Spectroscopy: Provides solution-state structures and information about protein dynamics. Three CASP14 targets were determined by NMR [2].
Each method has distinct advantages and limitations in resolution, size limitations, and representation of physiological conditions, creating a complementary validation framework.
Assessing Discrepancies: Computational vs. Experimental Error
With the dramatically improved accuracy of computational models in recent CASP experiments, analysis has shifted to carefully interpreting remaining discrepancies between predictions and experimental data:
Experimental Structure Uncertainty: Lower agreement between computation and experiment is observed for lower-resolution X-ray structures and for cryo-EM structures, suggesting experimental uncertainty may limit maximum achievable GDT_TS scores, particularly for values below 90 [2].
Dynamic Protein Structures: For some NMR targets, the best computed structures have been found to agree better with experimental NOE data than the deposited experimental structure, potentially representing legitimate conformations within the protein's dynamic ensemble [2].
Crystal Packing Effects: Minor differences in loop conformations between predictions and crystal structures are often attributable to crystal packing influences rather than computational errors [2].
Table 3: Interpretation of Common Discrepancies Between Models and Experimental Structures
| Discrepancy Type | Potential Computational Cause | Potential Experimental Cause | Validation Approach |
|---|---|---|---|
| Local backbone deviations | Inaccurate loop modeling | Crystal packing constraints | Compare multiple crystal forms or solution NMR data |
| Side chain rotamer errors | Limited rotamer sampling | Radiation damage in crystallography | Analyze B-factors and electron density maps |
| Domain orientation differences | Flexible hinge regions not captured | Solution vs. crystal state differences | SAXS or solution NMR validation |
| Disordered regions | Over-prediction of structure | Genuine structural flexibility | Biochemical proteolysis assays |
Diagram 1: CASP Validation Framework. This diagram illustrates the independent verification process where computational models are validated against experimental data using quantitative metrics, enabling research applications.
Case Study: AlphaFold2 Validation in CASP14
Unprecedented Accuracy Achievement
AlphaFold2's performance in CASP14 represented a watershed moment in protein structure prediction. The system achieved an average GDTTS of 92.4 across all targets, with predictions competitive with experimental accuracy for approximately two-thirds of targets [2] [18]. For comparison, the best system in CASP13 (2018) achieved approximately 60 GDTTS for the most difficult targets, while AlphaFold2 maintained approximately 85 GDT_TS even for the most challenging free-modeling targets [2].
The accuracy was particularly notable at the atomic level, with impressive agreement for both main chain and side chain atoms as demonstrated in the prediction of SARS-CoV-2 ORF8 (target T1064, FM category, GDT_TS 87) [2]. In many cases, the predicted models included structurally plausible conformations for regions that were disordered in the experimental structures due to crystal packing effects.
Independent Experimental Validation
Beyond the CASP competition, AlphaFold2 predictions have been validated through multiple independent experimental approaches:
Molecular Replacement in X-ray Crystallography: AlphaFold2 models successfully serve as search models for molecular replacement, enabling structure determination without traditional experimental phasing [18].
Cryo-EM Density Fitting: Predicted structures show excellent fit into experimental cryo-EM electron density maps, confirming their accuracy for large complexes [18].
Solution-State NMR Validation: AlphaFold2 models demonstrate strong agreement with NMR data obtained in solution, indicating they are not biased toward crystal states despite training predominantly on crystallographic data [18].
Cross-linking Mass Spectrometry: Experimental cross-linking data validates the accuracy of both single-chain predictions and protein-protein complexes in near-native conditions [18].
Research Reagents and Experimental Tools
Table 4: Essential Research Reagents and Tools for Structure Validation
| Reagent/Tool | Function in Validation | Application Context |
|---|---|---|
| CASP Targets Database | Provides standardized datasets for blind testing of prediction methods | Method development and benchmarking |
| GDT_TS Calculation Algorithm | Quantifies global model accuracy against reference structures | Objective model assessment |
| Molecular Replacement Software | Tests predictive models as phasing templates for crystallography | Practical utility assessment |
| Cryo-EM Density Fitting Tools | Evaluates model agreement with experimental electron density | Validation for large complexes |
| NMR Chemical Shift Prediction | Compares computed models with solution-state NMR data | Validation under physiological conditions |
| Cross-linking Mass Spectrometry | Provides experimental distance restraints in native environments | In situ validation of structural models |
Implications for Drug Discovery and Structural Biology
Practical Applications in Research
The independent verification of high-accuracy models through CASP has enabled numerous practical applications:
Accelerated Structure Determination: Four structures in CASP14 were solved using AlphaFold2 models for molecular replacement, demonstrating immediate practical utility for structural biologists [3].
Error Correction in Experimental Structures: In one CASP14 target, provision of computational models resulted in correction of a local experimental error [3].
Functional Insight: Accurate models enable reliable identification of binding sites and functional motifs, directly supporting drug discovery efforts [11].
Complex Assembly Prediction: Recent CASP experiments show enormous progress in modeling multimolecular protein complexes, with accuracy almost doubling in terms of Interface Contact Score between CASP14 and CASP15 [3].
Future Directions
The CASP framework continues to evolve to address new challenges:
Quaternary Structure Modeling: Increased emphasis on accurately predicting multimeric complexes and domain interactions [3] [2].
Refinement Methodologies: Developing methods to consistently improve initial models, with molecular dynamics approaches showing promise [11].
Sparse Data Integration: Effectively combining computational predictions with sparse experimental data from emerging techniques [11].
Accuracy Estimation: Improving methods to reliably estimate model accuracy at the residue level [11].
The independent verification framework established by CASP has transformed protein structure prediction from an intellectual challenge to a practical tool that routinely provides accurate structural models for biological and pharmaceutical research.
The Critical Assessment of protein Structure Prediction (CASP) is a biennial community-wide experiment established in 1994 to objectively assess the state of the art in computing three-dimensional protein structure from amino acid sequence [1]. CASP operates as a rigorous blind test: organizers provide participants with amino acid sequences of proteins whose structures have been experimentally determined but not yet publicly released, and predictors submit their computed models within a limited time frame [44] [1]. These predictions are then compared to the experimental ground truth through independent assessment, providing a benchmark for methodological progress [2]. For decades, CASP has served as the gold-standard assessment for protein structure prediction methods, catalyzing research and driving innovation in this fundamental biological problem [1] [14].
The protein folding problem—predicting a protein's native three-dimensional structure from its one-dimensional amino acid sequence—has stood as a grand challenge in biology for over 50 years [14]. The significance of this problem stems from the central role of protein structure in determining biological function. As noted by DeepMind, "Proteins are the complex, microscopic machines that drive every process in a living cell" [48]. Understanding their structure facilitates mechanistic understanding of their function, with profound implications for drug discovery, disease understanding, and environmental sustainability [10] [14]. The experimental determination of protein structures through techniques like X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy has been painstakingly slow and resource-intensive, creating a massive gap between known protein sequences and determined structures [10] [49]. This disparity highlighted the urgent need for accurate computational methods to predict protein structures at scale.
The CASP14 Experiment: Design and Assessment Methodology
Experimental Framework and Targets
CASP14 was conducted between May and August 2020, with the conference held virtually in November-December due to the COVID-19 pandemic [44] [2]. The experiment collected predictions for 52 proteins and protein complexes, determined primarily by X-ray crystallography (42 targets), cryo-electron microscopy (7 targets), and NMR (3 targets) [2]. These were divided into 68 tertiary structure modeling targets, which were further split into 96 evaluation units based on domain structure for assessment purposes [2]. CASP14 introduced several methodological refinements, including expanded assessment of inter-residue distance predictions and enhanced focus on modeling oligomeric proteins and protein-protein complexes [44].
Assessment Metrics and Criteria
The primary metric used for evaluating prediction accuracy in CASP is the Global Distance Test (GDTTS), which measures the percentage of amino acid residues within a threshold distance from their correct positions after optimal superposition [1] [14]. GDTTS scores range from 0-100, with scores around 90 considered competitively accurate compared to experimental methods [14]. According to CASP organizers, "a score of around 90 GDT is informally considered to be competitive with results obtained from experimental methods" [14]. Additional evaluation metrics included:
- Local Distance Difference Test (lDDT): A residue-wise quality measure that evaluates local structural consistency [50]
- Template Modeling Score (TM-score): A metric for measuring structural similarity [10]
- Root-mean-square deviation (RMSD): Measuring average atomic distances between predicted and experimental atomic positions [10]
Targets were categorized by difficulty into four classes: TBM-Easy (straightforward template modeling), TBM-Hard (difficult homology modeling), FM/TBM (remote structural homologies), and FM (free modeling, no detectable homology) [2].
Table 1: CASP14 Target Difficulty Classification
| Category | Description | Historical Performance (Pre-CASP14) |
|---|---|---|
| TBM-Easy | Straightforward template-based modeling | High accuracy (GDT_TS >85) |
| TBM-Hard | Challenging homology modeling | Moderate accuracy (GDT_TS 70-85) |
| FM/TBM | Remote structural homologies only | Lower accuracy (GDT_TS 50-70) |
| FM | Free modeling, no detectable homology to known structures | Lowest accuracy (GDT_TS <50 in CASP13) |
AlphaFold2: Technical Architecture and Methodological Innovations
AlphaFold2 represented a complete redesign from its predecessor used in CASP13, employing a novel end-to-end deep neural network trained to produce protein structures from amino acid sequence, multiple sequence alignments (MSAs), and homologous proteins [50]. The system directly predicts the 3D coordinates of all heavy atoms for a given protein using the primary amino acid sequence and aligned sequences of homologues as inputs [10]. The architecture comprises two main components: the Evoformer (a novel neural network block that processes inputs) and the structure module (which generates atomic coordinates) [10].
The neural network incorporates several key technical innovations that enabled its breakthrough performance:
- Joint embedding of multiple sequence alignments and pairwise features
- Equivariant attention architecture that respects the geometric symmetries of protein structures
- End-to-end differentiable structure learning that enables direct prediction of atomic coordinates
- Iterative refinement through recycling where outputs are recursively fed back into the network
- Novel loss functions that emphasize both positional and orientational correctness [10]
The Evoformer: Processing Evolutionary and Structural Information
The Evoformer constitutes the trunk of the AlphaFold2 network and represents a fundamental architectural innovation. It processes inputs through repeated layers to produce two key representations: an Nseq × Nres array representing a processed MSA and an Nres × Nres array representing residue pairs [10]. The Evoformer implements a graph-based inference approach where residues are treated as nodes and their relationships as edges [10]. Key operations within the Evoformer include:
- MSA-to-pair representation updates through element-wise outer products summed over the MSA sequence dimension
- Triangle multiplicative updates that enforce geometric consistency through triangular attention mechanisms
- Axial attention with biases projected from the pair representation to the MSA attention [10]
This architecture enables continuous information exchange between the evolving MSA representation and the pair representation, allowing the network to reason simultaneously about evolutionary constraints and physical interactions.
Structure Module and Output Representation
The structure module introduces an explicit 3D structure through a rotation and translation (rigid body frame) for each residue of the protein [10]. These representations are initialized trivially but rapidly develop into a highly accurate protein structure with precise atomic details. Innovations in this module include:
- Breaking the chain structure to allow simultaneous local refinement of all structure parts
- Equivariant transformer that implicitly reasons about unrepresented side-chain atoms
- Precise atomic modeling that extends beyond backbone atoms to include full heavy atom positions [10]
The network produces not only atomic coordinates but also auxiliary outputs including a distogram (pairwise distance distribution) and a predicted lDDT (pLDDT) confidence measure that reliably estimates the per-residue accuracy of the prediction [10] [50].
CASP14 Results: Quantitative Performance Analysis
AlphaFold2 achieved unprecedented accuracy in CASP14, with the assessors' ranking showing a summed z-score of 244.0 compared to 90.8 by the next best group [50]. The system predicted high-accuracy structures (GDTTS > 70) for 87 out of 92 domains, structures on par with experimental accuracy (GDTTS > 90) for 58 domains, and achieved a median domain GDTTS of 92.4 overall [50]. This performance marked a dramatic improvement over previous CASP experiments, with the CASP14 trend line starting at a GDTTS of about 95 for easy targets and finishing at about 85 for the most difficult targets [3] [2].
Perhaps the most significant achievement was AlphaFold2's performance on free modeling targets—those with no detectable homology to known structures. For these most challenging cases, AlphaFold2 achieved a median score of 87.0 GDT, dramatically outperforming all previous methods and demonstrating that high-accuracy prediction was possible even without evolutionary information from homologous structures [14].
Table 2: AlphaFold2 CASP14 Performance by Target Category
| Target Category | Median GDT_TS | Comparison to CASP13 Best | Experimental Competitiveness |
|---|---|---|---|
| Overall | 92.4 | ~40 point improvement | 2/3 of targets |
| TBM-Easy | ~95 | ~15 point improvement | Nearly all targets |
| TBM-Hard | ~90 | ~25 point improvement | Majority of targets |
| FM/TBM | ~87 | ~35 point improvement | Approximately half of targets |
| FM (Free Modeling) | 87.0 | ~40 point improvement | Significant portion of targets |
Atomic-Level Accuracy and Backbone Precision
Beyond global fold accuracy, AlphaFold2 achieved remarkable atomic-level precision. The system produced structures with a median backbone accuracy of 0.96 Å RMSD95 (Cα root-mean-square deviation at 95% residue coverage), compared to 2.8 Å for the next best method [10]. As noted in the Nature paper, "the width of a carbon atom is approximately 1.4 Å," highlighting that AlphaFold2's predictions were within atomic resolution [10]. All-atom accuracy was similarly impressive at 1.5 Å RMSD95 compared to 3.5 Å for the best alternative method [10].
The quality of side-chain predictions was exceptional when the backbone was accurately predicted, enabling realistic atomic models suitable for molecular docking and detailed functional analysis [10]. The system's internal confidence measure (pLDDT) proved highly correlated with actual accuracy, allowing researchers to identify reliable regions of predicted structures [10] [50].
Experimental Protocols and Methodological Details
AlphaFold2 was trained on publicly available data consisting of approximately 170,000 protein structures from the Protein Data Bank combined with large databases containing protein sequences of unknown structure [14]. The training incorporated novel procedures including:
- Intermediate losses to achieve iterative refinement of predictions
- Masked MSA loss to jointly train with the structure
- Learning from unlabeled protein sequences using self-distillation [10]
The computational resources required were substantial but relatively modest in the context of large state-of-the-art machine learning models: approximately 16 TPUv3s (equivalent to 128 TPUv3 cores or roughly 100-200 GPUs) run over a few weeks [14].
CASP14 Implementation and Model Selection
During CASP14, AlphaFold2 was operated with specific protocols to ensure optimal performance:
- Five predictions per target using five sets of model parameters, ranked by pLDDT for selection [50]
- Extensive testing and monitoring through unit tests and integration tests to detect regressions [50]
- Structural relaxation using gradient descent on Amber99sb with harmonic restraints to remove stereochemical violations [50]
- Visualization and quality checks using standardized notebooks to examine pLDDT, distance matrices, and structural diversity [50]
In most cases, predictions were generated automatically without manual intervention. For a small number of challenging targets (such as T1024, a transporter protein with multiple conformational states), limited manual interventions were applied to capture structural diversity, though subsequent improvements to AlphaFold2 automated these processes [50].
Table 3: Key Research Reagent Solutions for Protein Structure Prediction
| Resource Category | Specific Tools/Sources | Function in Prediction Pipeline |
|---|---|---|
| Sequence Databases | UniProt, Pfam, Conserved Domain Database (CDD) | Source of evolutionary information through multiple sequence alignments |
| Structure Databases | Protein Data Bank (PDB), Structural Classification of Proteins (SCOP) | Source of templates and structural fragments for modeling |
| MSA Generation Tools | HHblits, JackHMMER, HMMER | Identification of evolutionarily related sequences for covariance analysis |
| Template Identification | HHSearch, BLAST, PSI-BLAST | Detection of homologous structures for template-based modeling |
| Force Fields | Amber99sb, CHARMM, Rosetta | Energy functions for structural refinement and steric clash removal |
| Assessment Metrics | GDT_TS, lDDT, TM-score, RMSD | Quantitative evaluation of prediction accuracy relative to experimental structures |
| Visualization Tools | PyMOL, ChimeraX, NGL Viewer | Three-dimensional visualization and analysis of predicted structures |
Limitations and Future Directions
Despite its extraordinary performance, AlphaFold2 has certain limitations. The system shows reduced accuracy for proteins with low sequence complexity or intrinsic disorder, regions that may not adopt stable structures [49]. Prediction quality can also decrease for highly dynamic proteins that sample multiple conformational states, as the network typically produces a single structure [50] [2]. Additionally, while AlphaFold2 excels at single-chain prediction, modeling of protein complexes and interactions with other molecules initially remained challenging, though subsequent versions have addressed this limitation [48] [51].
The CASP14 assessment revealed that some disagreements between AlphaFold2 predictions and experimental structures stemmed from genuine experimental limitations rather than computational errors [2]. For lower-resolution X-ray structures and cryo-EM maps, the computational models sometimes provided more accurate representations than the experimental data, highlighting the potential for computational methods to complement experimental structural biology [2].
Future directions for the field include integrating protein language models that can extract evolutionary patterns directly from sequence databases without explicit alignment steps [49] [51]. There is also growing emphasis on incorporating physicochemical principles more explicitly to improve predictions for complex systems including membrane proteins, large complexes, and designed proteins [51]. The development of generative models for protein design represents another exciting frontier, with AlphaFold-inspired methods now being used to create novel protein structures not found in nature [48].
AlphaFold2's performance in CASP14 represented a watershed moment for structural biology and computational biophysics. The system's ability to predict protein structures with atomic accuracy has fundamentally changed the landscape of biological research, providing scientists with a powerful tool to explore protein function and design therapeutic interventions [48]. The subsequent release of the AlphaFold Protein Database in partnership with EMBL-EBI placed structural predictions for nearly all catalogued proteins into the hands of researchers worldwide, accelerating scientific discovery at unprecedented scale [48].
The breakthrough demonstrated that AI systems could solve fundamental scientific problems that had resisted solution for decades, establishing a template for how artificial intelligence can accelerate scientific progress more broadly [48] [14]. As noted by DeepMind, "Biology was our first frontier, but we view AlphaFold as the template for how AI can accelerate all of science to digital speed" [48]. The AlphaFold2 achievement in CASP14 thus represents not just a solution to a 50-year-old grand challenge, but a paradigm shift in how scientific research is conducted and how biological complexity can be understood through computational intelligence.
The Critical Assessment of Protein Structure Prediction (CASP) is a community-wide, biannual experiment that has served as the gold standard for objectively testing protein structure prediction methods since 1994 [1]. Operating as a rigorous blind test, CASP provides amino acid sequences for proteins with soon-to-be-solved but unpublished experimental structures. Predictors submit their models, and independent assessors evaluate them using established metrics, delivering an unbiased assessment of the state of the art [2] [1]. This framework provides the perfect backdrop for analyzing the seismic shift from classical physics-based methods to modern deep learning approaches, charting the progress that has fundamentally reshaped the field.
Classical Methods: Foundations in Physics and Homology
Classical computational methods for protein structure prediction can be broadly categorized into two paradigms: those based on physical principles and those leveraging evolutionary information.
2.1 Physical and Ab Initio Methods These methods attempt to predict protein structure from sequence alone by simulating the physical folding process. They rely on physics-based force fields that describe atomic interactions, bond angles, and torsions, and often use techniques like molecular dynamics or fragment assembly to search for the lowest-energy conformation [10]. While conceptually pure, these methods were historically limited by the computational intractability of simulating protein folding and the difficulty of producing sufficiently accurate energy functions. Success was largely confined to small proteins [3] [10].
2.2 Template-Based and Homology Modeling When a protein sequence shares significant similarity with a sequence of known structure (a "template"), comparative modeling can be highly effective. Classical methods use sequence alignment tools (e.g., BLAST, HHsearch) to identify templates and then build models by satisfying spatial restraints derived from the template structures [1] [52]. The accuracy of these models is heavily dependent on the sequence identity between the target and the template. Before the rise of deep learning, template-based modeling produced the most accurate predictions in CASP, but its performance dropped sharply for targets with only distant or no detectable homologs [2] [52].
The Deep Learning Revolution: Methodology and Mechanisms
The incorporation of deep learning, particularly since CASP13, has transformed protein structure prediction. These methods use vast amounts of data to learn complex relationships between sequence and structure.
3.1 Architectural Innovations Deep learning models like AlphaFold2 employ novel neural network architectures specifically designed for protein data.
- Evoformer and MSA Processing: A key innovation in AlphaFold2 is the Evoformer block, a neural network module that jointly processes a Multiple Sequence Alignment (MSA) and a representation of residue pairs [10]. The MSA provides evolutionary information, and the Evoformer uses attention mechanisms to reason about the relationships between sequences and residues, effectively inferring evolutionary couplings and structural constraints.
- Equivariant Networks and the Structure Module: The structure module in AlphaFold2 introduces an explicit 3D structure. It uses a special type of network called an equivariant transformer, which ensures that rotations and translations of the input data result in corresponding transformations of the output, a crucial property for handling 3D coordinates [10]. This module is initialized from scratch and iteratively refines the atomic coordinates to produce the final model.
3.2 Key Workflow and Information Flow The following diagram illustrates the core logical workflow of a deep learning system like AlphaFold2, highlighting how it integrates different types of information.
Quantitative Performance Comparison in CASP
The CASP experiments provide clear, quantitative evidence of the dramatic performance gap between classical and deep learning-era methods.
4.1 Overall Accuracy and the AlphaFold2 Breakthrough The table below summarizes the performance leap observed across key CASP rounds, using the Global Distance Test (GDT_TS), a primary CASP metric that measures the percentage of residues in a model placed within a threshold distance of their correct position in the experimental structure (a score of 100 is perfect) [1].
Table 1: Historical CASP Performance Trends
| CASP Round (Year) | Representative Method | Approach | Average GDT_TS (FM/TBM Targets) | Key Limitation |
|---|---|---|---|---|
| CASP12 (2016) | Leading Non-DL Methods | Template-Based & Ab Initio | ~40-60 [52] | Sharp accuracy decline for targets without templates. |
| CASP13 (2018) | AlphaFold1 | Deep Learning (Contacts) | ~65.7 [3] | Major improvement for Free Modeling (FM) targets. |
| CASP14 (2020) | AlphaFold2 | End-to-End Deep Learning | ~85-95 [3] [2] | Accuracy competitive with experiment for most single-chain proteins. |
CASP14 represented a paradigm shift. The trend line for the best models started at a GDT_TS of about 95 for easy targets and only fell to about 85 for the most difficult targets, a minor decline compared to the steep fall-off in pre-deep-learning CASPs [2]. Assessors concluded that for single protein chains, the structure prediction problem could be considered largely solved [2].
4.2 Performance Across Prediction Categories Deep learning has advanced all areas of structure prediction, as shown in the comparative table below.
Table 2: Method Performance by CASP Category
| Prediction Category | Classical Method Performance | Deep Learning Method Performance | Key Deep Learning Advance |
|---|---|---|---|
| Template-Free Modeling | Low accuracy (GDT_TS ~50-60); limited to small proteins [3]. | High accuracy (GDT_TS ~85) even for large proteins [3] [2]. | Use of predicted residue-residue distances and contacts from MSAs [3] [52]. |
| Template-Based Modeling | Highly dependent on template quality; minor improvements over direct copying [3]. | Significantly surpasses accuracy of simple template copying (Avg. GDT_TS=92) [3]. | Integration and refinement of template information within a deep learning framework. |
| Quaternary Structure | Models accurate only when templates existed for the whole complex [3]. | Accuracy nearly doubled (in terms of Interface Contact Score) in CASP15 [3]. | Extension of deep learning (e.g., AlphaFold-Multimer) to model multimeric interfaces [3] [24]. |
| Refinement | Molecular dynamics could provide modest, consistent improvements [3]. | Aggressive methods show dramatic improvements on some targets, but lack consistency [3]. | Potentially larger improvements but higher risk of model degradation. |
| Ligand Binding (Co-folding) | Docking tools (e.g., AutoDock Vina) were standard, with limited accuracy (~60% with known site) [41]. | New co-folding models (e.g., AF3, RFAA) show high initial accuracy (>90%) [41]. | Critical Limitation: Models show physical implausibilities in adversarial tests and may overfit training data [41]. |
Experimental Protocols for Validation
5.1 The CASP Evaluation Protocol
- Target Selection & Sequence Release: Experimental groups provide protein sequences for soon-to-be-public structures. The targets are categorized by difficulty (TBM-Easy, TBM-Hard, FM/TBM, FM) based on similarity to known structures [2] [1].
- Prediction Period: Participants (both automated servers and human expert groups) submit predicted 3D coordinate models within a set timeframe (e.g., 3 days for servers, 3 weeks for experts) [52].
- Blind Assessment: Independent assessors compare models to the newly released experimental structures using metrics like GDT_TS, LDDT, and RMSD without knowing the identity of the predictors [1] [52].
- Results & Analysis: All models, results, and assessments are made public, allowing for a comprehensive analysis of method capabilities and progress [3].
5.2 Protocol for Testing Physical Robustness Recent studies have developed specific protocols to test the physical understanding of deep learning co-folding models like AlphaFold3 and RoseTTAFold All-Atom [41]:
- Binding Site Mutagenesis: Select all binding site residues forming contacts with a ligand (e.g., ATP in Cyclin-dependent kinase 2).
- Adversarial Mutations: Systematically mutate these residues to glycine (removing side-chain interactions) or phenylalanine (sterically occluding the pocket).
- Prediction & Analysis: Run the co-folding models on the mutated sequences. A physically robust model should predict the ligand is displaced. Current models, however, often continue to place the ligand in the original site, revealing a lack of true physical understanding and potential overfitting [41].
The Scientist's Toolkit: Essential Research Reagents
The following table details key resources and their functions in modern protein structure prediction research.
Table 3: Key Resources for Protein Structure Prediction Research
| Resource Name | Type | Primary Function | Access |
|---|---|---|---|
| AlphaFold DB [24] | Database | Provides over 200 million pre-computed AlphaFold2 protein structure predictions. | Publicly Accessible |
| ColabFold [24] | Software Suite | Combines fast homology search (MMseqs2) with AlphaFold2 for accelerated protein structure and complex prediction. | Public Server / Local Install |
| Robetta [24] | Web Server | A protein structure prediction service offering both RoseTTAFold and AlphaFold2-based modeling. | Public Server |
| trRosetta [24] | Web Server | Provides protein structure prediction using the transform-restrained Rosetta protocol. | Public Server |
| ESMFold [24] | Software & Database | A rapid sequence-to-structure predictor; used to create the ESM Metagenomics Atlas. | Publicly Accessible |
| CAMEO [24] [52] | Evaluation Platform | A continuous, automated benchmarking platform for 3D structure prediction servers based on weekly PDB pre-releases. | Publicly Accessible |
The comparative analysis, framed by the CASP experiment, unequivocally demonstrates that deep learning methods have superseded classical approaches in accuracy and reliability for predicting single protein chains. However, the transition is not a complete victory. Deep learning models, particularly the latest co-folding systems, exhibit critical vulnerabilities when probed for their understanding of physical principles, showing overfitting and an inability to generalize in response to biologically plausible perturbations [41]. The future of the field lies not in choosing between classical and deep learning approaches, but in their synthesis. Integrating the data-driven power of deep learning with the robust, first-principles physics of classical methods will be essential to build models that are not only accurate but also truly generalizable and reliable for the most demanding applications in drug discovery and protein engineering.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind experiment that has been conducted every two years since 1994 to objectively assess the state of the art in computing protein three-dimensional structure from amino acid sequence [1]. CASP operates as a global benchmark where research groups worldwide test their structure prediction methods on proteins whose structures have been experimentally determined but are not yet publicly available [2] [1]. This double-blind experimental design ensures that predictors cannot have prior knowledge of the target structures, creating a rigorous testing environment that has catalyzed remarkable progress in the field of computational biology [1]. The primary goal of CASP is to help advance methods of identifying protein structure from sequence through independent assessment and blind testing, establishing current capabilities and limitations while highlighting where future efforts may be most productively focused [3].
CASP's significance extends far beyond a simple competition—it represents a unique scientific ecosystem that systematically drives innovation through standardized evaluation, community engagement, and collaborative knowledge sharing. By providing an objective framework for assessing methodological advances, CASP has created a fertile environment for both competition and cooperation among research groups, accelerating progress on one of biochemistry's most fundamental challenges. The experiment has evolved significantly from its early focus on establishing basic prediction capabilities to its current role in pushing the boundaries of atomic-level accuracy and expanding into new frontiers like protein complexes and RNA structures [26]. This evolution reflects both the tremendous progress made in computational structural biology and CASP's adaptive framework for nurturing innovation.
The CASP Experimental Framework
Core Methodology and Evaluation Metrics
The CASP experiment follows a meticulously designed protocol that maintains rigorous blind testing standards while accommodating the evolving needs of the research community. The process begins with target identification, where proteins with soon-to-be-solved structures are recruited from structural biology laboratories worldwide [1]. These targets are typically structures that have just been solved by X-ray crystallography, NMR spectroscopy, or cryo-electron microscopy and are kept on hold by the Protein Data Bank until the prediction season concludes [1]. During the prediction phase, participating research groups receive only the amino acid sequences of these targets and typically have three weeks to submit their computed structures, while automated servers must return models within 72 hours [2].
The evaluation methodology employs sophisticated comparative metrics to assess prediction accuracy:
- GDT_TS (Global Distance Test Total Score): The primary evaluation metric that measures the percentage of well-modeled residues in the prediction by calculating the largest set of Cα atoms that fall within a defined distance threshold from their correct positions [1]. This metric ranges from 0-100, with higher scores indicating better accuracy.
- GDT_HA (Global Distance Test High Accuracy): A more stringent variant that uses tighter distance thresholds to evaluate high-accuracy predictions [53].
- Local Distance Difference Test (lDDT): A local superposition-free score that evaluates the accuracy of local atomic environments [10].
- Interface Contact Score (ICS/F1): Used specifically for assessing the accuracy of multimeric complexes by measuring interface residue contacts [3].
Table 1: Key CASP Evaluation Metrics and Their Applications
| Metric | Full Name | Primary Application | Interpretation |
|---|---|---|---|
| GDT_TS | Global Distance Test Total Score | Overall backbone accuracy | Percentage of Cα atoms within distance thresholds (1-10Å) |
| GDT_HA | Global Distance Test High Accuracy | High-accuracy backbone assessment | More stringent distance thresholds (0.5-3Å) |
| lDDT | local Distance Difference Test | Local structure and side-chain accuracy | Evaluation without global superposition |
| ICS/F1 | Interface Contact Score | Quaternary structure assessment | Precision/recall for interface residue contacts |
| RMSD | Root Mean Square Deviation | Atomic-level accuracy | Atomic coordinate deviation after superposition |
Prediction Categories and Experimental Design
CASP organizes assessment into specialized categories that reflect the diverse challenges in structure prediction. These categories have evolved significantly over time, responding to both methodological advances and emerging research priorities:
- Tertiary Structure Prediction: The core category assessing overall protein structure, historically divided into Template-Based Modeling (TBM) for targets with detectable homology to known structures, and Free Modeling (FM) for novel folds without templates [11] [1]. CASP15 eliminated this distinction due to diminished relevance following the accuracy revolution [26].
- Assembly Modeling: Assessment of multimeric complexes, including domain-domain, subunit-subunit, and protein-protein interactions, conducted in collaboration with CAPRI (Critical Assessment of Predicted Interactions) [2] [26].
- Refinement: Evaluation of methods to improve initial models, typically using molecular dynamics approaches [3] [11].
- Contact and Distance Prediction: Assessment of residue-residue contact prediction accuracy, which was retired after CASP14 as contact prediction became integrated into end-to-end methods [26].
- Model Accuracy Estimation: Evaluation of methods for predicting the reliability of their own models [11] [26].
- Emerging Categories: CASP15 introduced new categories including RNA structures and complexes, protein-ligand complexes, and protein conformational ensembles, reflecting the expanding scope of computational structural biology [26].
The CASP timeline follows a biennial rhythm that structures the research cycle in the field. Target sequences are released from May through July, with predictions collected through August [26]. The following months are dedicated to independent assessment by appointed experts in each category, culminating in a conference where results are presented and discussed, followed by publication of proceedings in a special issue of the journal Proteins [26] [1]. This regular cycle creates a predictable framework that helps research groups coordinate their development efforts and provides consistent benchmarking for methodological advances.
Quantitative Assessment of Progress Through CASP
The impact of CASP on methodological advances is clearly demonstrated through the quantitative progress observed across successive experiments. The data reveal periods of incremental improvement punctuated by breakthrough advances, particularly with the introduction of deep learning methods.
Table 2: Historical Progress in CASP Backbone Accuracy (GDT_TS)
| CASP Edition | Year | Notable Methods | Average FM Performance | Key Advancements |
|---|---|---|---|---|
| CASP4 | 2000 | Early ab initio | Limited accuracy | First reasonable ab initio models for small proteins [3] |
| CASP7 | 2006 | Fragment assembly | GDT_TS~75 for best case [3] | First atomic-level structure determination using models [3] |
| CASP11 | 2014 | Co-evolution methods | GDT_TS~50 | Accurate contact prediction enables first large protein (256 residues) FM [3] [11] |
| CASP12 | 2016 | Improved contact prediction | GDT_TS~53 | Average precision of best contact predictor doubles to 47% [3] |
| CASP13 | 2018 | Deep learning contacts | GDT_TS~66 (20% increase) [3] | Widespread adoption of deep convolutional networks [53] |
| CASP14 | 2020 | AlphaFold2 end-to-end | GDT_TS~85 for difficult targets [2] | Atomic accuracy competitive with experiment for 2/3 of targets [10] [2] |
| CASP15 | 2022 | AlphaFold2 extensions | ICS doubled from CASP14 [3] | Major advances in multimeric complexes [3] |
The tabulated data demonstrates the remarkable acceleration in progress, particularly from CASP13 onward. Prior to the deep learning revolution, progress in the challenging Free Modeling category had been incremental, with the first accurate model of a larger protein (256 residues) not appearing until CASP11 [3] [11]. The incorporation of co-evolutionary information and early deep learning methods in CASP12 and CASP13 produced significant gains, but the extraordinary performance of AlphaFold2 in CASP14 represented a qualitative leap, with the trend line for model accuracy starting at GDT_TS of about 95 for easy targets and finishing at about 85 for the most difficult targets [2].
The impact of these advances is further illustrated by the changing relationship between prediction accuracy and target difficulty. Historically, model accuracy showed a sharp decline for targets without homologous templates, but CASP14 demonstrated only a minor fall-off in agreement with experiment despite decreasing evolutionary information [2]. This suggests that modern methods have substantially reduced dependence on homologous structure information, with models becoming only marginally more accurate when such information is available compared to cases where it is not [2].
Diagram 1: CASP Methodological Evolution
The visualization illustrates three distinct eras in CASP's history: the early period focused on establishing basic capabilities, the co-evolution era that dramatically improved contact prediction, and the current deep learning era that has revolutionized accuracy. Each transition represents not just incremental improvements but paradigm shifts in methodology, with the most recent era showing unprecedented acceleration in capabilities.
Methodological Innovations Catalyzed by CASP
The AlphaFold Breakthrough
The development of AlphaFold by DeepMind represents one of the most significant breakthroughs catalyzed by the CASP framework. AlphaFold's journey through CASP demonstrates how the experiment's competitive yet collaborative environment drives methodological innovation:
- CASP13 (2018): The first DeepMind entry, utilizing deep residual networks to predict discrete distance distributions and incorporating co-evolutionary analysis from multiple sequence alignments (MSAs). This approach placed first in the Free Modeling category by a significant margin [53].
- CASP14 (2020): AlphaFold2 introduced a completely redesigned end-to-end architecture that directly predicted atomic coordinates from sequence data, achieving median backbone accuracy of 0.96 Å RMSD95—approximately three times more accurate than the next best method and competitive with experimental structures in most cases [10] [54].
- Post-CASP14: The methods were made openly available, and by CASP15, virtually all high-ranking teams used AlphaFold or modifications of it, demonstrating how successful approaches rapidly disseminate through the community [1].
The technical innovations in AlphaFold2 were profound, centered on two key components:
- Evoformer: A novel neural network architecture that jointly embeds multiple sequence alignments and pairwise features through attention-based mechanisms, enabling direct reasoning about spatial and evolutionary relationships [10].
- Structure Module: An equivariant transformer that explicitly represents 3D structure through rotations and translations for each residue, enabling end-to-end learning from sequence to structure [10].
These innovations were validated not just through CASP evaluation but through real-world applications, with AlphaFold2 models helping to solve four crystal structures in CASP14 that would otherwise have been difficult to determine experimentally [3].
Collaborative Initiatives: The WeFold Project
Beyond individual methodological breakthroughs, CASP has actively fostered collaborative frameworks that amplify innovation through resource and expertise sharing. The WeFold project exemplifies this collaborative spirit, creating an "incubator" environment where researchers combine method components into hybrid pipelines [55].
WeFold was initiated in 2012 to address significant roadblocks in protein structure prediction, particularly the multi-step nature of the problem and the diversity of approaches to these steps [55]. The project established a flexible infrastructure where researchers could insert components of their methods—such as refinement and quality assessment—into integrated pipelines that participated in CASP as individual teams [55]. This cooperative competition or "coopetition" model allowed participants to benefit from shared expertise while still competing to advance the state of the art.
The impact of WeFold demonstrates the power of structured collaboration:
- WeFold2 (CASP11): Included 21 groups participating in 23 different pipelines combining four major decoy generators and multiple contact-prediction methods [55].
- WeFold3 (CASP12): Involved 16 groups participating in 12 pipelines, showing measurable improvement over the previous iteration [55].
- Knowledge Dissemination: The project created standardized interfaces between method components, making it easier to exchange elements between existing methods and lowering barriers for new researchers [55].
WeFold's infrastructure documented the entire information flow through prediction pipelines, creating valuable datasets for method development and enabling systematic analysis of which component combinations produced the best results [55]. This exemplifies how CASP fosters both competition and cooperation, accelerating progress through shared resources while maintaining rigorous independent evaluation.
The Scientist's Toolkit: CASP Research Reagents
The CASP ecosystem has generated and standardized numerous essential resources that constitute the fundamental toolkit for protein structure prediction research.
Table 3: Essential Research Resources in Protein Structure Prediction
| Resource Category | Specific Tools/Databases | Function in Research | CASP Role |
|---|---|---|---|
| Sequence Databases | UniProt/TrEMBL, GenBank | Provide evolutionary information via MSAs | Foundation for co-evolution methods [53] |
| Structure Databases | Protein Data Bank (PDB) | Template structures for comparative modeling | Source of experimental structures for validation [55] |
| Specialized Databases | AlphaFold Protein Structure Database | Over 200 million predicted structures | Enables template-based modeling for nearly all sequences [54] |
| Method Software | Rosetta, I-TASSER, MODELLER | Frameworks for structure prediction and refinement | Core methods tested and improved through CASP [11] [53] |
| Evaluation Software | LGA, TM-score, lDDT | Standardized metrics for model accuracy | CASP-provided tools for objective assessment [1] |
| Collaborative Platforms | WeFold pipelines, CASP Results Database | Infrastructure for method combination and analysis | Enables large-scale collaboration and data sharing [55] |
Experimental Protocols and Workflows
The methodological advances catalyzed by CASP have established sophisticated experimental protocols that represent the current state of the art in protein structure prediction. These workflows integrate multiple data sources and computational steps to generate accurate structural models.
Standard Protein Structure Prediction Workflow:
Input Sequence Analysis: Begin with the amino acid sequence of the target protein and search for homologous sequences across genomic databases to construct deep multiple sequence alignments (MSAs) [10] [53].
Template Identification: Search for structurally homologous templates in the Protein Data Bank using sequence alignment (BLAST, HHsearch) or protein threading methods [1].
Feature Extraction: Generate evolutionary coupling information from MSAs using statistical methods (e.g., GREMLIN) or deep learning approaches to identify residue-residue contacts and distance constraints [11] [53].
Model Generation:
Model Refinement: Apply molecular dynamics-based methods or specialized refinement algorithms to improve local geometry and steric clashes, particularly in regions with template bias or poor accuracy [3] [11].
Model Selection: Use quality assessment methods to identify the most accurate models from generated decoys, employing either standalone accuracy estimation methods or built-in confidence metrics (e.g., pLDDT in AlphaFold) [10] [11].
Validation: Compare final models against experimental structures using CASP metrics (GDT_TS, lDDT, RMSD) when available, or use internal validation measures for real-world applications [2] [1].
Diagram 2: Protein Structure Prediction Workflow
The workflow illustrates the integrated nature of modern structure prediction, combining evolutionary information from multiple sequence alignments with template-based modeling and de novo approaches. The refinement and selection stages highlight the importance of iterative improvement and quality control—areas that have received significant attention in recent CASP experiments.
Expanding Horizons: New Frontiers and Applications
Evolution of CASP Categories
As core protein structure prediction challenges have been substantially addressed, CASP has strategically expanded its assessment categories to focus on emerging frontiers in computational structural biology. This evolution reflects both the success of the CASP model and the changing needs of the research community:
- Retired Categories: After CASP14, several categories including contact prediction, refinement, and domain-level accuracy estimation were retired because they had either been effectively solved or become integrated into end-to-end methods [26].
- New Emphasis Areas: CASP15 introduced pioneering assessments for RNA structures and complexes, protein-ligand complexes, and protein conformational ensembles, representing the expanding scope of the field [26].
- Enhanced Collaboration: Strengthened partnerships with complementary initiatives including CAPRI (for protein complexes), CAMEO (continuous evaluation), and RNA-Puzzles (for RNA structures) [26].
This strategic refocusing demonstrates CASP's adaptive framework—maintaining rigorous assessment where it remains most needed while redirecting community effort toward unsolved challenges. The introduction of protein-ligand complex prediction addresses critical needs in drug discovery, while the new focus on conformational ensembles recognizes the importance of protein dynamics and alternative states in biological function [26].
Real-World Impact and Applications
The methodological advances catalyzed by CASP have translated into significant practical applications across biological research and drug development:
- Experimental Structure Determination: CASP models have progressively transitioned from theoretical exercises to practical tools for structural biologists. In early CASPs, models occasionally helped solve structures, but by CASP14, AlphaFold2 models were used to determine four crystal structures that would otherwise have been challenging, demonstrating real-world utility [3].
- Drug Discovery: Accurate protein structure predictions enable structure-based drug design for targets that lack experimental structures. The new protein-ligand category in CASP15 directly addresses this application [26].
- Functional Insight: Beyond structural accuracy, CASP has increasingly emphasized the ability of models to provide biological insights, particularly regarding molecular recognition and function-related features [2] [26].
- Database Generation: The AlphaFold Protein Structure Database, containing over 200 million predictions, represents an unprecedented resource for biological research, potentially saving "millions of dollars and hundreds of millions of years in research time" [54].
The broad adoption of these resources—with over 2 million researchers from 190 countries using the AlphaFold database—demonstrates how CASP-driven methodological advances have transcended the competition itself to become foundational tools across biological sciences [54].
The CASP experiment represents a uniquely effective framework for accelerating scientific progress through the strategic combination of blind assessment, competitive incentive, collaborative infrastructure, and community engagement. Over more than two decades, this ecosystem has systematically transformed protein structure prediction from a challenging theoretical problem to a practical tool that routinely produces models competitive with experimental determination. The dramatic progress observed through successive CASP experiments—particularly the revolutionary advances in CASP13 and CASP14—demonstrates how structured scientific assessment can catalyze breakthrough innovations.
The CASP model offers valuable insights for scientific progress more broadly. Its success derives from several key features: the double-blind experimental design that ensures rigorous evaluation, the regular biennial cycle that structures community effort, the categorical organization that focuses attention on specific challenges, the independent assessment that provides authoritative evaluation, and the open publication of results that facilitates knowledge dissemination. These features have created a virtuous cycle where methodological advances are rapidly validated, adopted, and extended across the research community.
As CASP looks toward future challenges—including protein complexes, RNA structures, ligand interactions, and conformational ensembles—its adaptive framework continues to guide the field toward the most pressing unsolved problems. The ecosystem of success cultivated by CASP provides a powerful model for how scientific communities can organize to accelerate progress on complex, foundational challenges with broad implications for biology, medicine, and human health.
The Critical Assessment of protein Structure Prediction (CASP) is a community-wide, blind experiment conducted every two years to objectively test and advance the state of the art in predicting protein three-dimensional structure from amino acid sequence [1]. While its primary goal is methodological assessment, a significant and growing impact of CASP is the demonstrated utility of its computational models in aiding experimental structure determination. This whitepaper details specific, real-world case studies from the CASP experiments where predicted models have transitioned from theoretical constructs to practical tools, directly assisting researchers in solving structures and correcting experimental errors. The advent of advanced deep learning methods, particularly since CASP14, has marked a paradigm shift, with computational models now achieving accuracy competitive with experimental methods for many targets [3] [26], thereby opening new avenues for synergistic computational-experimental approaches in structural biology.
Documented Case Studies in CASP
Early Pioneering Cases: Proof of Concept
The use of CASP models to solve experimental structures, while once rare, provided critical proof of concept that computational predictions could meaningfully assist the experimental process.
- CASP11 Target T0839 (Sla2 ANTH domain): The crystal structure of the Sla2 ANTH domain from Chaetomium thermophilum was determined using molecular replacement with CASP models. This was a notable early exception where a model generated through the blind prediction process successfully served as a phasing model, a technique that had been exceptionally rare in early CASPs [3].
The Inflection Point: CASP14 and the AlphaFold2 Revolution
CASP14 (2020) marked an extraordinary leap in model accuracy, which directly translated into a greater practical impact on experimental structure solution.
- Four Structures Solved with AlphaFold2 Models: During the CASP14 assessment, it was confirmed that four separate protein structures were solved with the direct aid of models generated by the deep learning method AlphaFold2. A post-CASP analysis further indicated that models from other groups would also have been effective in some of these cases. These targets were all considered "hard," with limited or no homology information available for at least some domains, demonstrating the power of the new methods for biologically challenging problems [3].
- Experimental Error Correction: For one other target in CASP14, the provision of highly accurate models directly led to the correction of a local experimental error in the determined structure, highlighting the role of prediction not just in phasing, but in the validation and refinement of experimental data [3].
The Advancement of Multimeric Modeling: CASP15
Progress has extended beyond single chains to complex protein assemblies, a key area for understanding cellular function.
- CASP15 Target T1113o: CASP15 demonstrated enormous progress in modeling multimolecular protein complexes. An impressive example, target T1113o, yielded a model with near-experimental accuracy (Interface Contact Score F1=92.2; LDDT=0.913). While not explicitly mentioned for molecular replacement, the high accuracy of these multimeric models indicates their potential for guiding the solution of complex structures [3].
Table 1: Summary of Documented Case Studies of CASP Models Aiding Experimental Structure Solution
| CASP Round | Target / Example | Reported Impact | Key Method |
|---|---|---|---|
| CASP11 | T0839 (Sla2 ANTH domain) | Structure solved by molecular replacement using a CASP model [3]. | Not Specified |
| CASP14 | Four unspecified targets | Structures solved using AlphaFold2 models [3]. | AlphaFold2 |
| CASP14 | One unspecified target | Provision of models led to the correction of a local experimental error [3]. | AlphaFold2 |
| CASP15 | T1113o | Exemplary high-accuracy model of an oligomeric complex (F1=92.2; LDDT=0.913), showcasing potential for guiding complex solution [3]. | Deep Learning Methods |
Experimental Protocols: Methodology for Utilizing Models
The Workflow of Molecular Replacement Using predicted models
Molecular replacement (MR) is a common method in X-ray crystallography for determining the phases of a protein's diffraction pattern. The typical workflow for employing a CASP-like predicted model in MR is outlined below and visualized in Figure 1.
- Target Identification & Crystallization: A protein target is selected, purified, and crystallized. X-ray diffraction data is collected from the crystal, yielding a diffraction pattern and unit cell parameters.
- Model Generation (Prediction): The amino acid sequence of the target is used to generate a 3D structural model. In a CASP-like blind scenario, this is done without access to the solved experimental structure. Modern pipelines like AlphaFold2 or RoseTTAFold are used for this high-accuracy prediction.
- Model Preparation for MR: The predicted model is prepared for use as a search model in MR software (e.g., Phaser). This involves:
- Trimming: Removing flexible loops and termini that are likely to be poorly modeled and could hinder phasing.
- Editing: Potentially dividing the model into smaller, rigid domains if the protein is large or has multiple domains.
- Molecular Replacement Search: The prepared search model is placed within the crystallographic unit cell of the target. The MR software performs a 6-dimensional search (3 rotational, 3 translational) to find the orientation and position that best explains the observed diffraction data.
- Phasing & Electron Density Map Calculation: Once correctly placed, the search model is used to calculate initial phases. These phases are combined with the experimental diffraction amplitudes to compute an initial electron density map.
- Model Building and Refinement: The initial map is used to build and refine an atomic model of the target protein against the experimental data using software like Coot and Phenix or REFMAC5. The final, refined experimental structure is then validated and deposited in the PDB.
Figure 1: Workflow for Molecular Replacement Using a Predicted Model. The process begins with a protein sequence and crystal, proceeds through model prediction and preparation, and culminates in phasing, model building, and refinement to produce a solved structure.
Data-Assisted Modeling with Sparse Experimental Data
Another emerging protocol involves integrating computational models with sparse, low-resolution experimental data to determine structures that are difficult to solve by conventional means. CASP experiments have begun to assess this hybrid approach [11].
- Sparse NMR Data: For larger proteins where traditional NMR is hindered, perdeuteration and selective labeling can provide ambiguous NOESY cross-peak assignments and chemical shifts. CASP11 demonstrated that sophisticated modeling techniques could utilize these sparse NMR restraints to generate accurate 3D models where conventional NMR methods would fail [11].
- Chemical Crosslinking: CASP11 introduced the use of simulated chemical crosslinking data as another form of sparse experimental constraint. Predictors were able to use these distance restraints to build improved models, showing the promise of this technique for hybrid modeling [3] [11].
Table 2: Key Research Reagents and Resources for Protein Structure Prediction and Validation
| Category | Item / Resource | Function and Relevance |
|---|---|---|
| Prediction Servers & Software | AlphaFold2, RoseTTAFold | Open-source deep learning systems for generating highly accurate protein structure models from sequence [1] [26]. |
| Molecular Replacement Software | Phaser | Leading software for performing molecular replacement to solve the phase problem in crystallography using a search model. |
| Model Building & Refinement | Coot, Phenix, REFMAC5 | Standard software for visually building atomic models into electron density maps and refining them against crystallographic data. |
| Model Accuracy Estimation | pLDDT (predicted Local Distance Difference Test) | Per-residue confidence score provided by AlphaFold2; crucial for assessing which model regions are reliable for molecular replacement [26]. |
| Experimental Data | Sparse NMR Restraints (NOESY, CS) | Ambiguous distance and chemical shift data from NMR experiments on partially labeled proteins, used as constraints in hybrid modeling [11]. |
| Experimental Data | Chemical Crosslinking Data | Low-resolution distance restraints between residues, used to guide and validate computational models [3] [11]. |
| Community Experiment | CASP Target Data | Publicly available sequences, predictions, and experimental structures for blind testing and benchmarking methods [3] [26]. |
The case studies documented through the CASP experiment provide compelling evidence of the tangible impact of protein structure prediction on experimental structural biology. The journey from the occasional, exceptional use of a model for molecular replacement in early CASPs to the routine generation of models in CASP14 and beyond that are competitive with experiment represents a fundamental shift. This progress, driven largely by deep learning, has transformed computational models from theoretical aids into practical tools that can actively accelerate structure solution, enable the determination of challenging targets, and even assist in the validation of experimental data. As the field continues to advance, particularly in areas like multimeric complexes, protein-ligand interactions, and conformational ensembles [26], the synergy between computation and experiment is poised to become even more deeply integrated, further expanding the real-world impact of protein structure prediction.
Conclusion
The CASP experiment has been instrumental in transforming protein structure prediction from a formidable challenge to a powerful, routine tool. The breakthrough achieved by deep learning methods like AlphaFold2, validated through CASP's rigorous blind tests, marks a paradigm shift for structural biology and drug discovery. However, CASP continues to prove its immense value by charting the path forward. The experiment's evolving focus on protein complexes, conformational ensembles, nucleic acids, and ligand binding identifies the next frontiers. For researchers and drug developers, this means that CASP-validated models are now reliable starting points for inquiry, yet the competition's ongoing work ensures the field will continue to tackle increasingly complex biological questions, ultimately accelerating the pace of biomedical innovation.
References
- 1. CASP [https://en.wikipedia.org/wiki/CASP]
- 2. Critical Assessment of Methods of Protein Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8726744/]
- 3. Protein Structure Prediction Center [https://predictioncenter.org/]
- 4. Engaging the Community: CASP Special Interest Groups [https://pmc.ncbi.nlm.nih.gov/articles/PMC12353253/]
- 5. Critical Assessment of Methods of Protein Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6927249/]
- 6. Critical assessment of methods of protein structure prediction ( CASP )... [https://pubmed.ncbi.nlm.nih.gov/34533838/]
- 7. Home - CASP16 [https://predictioncenter.org/casp16/]
- 8. Frontiers | An Overview of Alphafold's Breakthrough [https://www.frontiersin.org/journals/artificial-intelligence/articles/10.3389/frai.2022.875587/full]
- 9. CASP16 Call for Targets [https://www.rcsb.org/news/6610191172841524659883f5]
- 10. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 11. Critical Assessment of Methods of Protein Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5394799/]
- 12. Critical Assessment of Methods of Protein Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10843301/]
- 13. Global distance test [https://en.wikipedia.org/wiki/Global_distance_test]
- 14. AlphaFold: a solution to a 50-year-old grand challenge in ... [https://deepmind.google/blog/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology/]
- 15. Estimation of Uncertainties in the Global Distance Test ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0154786]
- 16. The future of pharmaceuticals: Artificial intelligence in drug ... [https://www.sciencedirect.com/science/article/pii/S2095177925000656]
- 17. The Protein Problem: The day AI unlocked a secret of life Folding [https://medicine.iu.edu/blogs/research-updates/the-protein-folding-problem-the-day-ai-unlocked-a-secret-of-life]
- 18. How have AlphaFold2's predictions of protein structure ... [https://www.ebi.ac.uk/training/online/courses/alphafold/validation-and-impact/how-have-alphafolds-predictions-of-protein-structure-been-validated/]
- 19. A glance into the evolution of template-free protein ... [https://www.sciencedirect.com/science/article/abs/pii/S0300908420300961]
- 20. Assessment of template-free modeling in CASP10 and ROLL [https://pubmed.ncbi.nlm.nih.gov/24343678/]
- 21. Template-based and free modeling by RAPTOR++ in CASP8 [https://pmc.ncbi.nlm.nih.gov/articles/PMC2785131/]
- 22. Template modeling score | 7 Publications | 7 Citations [https://scispace.com/topics/template-modeling-score-1lnjsiwo]
- 23. Overview of AlphaFold2 and breakthroughs in overcoming ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482524007054]
- 24. Unlocking the power of AI models: exploring protein folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11377126/]
- 25. Improving AlphaFold2-based protein tertiary structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10484931/]
- 26. Home - CASP15 [https://predictioncenter.org/casp15/]
- 27. Artificial intelligence for RNA–ligand interaction prediction [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000790]
- 28. Assessment of three-dimensional RNA structure prediction in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10168427/]
- 29. High-accuracy protein complex structure modeling based ... [https://www.nature.com/articles/s41467-025-65090-7]
- 30. Assessment of protein assembly prediction in CASP12 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5949145/]
- 31. Assessment of Pharmaceutical Protein-Ligand Pose and ... [https://pubmed.ncbi.nlm.nih.gov/41045049/]
- 32. Soft disorder modulates the assembly path of protein complexes [https://pmc.ncbi.nlm.nih.gov/articles/PMC9714922/]
- 33. Critical assessment of methods of protein structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4394854/]
- 34. Critical Assessment of protein Structure Prediction (CASP) [https://elearning.vib.be/courses/alphafold/lessons/introduction-to-alphafold/topic/critical-assessment-for-structure-prediction-casp/]
- 35. Progress at protein structure prediction, as seen in CASP15 [https://www.sciencedirect.com/science/article/pii/S0959440X23000684]
- 36. Estimation of model accuracy by a unique set of features ... [https://www.nature.com/articles/s41598-022-17097-z]
- 37. Protein model accuracy estimation empowered by deep ... [https://www.nature.com/articles/s41598-021-90303-6]
- 38. Functional Relevance of CASP16 Nucleic Acid Predictions as ... [https://pubmed.ncbi.nlm.nih.gov/40905273/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=pubmed-2&utm_content=1XoGOxMvv5pNH9lSyhbWgYl81XwaZJ8xxi_7in25wjc_0FRIM2&fc=20250727100458)&ff=20250904072625&v=2.18.0.post9+e462414]
- 39. Deep learning techniques have significantly impacted protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8222070/]
- 40. Progress and Bottlenecks for Deep Learning in Computational ... [https://pubmed.ncbi.nlm.nih.gov/41178755/]
- 41. Investigating whether deep learning models for co-folding ... [https://www.nature.com/articles/s41467-025-63947-5]
- 42. Successes and challenges in simulating the folding of ... [https://www.sciencedirect.com/science/article/pii/S0021925817495470]
- 43. Role of environmental specificity in CASP results [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05559-8]
- 44. Home - CASP14 [https://predictioncenter.org/casp14/]
- 45. AlphaFold [https://en.wikipedia.org/wiki/AlphaFold]
- 46. After AlphaFold, how CASP competition is staying relevant [https://www.statnews.com/2025/01/07/casp-protein-structure-prediction-competition-after-alphafold/]
- 47. Critical assessment of AI-based protein structure prediction [https://www.sciencedirect.com/science/article/pii/S2950363925000353]
- 48. AlphaFold: Five Years of Impact [https://deepmind.google/blog/alphafold-five-years-of-impact/]
- 49. Before and after AlphaFold2: An overview of protein ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2023.1120370/full]
- 50. Applying and improving AlphaFold at CASP14 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9299164/]
- 51. Advancing protein structure prediction beyond AlphaFold2 [https://www.sciencedirect.com/science/article/abs/pii/S0959440X2500003X]
- 52. Critical Assessment of Methods of Protein Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5897042/]
- 53. AlphaFold at CASP13 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6907002/]
- 54. AlphaFold [https://deepmind.google/science/alphafold/]
- 55. An analysis and evaluation of the WeFold collaborative for ... [https://www.nature.com/articles/s41598-018-26812-8]