ChIP-Seq and Beyond: Decoding Histone Modifications in Gene Regulation, Disease, and Drug Discovery
This article provides a comprehensive resource for researchers exploring the epigenetic landscape through histone modifications.
ChIP-Seq and Beyond: Decoding Histone Modifications in Gene Regulation, Disease, and Drug Discovery
Abstract
This article provides a comprehensive resource for researchers exploring the epigenetic landscape through histone modifications. It covers the foundational principles of how specific histone marks regulate gene transcription and connects them to their roles in neurodevelopment and disease. The piece offers a detailed, practical guide to ChIP-seq methodology, from experimental design to data analysis, including troubleshooting and optimization strategies. Furthermore, it presents a critical comparison of established (ChIP-seq) and emerging (CUT&Tag) techniques, empowering scientists to select the appropriate tools for their epigenetic studies and translate findings into therapeutic insights for conditions like addiction and neurodegenerative disorders.
The Histone Code: Linking Specific Modifications to Transcriptional Outcomes
The eukaryotic genome is packaged into chromatin, a dynamic complex of DNA and proteins. The fundamental repeating unit of chromatin is the nucleosome, which serves as a critical scaffold for epigenetic regulation. Each nucleosome consists of 147 base pairs of DNA wrapped around a core histone octamer composed of two copies each of histones H2A, H2B, H3, and H4 [1]. Linker histone H1 associates with internucleosomal DNA, further promoting chromatin compaction [1]. Post-translational modifications (PTMs) of histones—chemical alterations to the N-terminal tails and core domains of these proteins—constitute a primary mechanism for regulating DNA accessibility without changing the underlying DNA sequence [2]. These modifications form the biochemical basis of the "histone code" hypothesis, which posits that specific combinations of histone modifications dictate unique chromatin states and downstream functional outcomes [3] [1]. In the context of gene regulation research, particularly Chromatin Immunoprecipitation followed by sequencing (ChIP-seq), understanding this code is paramount for interpreting genome-wide epigenetic landscapes.
Nucleosome Structure and Histone Modifications
The nucleosome core particle provides a versatile platform for epigenetic regulation. The octameric core organizes DNA into a higher-order structure that can be altered through several mechanisms, including ATP-dependent chromatin remodeling and histone PTMs [4]. Histone modifications regulate nucleosome dynamics—affecting their mobility, stability, and turnover—which in turn influences essentially every cellular process requiring DNA access, including transcription, replication, and repair [4].
These PTMs occur predominantly on the N-terminal tails of histones that protrude from the nucleosome core [1]. The major classes of histone modifications include acetylation, methylation, phosphorylation, and ubiquitylation, with more recent discoveries including GlcNAcylation, citrullination, crotonylation, sumoylation, and isomerization [1] [5]. These modifications function through two primary mechanisms:
- Disrupting chromatin contacts by altering the charge of histone proteins (e.g., acetylation neutralizes the positive charge of lysines).
- Recruiting nonhistone effector proteins ("readers") that recognize specific modifications and initiate downstream functional consequences [2].
The combinatorial nature of these modifications allows for precise regulation of chromatin structure, creating transcriptionally permissive euchromatin or repressive heterochromatin [1].
Major Classes of Histone Modifications
The following table summarizes the key properties, functions, and genomic locations of the most extensively studied histone modifications.
Table 1: Major Histone Modifications: Functions, Locations, and Regulatory Enzymes
| Modification | Function | Genomic Location | Writer Enzymes | Eraser Enzymes |
|---|---|---|---|---|
| H3K4me3 | Transcriptional activation [1] | Promoters, bivalent domains [1] | SET1, MLL, ALL-1 [6] | LSD1, JmjC family [7] |
| H3K27ac | Transcriptional activation [1] | Enhancers, promoters [1] | p300/CBP [6] | Histone Deacetylases (HDACs) [5] |
| H3K36me3 | Transcriptional activation [1] | Gene bodies [1] | SET2 [6] | KDM4A [7] |
| H3K9ac | Transcriptional activation [1] | Enhancers, promoters [1] | Gcn5, PCAF [6] | Histone Deacetylases (HDACs) [5] |
| H3K27me3 | Transcriptional repression, controls developmental regulators [8] [1] | Promoters in gene-rich regions [1] | EZH2 (PRC2) [7] [6] | UTX (KDM6A) [7] |
| H3K9me3 | Transcriptional repression, heterochromatin formation [8] [1] | Satellite repeats, telomeres, pericentromeres [1] | Suv39h1, SETDB1, G9a [7] [6] | LSD1, KDM4A [7] |
| γH2A.X (H2A.X S139ph) | DNA damage response [1] | DNA double-strand breaks [1] | ATR, ATM, DNA-PK [6] | Protein Phosphatases [5] |
Acetylation
Histone acetylation involves the addition of an acetyl group to lysine residues by histone acetyltransferases (HATs), and is removed by histone deacetylases (HDACs) [5]. This modification neutralizes the positive charge of lysine residues, weakening histone-DNA interactions and promoting an open chromatin structure (euchromatin) conducive to transcription [1]. Key acetylation marks include H3K9ac, H3K27ac, and H4K16ac [1]. Beyond transcription, histone acetylation is implicated in cell cycle regulation, proliferation, and apoptosis [1].
Methylation
Histone methylation is a stable mark added by histone methyltransferases (HMTs) and removed by histone demethylases (HDMs) [1] [5]. Unlike acetylation, methylation does not alter histone charge but functions as a docking site for reader proteins [1]. Lysines can be mono-, di-, or tri-methylated, with functional outcomes dependent on both the modified residue and methylation status [1]. For example, H3K4me3 is an activation mark at promoters, while H3K9me3 and H3K27me3 are repressive, albeit with distinct genomic contexts and propagation mechanisms [1]. Recent research highlights that the functional effects of individual modifications like H3K27me3 are highly dependent on interplay with the existing chromatin environment, such as H3K4me3 status [8].
Phosphorylation
Histone phosphorylation occurs on serine, threonine, and tyrosine residues, catalyzed by protein kinases and reversed by protein phosphatases [5]. This modification often serves as a platform for effector proteins, triggering downstream cascades [1]. Key functions include:
- Chromosome Condensation: Phosphorylation of H3S10 and H3S28 is crucial during mitosis [1].
- DNA Damage Response: Phosphorylation of H2A.X at S139 (γH2A.X) is one of the earliest events after DNA double-strand breaks, recruiting repair proteins [1] [6].
Ubiquitylation
Histone ubiquitylation involves the covalent attachment of ubiquitin. While polyubiquitylation typically targets proteins for degradation, monoubiquitylation of H2A and H2B regulates transcription and DNA repair [1]. Monoubiquitylated H2A (H2AK119ub) is associated with gene silencing, while H2B ubiquitylation (H2BK120ub in vertebrates) is linked to transcriptional activation [1] [6].
Experimental Methods for Studying Histone Modifications
Chromatin Immunoprecipitation Sequencing (ChIP-seq) and CUT&Tag
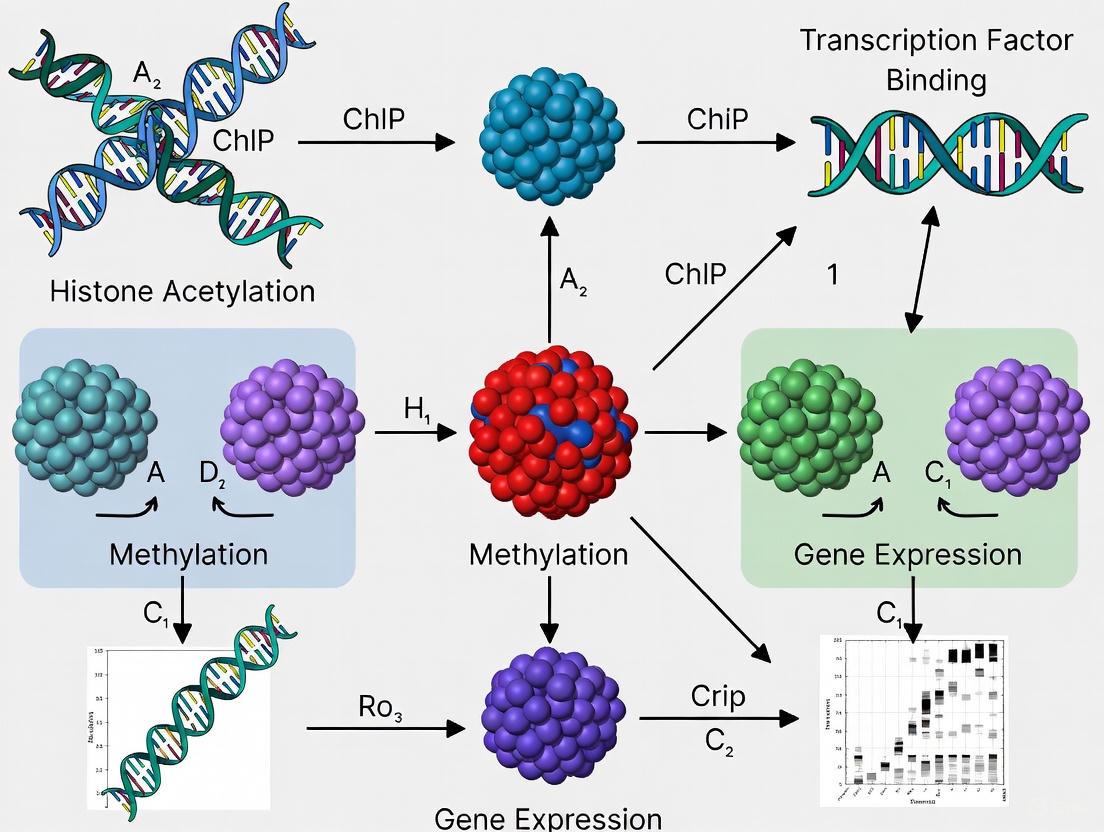
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has been the gold standard for genome-wide mapping of histone modifications and transcription factor binding sites [9]. The fundamental steps of the ChIP-seq protocol are as follows [1]:
- Cross-linking: Covalently link proteins to DNA in living cells, typically using formaldehyde.
- Chromatin Fragmentation: Sonicate or enzymatically digest chromatin into small fragments.
- Immunoprecipitation: Incubate with a high-specificity antibody against the histone modification of interest to pull down bound DNA fragments.
- Decrosslinking and Purification: Reverse crosslinks and purify the immunoprecipitated DNA.
- Library Preparation and Sequencing: Prepare a sequencing library from the purified DNA and sequence on a high-throughput platform.
- Data Analysis: Map sequenced reads to a reference genome to identify enriched regions ("peaks").
A newer technique, CUT&Tag (Cleavage Under Targets and Tagmentation), is gaining popularity as a sensitive and efficient alternative [9]. CUT&Tag uses a protein A-Tn5 transposase fusion protein targeted to the histone mark of interest by an antibody. Upon activation, the transposase simultaneously cleaves and inserts adapters into adjacent DNA in situ, streamlining library preparation [9]. A 2025 systematic comparison found that while CUT&Tag recovers approximately half of the peaks identified by ENCODE ChIP-seq datasets, it robustly captures the most significant signals and shows similar functional enrichments [9].
Mass Spectrometry-Based Analysis
Mass spectrometry (MS) provides a unbiased, antibody-free approach for identifying and quantifying histone PTMs, including rare modifications and combinatorial PTM patterns [3]. The standard "bottom-up" workflow involves [3]:
- Histone Extraction: Acid extraction of histone proteins from isolated cell nuclei.
- Chemical Derivatization: Propionic anhydride derivatization blocks unmodified and monomethylated lysines to improve tryptic digestion.
- Trypsin Digestion: Enzymatic cleavage generates short peptides suitable for LC-MS/MS.
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Peptides are separated by reversed-phase chromatography and analyzed by MS.
- Data Analysis: Computational pipelines identify and quantify modified peptides based on mass-to-charge ratios and fragmentation spectra.
This protocol can be adapted for various cell culture models, including 3D spheroids that more accurately mimic in vivo chromatin states compared to traditional 2D cultures [3].
Workflow Comparison
The following diagram illustrates the key decision points and parallel paths for the major methodologies discussed.
The Scientist's Toolkit: Research Reagent Solutions
Successful investigation of histone modifications relies on a suite of specialized reagents and tools. The following table outlines essential materials for key experimental steps.
Table 2: Essential Research Reagents for Histone Modification Studies
| Product Category | Specific Example | Function & Application |
|---|---|---|
| Histone Extraction Kits | EpiQuik Total Histone Extraction Kit [10] | Rapid isolation of histone proteins from mammalian cells or tissues with PTMs intact, for downstream assays. |
| Nuclear Extraction Kits | EpiQuik Nuclear Extraction Kit [10] | Isolation of nuclear proteins from cells/tissues, maintaining enzymatic activity for HAT, HDAC, HMT, HDM assays. |
| Antibodies for Specific Marks | Anti-H3K4me3, Anti-H3K27me3, Anti-H3K9ac [10] | Critical for immunodetection methods including ChIP-seq, CUT&Tag, western blot, and immunofluorescence. |
| Histone Modification Quantification Kits | Fluorometric H3K9me3 Quantification Kit [10] | ELISA-like, plate-based quantitation of specific histone modifications in a colorimetric or fluorometric format. |
| Histone H3/H4 Multiplex Assays | EpiQuik Histone H3 Modification Multiplex Assay Kit [10] | Simultaneous measurement of 21 different H3 modification patterns from a single sample on one plate. |
| Enzyme Activity Assays | HDAC/HAT Activity Assay Kits [10] | Measure activity of histone-modifying enzymes (e.g., HDACs, HATs) in nuclear extracts or purified preparations. |
| Cell Culture Models | 3D Spheroid Culture Systems [3] | Advanced in vitro models that more accurately mimic in vivo chromatin states and histone modification profiles. |
The intricate relationship between nucleosome structure and histone modifications forms the cornerstone of epigenetic gene regulation. The major classes of PTMs—acetylation, methylation, phosphorylation, and ubiquitylation—each exert distinct effects on chromatin architecture and function through defined biochemical mechanisms. For researchers employing ChIP-seq and related technologies, a deep understanding of this "histone code" is essential for interpreting genomic data. The ongoing development of sophisticated tools, from highly specific antibodies and quantitative assays to improved cell culture models and sequencing techniques like CUT&Tag, continues to empower scientists to decrypt the complex language of histone modifications. This knowledge is pivotal for advancing our understanding of development, disease mechanisms, and the discovery of novel epigenetic therapeutics.
Histone modifications represent a fundamental layer of epigenetic control that dynamically regulates gene expression and chromatin structure without altering the underlying DNA sequence. These post-translational modifications function as crucial regulators of genomic function, influencing cellular processes ranging from development and differentiation to stress response and disease pathogenesis. Within the context of modern ChIP-seq research, understanding the distinct functions and genomic distributions of specific activating marks is essential for interpreting genome-wide epigenetic datasets and elucidating mechanisms of transcriptional regulation. This technical guide provides an in-depth examination of four key activating histone modifications—H3K4me3, H3K9ac, H3K27ac, and H3K36me3—detailing their molecular functions, genomic distributions, experimental methodologies for investigation, and interplay within the epigenetic landscape. The information presented serves as a critical resource for researchers, scientists, and drug development professionals working to understand and target epigenetic mechanisms in both basic and translational research contexts.
Histone Modification Fundamentals
Molecular Functions and Genomic Distributions
Table 1: Core Characteristics of Activating Histone Modifications
| Histone Mark | Primary Genomic Location | Molecular Function | Associated Biological Processes | Enzyme Writers |
|---|---|---|---|---|
| H3K4me3 | Transcription start sites (TSS) of active genes [11] [12] | Facilitates RNA polymerase II activity, transcription initiation, and pause-release [11] [12] | Cell differentiation, development, meiotic recombination [11] [12] | SET1/MLL complexes, SDG2 (plants) [12] [13] |
| H3K9ac | Active coding regions [14] | Promotes open chromatin configuration, gene activation [14] | Heat shock response, stress adaptation [14] | Gcn5, CBP/p300 [14] |
| H3K27ac | Active enhancers and promoters [15] | Distinguishes active from poised enhancers, recruits transcriptional coactivators [15] | Enhancer activation, cell-type specific gene expression [15] | CBP/p300 [15] |
| H3K36me3 | Gene bodies of actively transcribed genes [16] | Suppresses spurious transcription initiation, ensures transcription fidelity [16] | Transcriptional elongation, neural differentiation [16] | SETD2 [16] |
Genomic Distribution and Functional Relationships
The following diagram illustrates the typical genomic distribution and functional relationships between the four activating histone marks relative to a generic gene structure:
Figure 1. Genomic distribution of activating histone marks across a typical gene locus. H3K27ac marks active enhancers that regulate promoter activity; H3K4me3 and H3K9ac are enriched at promoters/transcription start sites (TSS); H3K36me3 is deposited across gene bodies during transcriptional elongation.
Detailed Functions and Mechanisms
H3K4me3: The Promoter Activation Mark
H3K4me3 is one of the most extensively studied histone modifications, characterized by its pronounced enrichment at transcription start sites (TSS) of actively transcribed genes [11]. Beyond its correlation with active transcription, recent research utilizing CRISPR-based epigenome editing has demonstrated an instructive role for H3K4me3 in transcriptional activation. Studies in both mammalian systems and plants have shown that targeted deposition of H3K4me3 at specific genomic loci is sufficient to drive gene expression, confirming its causal rather than merely correlative relationship with transcription [12].
The molecular mechanisms through which H3K4me3 facilitates transcription involve interactions with specific reader proteins and chromatin remodelers. H3K4me3 directly interacts with transcriptional cofactor TAF3 and recruits chromatin remodeling complexes such as BPTF and CHD1, which facilitate RNA polymerase II activity [11]. Recent evidence suggests that H3K4me3 plays a particularly important role in RNA polymerase II pause-release and elongation rather than initial recruitment to promoters [11] [12]. Interestingly, H3K4me3 also functions in intergenic regions, where it can amplify transcription at active cis-regulatory elements independent of enhancer function or target gene proximity [11].
Beyond transcription, H3K4me3 plays critical roles in other genome-related processes. In plants, targeted deposition of H3K4me3 using SunTag-SDG2 systems has been shown to unlock meiotic crossover recombination in typically suppressed centromere-proximal regions, demonstrating its potential for agricultural applications [12]. The maintenance of balanced H3K4me3 deposition is crucial for transcriptional stability, with H3K4 methyltransferases and demethylases being frequently mutated in cancers, underscoring the mark's importance in disease contexts [11].
H3K9ac: The Transcriptional Activation Mark
H3K9ac is a histone modification predominantly associated with active coding regions and represents a central epigenetic modification for gene activation [14]. Unlike methylated forms of H3K9 which are repressive, acetylation at this residue neutralizes the positive charge on histones, weakening histone-DNA interactions and promoting an open chromatin configuration that facilitates transcription factor binding and transcriptional activation.
Research across diverse organisms has demonstrated the involvement of H3K9ac in stress response pathways. In the sea cucumber (Apostichopus japonicus), genome-wide ChIP-seq analysis revealed that H3K9ac is extensively involved in heat shock response, with differential H3K9ac regions identified under thermal stress conditions [14]. Notably, various transcription factor families showed significant H3K9ac modification changes under stress conditions, suggesting this mark plays a regulatory role in coordinating transcriptional responses to environmental challenges.
Integration of H3K9ac ChIP-seq data with transcriptomic analysis (RNA-seq) has demonstrated a generally positive correlation between H3K9ac enrichment in promoter regions and increased transcriptional output [14]. However, exceptions to this pattern have been observed, indicating that H3K9ac functions within a broader epigenetic context and may have context-dependent effects on gene expression. The dynamic regulation of H3K9ac by histone acetyltransferases (HATs) and histone deacetylases (HDACs) positions it as a key mediator of rapid epigenetic responses to environmental stimuli across eukaryotic species.
H3K27ac: The Enhancer Activation Mark
H3K27ac serves as a definitive marker of active enhancers and promoters, distinguishing them from their poised or inactive counterparts [15]. This modification is catalyzed by the histone acetyltransferases CBP/p300, which weaken histone-DNA interactions to promote chromatin accessibility and facilitate the recruitment of transcriptional coactivators and RNA polymerase II.
The significance of H3K27ac in defining cell-type-specific gene expression programs is particularly evident in disease contexts such as cancer. In glioblastoma stem cells (GSCs), which exhibit considerable transcriptomic and phenotypic heterogeneity, H3K27ac demonstrates remarkably consistent distribution patterns across patient samples [15]. Machine learning approaches have revealed that H3K27ac alone is sufficient to accurately predict gene expression in GSCs across different patients, suggesting that a common enhancer activation landscape, characterized by H3K27ac patterning, defines the underlying transcriptomic expression pattern in these heterogeneous cell populations [15].
The predictive power of H3K27ac for gene expression highlights its central role in transcriptional regulation and positions it as a particularly informative epigenetic mark for interpreting regulatory genomics data. Enhancers marked by H3K27ac are critical for context-dependent gene activation during development, differentiation, and cellular responses to environmental signals, with dysregulation of H3K27ac-associated enhancers frequently observed in diseases like cancer, where aberrant enhancer activity leads to misexpression of oncogenes and transcriptional reprogramming [15].
H3K36me3: The Elongation Fidelity Mark
H3K36me3 is primarily enriched across the gene bodies of actively transcribed genes, where it is deposited co-transcriptionally by the histone methyltransferase SETD2 in association with the phosphorylated C-terminal domain (CTD) of RNA polymerase II [16]. This modification plays a crucial role in maintaining transcriptional fidelity by suppressing spurious initiation events within coding regions and ensuring proper mRNA processing.
Recent research has revealed fascinating functional synergies between H3K36me3 and other histone modifications. Simultaneous catalytic inactivation of DOT1L (responsible for H3K79me) and SETD2 in embryonic stem cells leads to synergistic effects, including hyperactive transcription and failures in neural differentiation—phenotypes not observed with individual inactivation of either enzyme [16]. This functional collaboration between H3K36me3 and H3K79me demonstrates how different histone modifications can work cooperatively to fine-tune gene expression programs essential for cellular differentiation.
The mechanism by which H3K36me3 suppresses spurious transcription involves recruitment of factors that maintain chromatin in a repressive state for internal initiation. H3K36me3 recruits the de novo DNA methyltransferase DNMT3B, which deposits intragenic DNA methylation to further suppress illegitimate transcription initiation within gene bodies [16]. Loss of H3K36me3 results in increased chromatin accessibility at enhancers and aberrant recruitment of transcription factors like TEAD4 and its coactivator YAP1, leading to misregulated gene expression [16].
Experimental Approaches in ChIP-seq Research
Standardized ChIP-seq Methodologies
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) represents the gold standard technique for genome-wide mapping of histone modifications. The ENCODE consortium has established comprehensive standards and processing pipelines for histone ChIP-seq experiments to ensure data quality and reproducibility [17].
Table 2: ENCODE ChIP-seq Experimental Standards and QC Metrics
| Parameter | Histone Mark Type | Minimum Requirements | Optimal Standards |
|---|---|---|---|
| Biological Replicates | All marks | 2 replicates | 2+ isogenic or anisogenic replicates |
| Input Controls | All marks | Required, with matching run type and replicate structure | Matching input control for each experimental condition |
| Read Depth | Narrow marks (H3K4me3, H3K9ac, H3K27ac) | 10 million fragments per replicate | 20 million usable fragments per replicate |
| Read Depth | Broad marks (H3K36me3) | 20 million fragments per replicate | 45 million usable fragments per replicate |
| Library Complexity | All marks | NRF > 0.9, PBC1 > 0.9, PBC2 > 3 | NRF > 0.9, PBC1 > 0.9, PBC2 > 10 |
| Antibody Validation | All marks | Characterization per ENCODE standards | Target-specific validation with knockout controls |
The basic ChIP-seq workflow involves: (1) crosslinking proteins to DNA, (2) chromatin fragmentation, (3) immunoprecipitation with specific antibodies, (4) reverse crosslinking and DNA purification, and (5) library preparation and sequencing. For histone modifications, the ENCODE pipeline processes sequencing reads through standardized mapping steps followed by peak calling algorithms optimized for either punctate (narrow) or broad chromatin domains [17].
Advanced Integrative Approaches
Beyond standard ChIP-seq, advanced multi-omics approaches are providing unprecedented insights into the functional relationships between histone modifications and gene expression. Parallel-seq technology enables simultaneous measurement of chromatin accessibility and gene expression in the same single cells, allowing direct correlation of epigenetic states with transcriptional outputs [18]. This approach has been successfully applied to profile tens of thousands of cells from clinical lung tumor samples, revealing cancer-specific regulatory programs and epigenetic heterogeneity.
Machine learning frameworks are increasingly being deployed to integrate multi-epigenomic datasets and predict gene expression patterns. The CIPHER (Cross patient-Informed Prediction of Human Epigenetic Regulation) pipeline employs XGBoost algorithms to integrate ATAC-seq, CTCF ChIP-seq, RNAPII ChIP-seq, and H3K27ac ChIP-seq data for cross-patient gene expression prediction in glioblastoma stem cells [15]. These approaches demonstrate that H3K27ac alone shows remarkable predictive power for gene expression across patient samples, highlighting its central role in transcriptional regulation.
Integrative analysis of multiple histone modifications has revealed complex interplay and cross-talk between different epigenetic marks. Studies in the fungus Pyricularia oryzae have shown that loss of specific histone modifications (H3K4me2/3, H3K9me3, or H3K27me3) leads to redistribution of other modifications and altered gene expression in a compartment-specific manner [13]. Such research demonstrates how histone modifications exist within a networked regulatory system rather than functioning in isolation.
Cross-Talk and Integrative Functions
Inter-Modification Relationships and Functional Synergies
Histone modifications do not function in isolation but rather exhibit complex cross-talk and functional interdependencies. Different modifications can work synergistically or antagonistically to fine-tune gene expression outcomes. For instance, simultaneous loss of H3K79me and H3K36me3 leads to hyperactive transcription and differentiation defects that are not observed with individual inactivation, demonstrating synergistic repression of gene expression by these two marks [16].
Genomic compartment analysis has revealed distinct domains of facultative heterochromatin defined by specific combinations of histone modifications. In Pyricularia oryzae, two distinct subcompartments of facultative heterochromatin have been identified: K4-fHC (adjacent to euchromatin and influenced by H3K4 methylation) and K9-fHC (adjacent to constitutive heterochromatin and influenced by H3K9 methylation) [13]. These compartments harbor different functional elements and respond differently to environmental cues, illustrating how the genomic context of histone modifications contributes to their regulatory specificity.
Cross-talk between histone modifications and other epigenetic layers represents another important regulatory mechanism. In plants, m6A methylation in the 5' UTR of mRNAs triggers a downstream shift in H3K4me3 positioning, creating a regulatory circuit between RNA modification and histone methylation [19]. This m6A-mediated H3K4me3 shift is conserved across plant species and influences gene expression patterns, particularly during developmental processes like leaf senescence.
Research Reagent Solutions
Table 3: Essential Research Reagents and Experimental Tools
| Reagent/Tool | Specific Function | Application Examples |
|---|---|---|
| dCas9-SunTag Systems | Targeted epigenetic editing | Recruitment of histone methyltransferases to specific loci [12] |
| PRDM9 methyltransferase | Orthogonal H3K4me3 deposition | Targeted H3K4me3 editing with reduced off-target effects [12] |
| Anti-H3K27ac antibodies | Immunoprecipitation of H3K27ac-marked chromatin | ChIP-seq for active enhancer mapping [15] |
| SETD2 catalytic domain mutants | Dissection of catalytic vs. non-catalytic functions | Functional studies of H3K36me3 [16] |
| DOT1L inhibitors | Pharmacological inhibition of H3K79 methylation | Investigation of H3K79me-H3K36me3 synergism [16] |
| CIPHER pipeline | Machine learning prediction of gene expression | Integration of multi-epigenomic features [15] |
| Parallel-seq | Joint scATAC-seq and scRNA-seq profiling | Mapping regulatory programs in heterogeneous samples [18] |
The activating histone modifications H3K4me3, H3K9ac, H3K27ac, and H3K36me3 represent distinct but interconnected components of the epigenetic regulatory machinery. Each mark exhibits characteristic genomic distributions and molecular functions: H3K4me3 at promoters facilitating transcription initiation; H3K9ac promoting open chromatin configurations in active coding regions; H3K27ac defining active enhancers and predicting gene expression patterns; and H3K36me3 ensuring transcriptional fidelity across gene bodies. Advanced ChIP-seq methodologies and integrative multi-omics approaches continue to reveal the complex cross-talk and functional synergies between these modifications, highlighting their collective role in fine-tuning gene expression programs in development, homeostasis, and disease. As epigenetic therapies gain traction in clinical contexts, particularly for cancer treatment, understanding the specific functions and interactions of these activating marks will be essential for developing targeted epigenetic interventions with improved efficacy and specificity.
Gene expression regulation in eukaryotes depends on epigenetic mechanisms, with post-translational histone modifications serving as a fundamental layer of control. Among these, methylation of specific histone lysine residues is critical for establishing transcriptionally repressive chromatin states. This technical guide focuses on three key repressive marks—H3K27me3, H3K9me3, and H3K79me—detailing their molecular effectors, genomic distributions, functional consequences, and methodologies for their investigation in the context of ChIP-seq research. Understanding these marks provides crucial insights into developmental processes, cellular identity maintenance, and disease mechanisms, particularly in cancer and other disorders where epigenetic regulation is disrupted.
Molecular Mechanisms and Genomic Distribution
The table below summarizes the core characteristics, genomic distributions, and functional roles of the three repressive histone marks.
Table 1: Key Characteristics of Repressive Histone Modifications
| Feature | H3K27me3 | H3K9me3 | H3K79me |
|---|---|---|---|
| Primary Role | Facultative heterochromatin; temporary repression of developmental genes [20] | Constitutive heterochromatin; permanent repression of repeats & stable silencing [20] | Transcriptional regulation; mixed roles in silencing & activation [21] |
| Writer Complex/Enzyme | PRC2 (EZH2/EZH1, EED, SUZ12, RbAp46/48) [20] [22] | Multiple: SETDB1, SUV39H1/2, EHMT1/2 (G9a/GLP) [20] [23] | Dot1 (KMT4) [21] |
| Reader Proteins | PRC1 components (CBX family) [22] | HP1 family (HP1α, HP1β, HP1γ) [23] [24] | Information limited in search results |
| Eraser Enzymes | UTX (KDM6A), JMJD3 (KDM6B) [22] | JMJD2/KDM4, JMJD1/KDM3 families [23] | Information limited in search results |
| Genomic Context | Gene-rich regions; CpG-rich promoters of developmental regulators [20] | Gene-poor regions; telomeres, pericentromeres, retrotransposons [20] | Preferentially in euchromatin; role in telomeric silencing in yeast [21] |
| Developmental Dynamics | Prevalent in embryonic stages; reprogrammed during development [20] [22] | Prevalent in embryonic stages; transitions to DNA methylation [20] | Required for reporter gene silencing in yeast [21] |
| Relationship with DNA Methylation | Antagonistic; protected from DNA methylation [20] | Promotive; regions often become DNA methylated in somatic cells [20] | Information limited in search results |
H3K27me3: Master Regulator of Developmental Genes
H3K27me3 is deposited by the multi-subunit Polycomb Repressive Complex 2 (PRC2), whose core components include EZH2 (or its homolog EZH1), EED, SUZ12, and RbAp46/48 [20] [22]. EZH2 serves as the catalytic subunit, while other components enhance enzymatic activity and complex stability. PRC2 recruitment involves accessory proteins like AEBP2, JARID2, and DNA-binding transcription factors such as YY1 [20]. This mark is characteristic of facultative heterochromatin, which is dynamically regulated in a cell-type-specific manner.
H3K27me3 is enriched at CpG-rich promoters of approximately 500 developmental regulators in embryonic stem cells, including HOX, PAX, and SOX gene family members [20]. These genes are silenced in pluripotent cells but poised for activation upon differentiation. Genome-wide, H3K27me3 can form large repressive domains spanning hundreds of kilobases, known as Large Organized Chromatin K27 domains (LOCKs) [25]. These domains exhibit stronger repression and are strongly associated with developmental functions.
H3K9me3: Guardian of Genomic Stability
In contrast to H3K27me3, H3K9me3 is a hallmark of constitutive heterochromatin, which is more stable and present in all cell types [23]. This mark is catalyzed by several enzymes with non-redundant functions: SUV39H1/2 primarily target pericentromeric and telomeric repeats; SETDB1 modifies transposable elements and euchromatic regions; and G9a/GLP regulate H3K9 mono- and di-methylation in euchromatin [20] [23].
The effector protein HP1 recognizes H3K9me3 through its chromodomain and promotes chromatin condensation through chromoshadow domain-mediated oligomerization [23] [24]. H3K9me3 is preferentially detected in gene-poor regions with tandem repeat structures, including satellite repeats in telomeres and pericentromeres, as well as retrotransposons like LINEs and LTRs [20]. This positioning underscores its fundamental role in maintaining genomic stability by silencing repetitive elements and preventing illegitimate recombination.
H3K79me: A Conserved Modification with Context-Dependent Functions
H3K79 methylation is catalyzed by the Dot1 (KMT4) enzyme and differs from the other marks as it occurs on the histone globular domain rather than the tail [21]. In S. cerevisiae, Dot1 and H3K79 methylation are required for silencing reporter genes placed near telomeres (TPEV) and at the silent mating-type loci [21]. Mammalian Dot1 complements yeast Dot1, suggesting functional conservation.
The prevailing model suggests that H3K79 methylation, which is enriched in euchromatin, acts as a barrier to the binding of Sir silencing proteins, thereby restricting heterochromatin formation [21]. However, this role appears context-dependent, as H3K79 methylation is also required for the silencing of certain reporter constructs.
Functional Consequences in Gene Silencing and Cellular Identity
Distinct Modes of Transcriptional Repression
H3K27me3 and H3K9me3 employ fundamentally different mechanisms to silence genes. H3K27me3-mediated repression is generally reversible and dynamic, allowing genes to remain poised for activation during differentiation [20] [26]. This mark can repress transcription by blocking initiation through mechanisms that may involve inhibiting transcription factor binding or RNA polymerase II recruitment [24].
In contrast, H3K9me3 promotes stable long-term silencing through the formation of condensed heterochromatic structures that are largely inaccessible to the transcription machinery [23] [24]. This creates a physical barrier that prevents transcription factor binding, effectively locking genes in a silent state.
Roles in Cell Fate Specification and Stability
Both H3K27me3 and H3K9me3 play critical but distinct roles in maintaining cellular identity. H3K27me3 represses lineage-specific genes in stem and progenitor cells, allowing for their controlled activation during differentiation [26]. Notably, H3K27me3-rich regions can function as silencers that repress gene expression over long genomic distances through chromatin looping [26] [27]. CRISPR excision of these silencer elements leads to upregulation of interacting genes, loss of H3K27me3, gain of active marks like H3K27ac, and altered cell phenotypes [26].
H3K9me3 establishes a barrier to cell fate changes that must be overcome during cellular reprogramming [24]. Differentiated cells show large H3K9me3 domains at lineage-specific genes that are not expressed in that cell type. These domains impede transcription factor binding during reprogramming to pluripotency, and reducing H3K9me3 levels significantly enhances reprogramming efficiency [24].
Interplay Between Repressive Marks
Emerging evidence reveals complex crosstalk between different repressive marks. In the fungus Fusarium proliferatum, deletion of the H3K9 methyltransferase Kmt1 results in replacement of H3K9me3 with H3K27me3 at most genomic loci, indicating compensatory mechanisms between these silencing pathways [28]. Similarly, in cancer cells, H3K27me3 can compensate for the loss of H3K9me3 within specific genomic domains [25].
Table 2: Functional Roles in Biological Processes
| Biological Process | H3K27me3 Role | H3K9me3 Role | H3K79me Role |
|---|---|---|---|
| Embryonic Development | Defines bivalent promoters in ESCs; reprogrammed after fertilization [22] | Essential for embryogenesis; maintains heterochromatin [23] | Information limited |
| Cellular Differentiation | Silences developmental regulators; resolved upon lineage commitment [20] [26] | Stabilizes differentiated state; barrier to reprogramming [24] | Information limited |
| Nuclear Architecture | Forms repressive domains; mediates long-range chromatin interactions [26] [25] | Establishes constitutive heterochromatin at nuclear periphery [23] | Information limited |
| Dysregulation in Disease | Common in cancer; silences tumor suppressors [26] [25] | Loss in cancer leads to genomic instability; altered in heterochromatin [24] [25] | Information limited |
Investigating Repressive Marks: ChIP-seq and Advanced Methodologies
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) is the gold standard for genome-wide mapping of histone modifications. The fundamental workflow involves: crosslinking proteins to DNA, chromatin fragmentation, antibody-based immunoprecipitation of protein-DNA complexes, reversal of crosslinks, and high-throughput sequencing of enriched DNA fragments.
Advanced Methods for Low-Input Samples
Recent methodological advances address the challenge of profiling histone modifications in small cell populations, such as early embryos or rare cell types.
CUT&Tag (Cleavage Under Targets and Tagmentation) utilizes a protein A-Tn5 transposase fusion protein targeted to specific chromatin features by antibodies [29]. When activated by Mg²⁺, the tethered Tn5 simultaneously cleaves DNA and inserts adapters for PCR amplification. The NON-TiE-UP CUT&Tag (NTU-CAT) variant eliminates the solid-phase magnetic beads used in conventional protocols, allowing the entire procedure to be performed in solution with single mammalian blastocysts [29].
Key advantages of CUT&Tag include:
- Lower cell input requirements (as few as single embryos)
- Reduced background signal
- Higher sensitivity compared to ChIP-seq
- Faster protocol with simplified workflow
However, limitations include potential bias from Tn5 transposase preference for open chromatin regions, which can result in false positive rates of 10-25% for H3K4me3 and H3K27me3, and false negative rates of 21-32% for H3K4me3 [29].
The following diagram illustrates the key procedural differences between traditional ChIP-seq and modern CUT&Tag approaches:
Analyzing 3D Chromatin Architecture
Understanding how repressive marks function through long-range interactions requires methods that capture 3D chromatin architecture. Chromatin Interaction Analysis with Paired-End Tag sequencing (ChIA-PET) combines chromatin immunoprecipitation with proximity ligation to identify long-range interactions mediated by specific protein factors [26] [27].
In rice, H3K27me3 ChIA-PET revealed that H3K27me3-marked regions can function as silencer-like elements that interact with distal target genes through chromatin looping [27]. Deletion of these silencers disrupted loops and led to gene upregulation, demonstrating their functional importance in gene silencing.
Table 3: Key Research Reagents and Experimental Tools
| Reagent/Tool | Primary Function | Examples/Specifications | Key Applications |
|---|---|---|---|
| H3K27me3 Antibodies | Immunoprecipitation of H3K27me3-marked chromatin | Validation of specificity essential; multiple commercial sources available | ChIP-seq, CUT&Tag, immunofluorescence |
| H3K9me3 Antibodies | Immunoprecipitation of H3K9me3-marked chromatin | Specificity for tri-methylated form crucial | ChIP-seq, CUT&Tag, Western blot |
| EZH2 Inhibitors | Pharmacological inhibition of H3K27me3 deposition | GSK126, UNC1999; target PRC2 catalytic activity | Functional studies of H3K27me3, cancer research |
| pA-Tn5 Transposase | Enzyme-antibody fusion for tagmentation | Recombinant protein for CUT&Tag protocols | CUT&Tag, NTU-CAT for low-input samples |
| HP1 Mutants/Inhibitors | Disruption of H3K9me3 reading capability | Mutant HP1 constructs, small molecule inhibitors | Studying heterochromatin formation & maintenance |
| CREAM Software | Identification of LOCKs from ChIP-seq data | R package for domain calling | Analysis of broad histone modification domains [25] |
H3K27me3, H3K9me3, and H3K79me represent distinct repressive systems with specialized functions in gene silencing. H3K27me3 provides dynamic, developmentally-regulated repression of protein-coding genes through PRC2. H3K9me3 establishes stable, long-term silencing of repetitive elements and maintains cellular identity. H3K79me plays context-dependent roles in silencing, particularly in lower eukaryotes. Advanced genomic technologies like CUT&Tag and ChIA-PET continue to reveal new insights into how these marks orchestrate gene silencing through 3D genome organization. Their differential patterns in disease states, particularly cancer, highlight their potential as therapeutic targets and biomarkers for future drug development.
The establishment and maintenance of cellular identity are fundamental processes in development, tissue homeostasis, and disease pathogenesis. At the heart of these processes lies the precise regulation of gene expression through epigenetic mechanisms, with histone post-translational modifications (PTMs) serving as a central regulatory layer. These modifications constitute a complex "histone code" that dynamically controls chromatin structure and function [30]. In developmental biology and stem cell research, histone modifications enable plasticity while maintaining transcriptional programs, with distinct modification patterns marking pluripotent states and lineage commitment [31]. The core histone proteins (H2A, H2B, H3, and H4) contain unstructured N-terminal tails that protrude from the nucleosome surface and are subject to at least 20 different types of chemical modifications, including acetylation, methylation, phosphorylation, and ubiquitination [30]. These PTMs regulate chromatin structure and function through two primary mechanisms: directly altering chromatin packaging through charge modifications or internucleosomal interactions, and recruiting PTM-specific "reader" proteins that execute downstream functions [30]. The dynamic interplay between different histone modifications creates an epigenetic landscape that can be maintained through cell divisions, providing a molecular memory of cellular identity [30].
Table 1: Major Histone Modifications and Their Functional Roles in Stem Cells and Development
| Modification | General Function | Role in Stem Cells/Development | Enzymes |
|---|---|---|---|
| H3K4me3 | Transcriptional activation | Marks promoters in pluripotent cells; bivalent with H3K27me3 | SET1A/MLL complexes |
| H3K27me3 | Transcriptional repression | Maintains developmental genes in poised state; PRC2-mediated | EZH1/2 (PRC2) |
| H3K9me3 | Heterochromatin formation | Reprogramming barrier; re-established after fertilization | SUV39H1/2 |
| H3K36me3 | Transcriptional elongation | Gene body marking; prevents spurious transcription | SETD2 |
| H3K27ac | Active enhancers | Marks active regulatory elements | p300/CBP |
| H3K9me1 | Transcriptional activation | Early replication marking | Unknown |
| H3K27me1 | Transcriptional activation | Early replication marking | Unknown |
Quantitative Dynamics of Histone Modification Landscapes
Technological Advances in Histone Modification Mapping
The quantitative analysis of histone modifications has revolutionized our understanding of epigenetic regulation in development and disease. Traditional chromatin immunoprecipitation followed by sequencing (ChIP-seq) has been widely deployed for studying DNA-associated protein location across genomes, though it has historically been perceived as non-quantitative [32]. Recent methodological advances have addressed this limitation through approaches like sans spike-in quantitative ChIP (siQ-ChIP), which establishes an absolute quantitative scale by leveraging the equilibrium binding reaction in chromatin immunoprecipitation [32]. This method connects sequenced fragments to the total number of fragments in the immunoprecipitation product, allowing direct quantitative comparison across samples and cellular perturbations. Simultaneously, the development of CUT&Tag has provided a more sensitive alternative to ChIP-seq, enabling epigenetic profiling with lower cell numbers and single-cell resolution [9]. Systematic comparisons reveal that while CUT&Tag captures approximately half of the DNA-protein interaction sites identified by ChIP-seq, it recovers the most significant signals and shows similar enrichments in regulatory elements and functional annotations [9].
Most recently, the development of target chromatin indexing and tagmentation (TACIT) has enabled genome-coverage single-cell profiling of multiple histone modifications simultaneously [33]. This approach generates a 41-fold increase in non-duplicated reads per cell compared to previous methods and can profile seven core histone modifications across thousands of individual cells during embryonic development [33]. For simultaneous mapping of multiple modifications in the same cell, combined TACIT (CoTACIT) performs several rounds of antibody binding, protein A-Tn5 transposon incubation, and tagmentation, effectively measuring combinatorial histone modification states at single-cell resolution [33].
Table 2: Quantitative Changes in Histone Modifications During Early Embryonic Development
| Developmental Stage | H3K4me3 (reads/cell) | H3K27ac (reads/cell) | H3K36me3 (reads/cell) | Key Features |
|---|---|---|---|---|
| Zygote | 233,164 | 98,559 | 100,594 | Broad H3K4me3 domains |
| 2-cell | 261,716 (H3K4me1) | 98,559 | 100,594 | H3K27ac heterogeneity emerges |
| 4-cell | - | - | - | Increased cellular variation |
| 8-cell | - | - | - | Progressive restriction |
| Morula | - | - | - | Lineage priming |
| Blastocyst | 23,272 | 53,563 | 49,146 | Sharp H3K4me3 peaks |
Quantitative Histone Modification Signatures in Gene Regulation
Advanced quantitative approaches have revealed precise relationships between histone modification levels and transcriptional states. In highly expressed genes in human CD4+ T cells, most histone acetylation marks increase dramatically (>5-fold) during transcriptional activation [34]. This quantitative analysis also revealed that the first nucleosome upstream of the transcription start site shifts in the 5' direction, forming a broad nucleosome-free region (NFR) bound by transcription factors like YY1 and histone acetyltransferases in highly expressed genes [34]. Mass spectrometry-based proteomic approaches have further enabled comprehensive quantification of histone modification abundances across cell types, revealing cancer-specific histone modification signatures [35]. For example, combinatorial PTMs containing H3K27 methylation are especially enriched in breast cancer cell lines, and knockdown of the H3K27 methyltransferase EZH2 in a mouse mammary xenograft model significantly reduces tumor burden [35].
Beyond modification patterns, the total histone content itself represents an underappreciated layer of epigenetic regulation in stem cells and development. Mouse embryonic stem cells (ESCs) contain approximately 30% fewer histones than their differentiated counterparts, including embryoid bodies, neuronal cells, endodermal cells, and mouse embryonic fibroblasts [36]. This reduced histone content in ESCs contributes to their more decondensed chromatin architecture and increased transcriptional plasticity, representing an additional hallmark of pluripotency alongside specific histone modifications [36].
Histone Modification Networks in Stem Cell Pluripotency and Lineage Commitment
The Bivalent Chromatin Signature in Pluripotency
Pluripotent stem cells possess a distinctive epigenetic landscape characterized by a more "active" chromatin conformation with higher acetylation and lower methylation levels compared to differentiated cells [31]. A hallmark feature of embryonic stem cells is the presence of bivalent domains at promoters of developmentally important genes, which contain both the activating mark H3K4me3 and the repressive mark H3K27me3 [31]. In mouse ESCs, these bivalent domains mark genes that are repressed but poised for activation during lineage commitment, while in human ESCs, bivalency appears to be the default chromatin state at key developmental control genes marked by H3K27me3 [31]. The deposition of H3K27me3 is mediated by Polycomb repressive complex 2 (PRC2) via its catalytic subunit EZH1/2, while trimethylation of H3K4 is mediated by SETD1 (COMPASS) and MLL-containing complexes [31]. Mutations in either H3K27 or H3K4 methyltransferases result in severe defects in ESC growth and self-renewal capacity, underscoring their critical role in maintaining pluripotency [31].
The mechanisms governing the recruitment of chromatin-modifying complexes to specific genomic targets are not fully understood, but appear to involve DNA shape, nucleosome density, and overall chromatin conformation rather than specific sequences alone [31]. Despite similar recruitment mechanisms, H3K4me3 and H3K27me3 show markedly different genomic distributions, with H3K4me3 concentrated at promoter regions and H3K27me3 covering larger genomic domains through spreading mechanisms [31]. This differential distribution reflects the substantially greater amount of histone H3 methylated at K27 compared to K4 in eukaryotic cells [31].
Functional Specificity of Repressive Modifications
While histone modifications are broadly categorized as activating or repressive, recent evidence suggests they play non-redundant roles in gene regulation. In H3K27me3 null mouse embryonic stem cells, experimental substitution of the H3K27me3 pattern with other histone modifications revealed functional differences between repressive marks [8]. When H3K36me3 was directed to PRC2 target genes, it failed to substitute for H3K27me3-mediated repression despite accurate genome-wide recruitment and reduction in H3K4me3 levels, because residual H3K4me3 prevented H3K36me3 from recruiting sufficient DNA methylation [8]. In contrast, H3K9me3 demonstrated greater efficiency in repressing H3K27me3-regulated genes, though this repression remained contingent on H3K4me3 status [8]. These findings highlight the unique repressive functions of H3K27me3 and demonstrate that the functional effects of individual PTMs are highly dependent on interplay with the existing chromatin environment [8].
The functional specificity of histone modifications extends to their roles in chromatin structure. H3K27me3 and H3K9me3 represent distinct repression mechanisms, with H3K9me3 typically associated with constitutive heterochromatin and H3K27me3 with facultative heterochromatin [8]. This specificity has important implications for developmental gene regulation, as the inappropriate substitution of one repressive mark for another could disrupt normal differentiation processes.
Metabolic and Cell Cycle Regulation of Histone Modifications
The establishment and maintenance of histone modification landscapes are influenced by global cellular processes, including metabolism and cell cycle progression. Histone-modifying enzymes rely on key metabolites such as acetyl-CoA, S-adenosyl methionine (SAM), NAD, and 2-oxoglutarate as cofactors or substrates [31]. The intracellular concentrations of these metabolites are tightly linked to the physiological status and nutrient availability within stem cells, which typically exhibit specialized metabolism with greater dependence on glycolysis than oxidative phosphorylation [31]. Mouse ESCs depend on threonine and human ESCs on methionine to maintain pluripotency, and removal of these amino acids results in decreased SAM levels and concomitant reduction in specific histone methylation sites such as H3K4me3 [31]. Similarly, histone acetylation is influenced by stem cell-specific metabolic pathways, as evidenced by the finding that blockage of glycolysis-derived cytosolic acetyl-CoA production decreases histone acetylation and promotes early differentiation of human ESCs [31].
The cell cycle imposes another layer of regulation on histone modification states. With each cell division, newly synthesized, largely unmodified histones are incorporated into chromatin, leading to dilution of existing modifications [31]. Newly replicated chromatin carries specific modification patterns on histone H3 (H3K14ac, H3K18/K23ac, and H3K9me1) and H4 (H4K5ac and H4K12ac) that mirror the modification state of soluble histones in pre-deposition complexes [31]. During chromatin maturation, additional modifications including K27me1, K36me1, and K27me2 are imposed on new histones shortly after deposition, while further methylation occurs with slower kinetics similar to the cell cycle duration [31]. This kinetic regulation makes cell cycle length an important determinant of global chromatin methylation states, with slowly dividing cells accumulating more histone methylation than rapidly cycling cells [31]. The correlation between cell cycle length and cellular plasticity suggests that the inherent dilution of modifications during DNA synthesis may facilitate epigenetic reprogramming during differentiation [31].
Experimental Approaches and Methodological Guidelines
Quantitative ChIP-seq Methodologies
The siQ-ChIP methodology represents a significant advance in quantitative epigenomics by establishing an absolute physical scale for ChIP-seq measurements without requiring spike-in reagents [32]. This approach is grounded in the theoretical framework that the captured immunoprecipitated (IP) mass follows a sigmoidal binding isotherm governed by classical mass conservation laws. The fundamental relationship is described by:
[ \alpha = \frac{v{\textrm{in}}}{V-v{\textrm{in}}}\frac{m{\textrm{IP}}}{m{\textrm{in}}}\frac{m{\textrm{loaded,in}}}{m{\textrm{loaded}}} ]
where (v{\textrm{in}}) is the input sample volume, (V-v{\textrm{in}}) is the IP reaction volume, (m{\textrm{IP}}) and (m{\textrm{in}}) are the IP and input masses, and (m{\textrm{loaded}}) and (m{\textrm{loaded,in}}) are the masses loaded for sequencing [32]. The resulting siQ-ChIP scaled sequencing track represents the IP reaction efficiency projected across the genome, enabling direct quantitative comparison between samples and experimental conditions [32].
A critical implication of this quantitative framework is the normalization constraint that requires tracks to be interpreted as probability distributions rather than arbitrary signals [32]. This constraint has practical implications for data interpretation, as demonstrated by the reanalysis of p300/CBP inhibition studies where conventional non-quantitative approaches led to misinterpretation of chromatin changes [32]. The siQ-ChIP methodology enables novel modes of whole-genome analysis that automatically visualize and compare the effects of cellular perturbations on histone PTM distribution and abundance [32].
Single-Cell Epigenomic Profiling
The TACIT method enables genome-wide single-cell profiling of histone modifications with high coverage and low noise [33]. The experimental workflow involves:
- Cell Permeabilization: Treatment with digitonin to permit antibody access while maintaining nuclear structure
- Antibody Incubation: Specific primary antibodies against target histone modifications
- PAT Complex Binding: Protein A-Tn5 transposome binding to antibody-target complexes
- Tagmentation: Targeted fragmentation and adapter insertion by activated Tn5
- Library Amplification: PCR amplification with barcoded primers for multiplexing
- Sequencing: High-throughput sequencing on Illumina platforms
For simultaneous profiling of multiple modifications in the same cell (CoTACIT), steps 2-4 are repeated with different antibodies in sequential rounds [33]. This approach has been successfully applied to profile seven core histone modifications (H3K4me1, H3K4me3, H3K27ac, H3K27me3, H3K36me3, H3K9me3, and H2A.Z) across 3,749 individual cells during mouse early embryonic development [33]. The method generates up to 500,000 non-duplicated reads per cell, with higher coverage in blastocysts compared to zygotes for active marks like H3K4me3 (233,164 vs 23,272 reads/cell) [33].
Integration of single-cell histone modification data with transcriptomic profiles enables comprehensive mapping of the epigenetic and regulatory landscape during development [33]. Machine learning approaches applied to these multimodal datasets can identify totipotency gene regulatory networks, including stage-specific transposable elements and putative transcription factors [33]. CRISPR activation of identified transcription factor combinations has successfully induced totipotency activation in mouse embryonic stem cells, validating the predictive power of these integrated approaches [33].
Table 3: Essential Research Reagents and Experimental Tools
| Category | Specific Reagents/Tools | Application | Key Features |
|---|---|---|---|
| Antibodies | H3K4me3, H3K27me3, H3K9me3, H3K27ac, H3K36me3, H3K4me1 | Histone modification mapping | Specificity validated for ChIP-seq/CUT&Tag |
| Enzymes | Protein A-Tn5 transposase (PAT) | CUT&Tag/TACIT | Fusion protein for antibody-directed tagmentation |
| Cell Lines | Mouse ESCs (CCE, R1), Human ESCs | In vitro differentiation models | Well-characterized pluripotency |
| Kits | siQ-ChIP kit, TACIT workflow reagents | Quantitative epigenomics | Standardized protocols |
| Bioinformatics | Seurat, ChromVAR, ArchR | Single-cell data analysis | Multimodal integration |
The comprehensive analysis of histone modifications in stem cells and developmental models has revealed fundamental principles of epigenetic regulation in cellular identity establishment and maintenance. The quantitative dynamics of histone modification deposition, removal, and inheritance create a complex regulatory network that integrates metabolic signals, cell cycle progression, and lineage-specific transcription factors. Recent technological advances in quantitative epigenomics, particularly single-cell multiomics approaches, have enabled unprecedented resolution in mapping these dynamics during critical developmental transitions.
Future research directions will likely focus on several key areas: First, understanding the mechanistic basis of histone modification crosstalk and how combinatorial codes are read, interpreted, and maintained through cell divisions. Second, elucidating how metabolic and signaling pathways interface with the epigenetic machinery to coordinate cell fate decisions. Third, developing more precise tools for manipulating specific histone modifications at defined genomic locations to establish causal relationships between epigenetic states and cellular identities. Finally, translating basic insights from developmental epigenetics into therapeutic strategies for regenerative medicine and cancer treatment, particularly through targeting of histone-modifying enzymes that are frequently dysregulated in disease.
The continued refinement of quantitative epigenomic technologies will further enhance our ability to decode the histone language and its role in cellular identity, ultimately enabling more precise control of cell fate for both basic research and clinical applications.
The epigenome comprises dynamic, reversible molecular modifications that regulate DNA-related processes such as gene transcription, chromatin organization, and DNA repair without altering the underlying DNA sequence [37] [38]. In the nervous system, these mechanisms—including DNA methylation, histone modifications, and non-coding RNA regulation—orchestrate complex neurodevelopmental processes and maintain neuronal function throughout life [39] [37]. The field of neuroepigenetics investigates how these chromatin-based mechanisms mediate brain development, neural plasticity, and cognitive function, and how their dysregulation contributes to a spectrum of neurological and psychiatric disorders [39]. Unlike the relatively static genome, the epigenome remains highly plastic and responsive to environmental influences, serving as a molecular interface between genetic predisposition, environmental exposure, and disease pathogenesis in the brain [39] [40] [37].
The postmitotic nature of neurons once suggested that epigenetic regulation might be limited in the mature brain. However, research over the past decade has revealed that neurons possess remarkably dynamic and complex epigenetic machinery that directs lifelong neural adaptation [39]. This epigenetic plasticity enables the brain to respond to experiential cues, from learning and memory to stress and drug exposure, while also creating vulnerabilities when these mechanisms become dysregulated [40] [38]. The investigation of epigenetic processes in neurological contexts has been transformed by advanced technologies such as ChIP-seq (Chromatin Immunoprecipitation followed by sequencing), which enables genome-wide mapping of histone modifications and transcription factor binding sites in neural tissues [41] [42]. This technical advancement has revealed that distinct epigenetic signatures characterize different neural cell types, developmental stages, and disease states, providing unprecedented insights into the molecular basis of neurodevelopmental, neurodegenerative, and neuropsychiatric disorders [41] [39].
Fundamental Epigenetic Mechanisms
DNA Methylation and Hydroxymethylation
DNA methylation involves the covalent addition of a methyl group to the 5-position of cytosine bases, primarily within cytosine-guanine (CpG) dinucleotides, to form 5-methylcytosine (mC5) [43] [39]. This modification is catalyzed by DNA methyltransferases (DNMTs), including DNMT1, which maintains methylation patterns during DNA replication, and DNMT3A and DNMT3B, which mediate de novo methylation [43] [40]. In the brain, a significant proportion of methylation also occurs at non-CG sites (mCH), particularly in neurons [43]. DNA methylation typically leads to gene repression when it occurs in promoter regions, often by preventing transcription factor binding or recruiting methyl-binding proteins that promote chromatin condensation [39] [37].
An oxidation product of 5-methylcytosine, 5-hydroxymethylcytosine (5hmC), is generated by the ten-eleven translocation (TET) family of enzymes (TET1, TET2, TET3) and is particularly abundant in the brain [43] [39]. While 5hmC can serve as an intermediate in active DNA demethylation pathways, it also functions as a stable epigenetic mark associated with active transcription, especially at gene promoters and enhancers [39]. The distribution of 5hmC undergoes dramatic changes during brain development and aging, with studies showing a 10-fold increase in cerebellar 5hmC from postnatal week 1 to adulthood, highlighting its importance in neuronal maturation and function [39].
Histone Modifications
Histone modifications represent a diverse array of post-translational modifications to histone proteins that package DNA into chromatin [42] [38]. The nucleosome, chromatin's basic repeating unit, consists of 146 base pairs of DNA wrapped around an octamer of core histone proteins (H2A, H2B, H3, and H4) [40] [38]. Histone N-terminal tails extend from the nucleosome core and undergo numerous modifications, including acetylation, methylation, phosphorylation, ubiquitination, and ADP-ribosylation [42] [38]. These modifications influence chromatin structure and function through two primary mechanisms: by altering the electrostatic charge of histones and consequently their affinity for DNA, or by creating binding sites for protein recognition modules that mediate downstream functional effects [42].
Table 1: Major Histone Modifications and Their Functional Consequences in the Nervous System
| Modification | Associated Function | Writer Enzymes | Eraser Enzymes |
|---|---|---|---|
| H3K4me3 | Transcriptional activation | KMT2 family, SET1 | KDM5 family |
| H3K9me3 | Heterochromatin formation, transcriptional repression | KMT1 family (e.g., G9a) | KDM4 family, KDM3B |
| H3K27me3 | Facultative heterochromatin, developmental gene repression | PRC2 (EZH2) | KDM6 family |
| H3K36me3 | Transcriptional elongation | SETD2 | KDM2 family |
| H3K9ac | Transcriptional activation | HATs (e.g., CBP/p300) | HDACs |
| H3K14ac | Transcriptional activation | HATs (e.g., CBP/p300) | HDACs |
Histone modifications are dynamically regulated by opposing enzyme families: "writer" enzymes that add modifications (e.g., histone acetyltransferases [HATs], histone methyltransferases [HMTs]) and "eraser" enzymes that remove them (e.g., histone deacetylases [HDACs], histone demethylases [KDMs]) [40] [38]. These enzymes are further regulated by "reader" proteins that recognize specific modifications and mediate functional outcomes through recruitment of additional effector complexes [39] [38]. The complexity of this regulatory system is staggering, with dozens of writer, eraser, and reader proteins targeting more than 50 distinct modification sites across the histone code [38].
Chromatin Remodeling and Non-Coding RNAs
ATP-dependent chromatin remodeling complexes use ATP hydrolysis to reposition nucleosomes, altering DNA accessibility for transcription factors and RNA polymerase [44]. These complexes play crucial roles in neural development by dynamically regulating gene expression programs during cell fate specification and differentiation [44]. Additionally, non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), contribute to epigenetic regulation by targeting chromatin-modifying complexes to specific genomic loci or by regulating the stability and translation of mRNAs encoding epigenetic regulators [37].
Epigenetics in Neurodevelopment and Neurodevelopmental Disorders
Epigenetic Regulation of Normal Neurodevelopment
The formation of the mammalian cerebral cortex requires precisely orchestrated processes including neural progenitor proliferation, neuronal differentiation, migration, and circuit formation [44]. Epigenetic mechanisms govern each of these developmental milestones, ensuring the proper spatial and temporal expression of gene networks that dictate neural cell fate and function [44]. During corticogenesis, radial glial cells in the ventricular zone give rise to excitatory projection neurons through asymmetric divisions, while inhibitory interneurons originate primarily in the ganglionic eminences of the basal telencephalon [44]. The generation of diverse neuronal subtypes with distinct morphological, connectional, and neurochemical properties depends on tightly regulated epigenetic programs that establish and maintain cell-type-specific gene expression patterns [44].
Key epigenetic regulators in neurodevelopment include the Polycomb repressive complexes (PRC1 and PRC2), which maintain developmental genes in a transcriptionally repressed but poised state through deposition of H3K27me3, and trithorax group proteins, which promote gene activation through H3K4me3 [44]. The dynamic balance between these opposing chromatin states ensures proper temporal activation of developmental gene programs while maintaining cellular commitment to specific lineages once established [44]. Disruption of these epigenetic pathways can lead to malformations of cortical development (MCDs), which underlie approximately 75% of cases of epileptic seizures and 40% of medication-resistant childhood epilepsies [44].
Neurodevelopmental Disorders
Autism spectrum disorders (ASD) have been strongly linked to epigenetic dysregulation, with studies identifying aberrant DNA methylation patterns in genes involved in synaptic function, neuronal signaling, and immune response [37]. Mutations in genes encoding chromatin regulators, including MECP2, CHD8, and ADNP, contribute to ASD pathogenesis by disrupting the normal epigenetic landscape during critical periods of brain development [44] [39]. Similarly, Rett syndrome, caused primarily by mutations in the MECP2 gene, provides a compelling example of the consequences of epigenetic dysregulation in neurodevelopment [45] [39]. MECP2 functions as a reader of methylated DNA, interpreting DNA methylation marks and recruiting additional chromatin-modifying complexes to regulate gene expression [39]. Loss of MECP2 function leads to widespread transcriptional dysregulation and progressive neurological decline, highlighting the critical importance of proper epigenetic interpretation for normal brain function [39].
Schizophrenia has also been associated with epigenetic alterations, particularly in genes involved in glutamatergic and GABAergic neurotransmission, synaptic plasticity, and stress response pathways [37]. DNA methylation changes in genes such as RELN, which encodes a protein critical for neuronal migration and synaptic function, and GAD1, which encodes the GABA-synthesizing enzyme glutamic acid decarboxylase, have been implicated in the pathophysiology of schizophrenia [44] [37]. These epigenetic alterations may underlie the disrupted cortical connectivity and impaired information processing that characterize the disorder [44].
Epigenetics in Addiction
Drug-Induced Epigenetic Adaptations in the Reward Pathway
Addiction to drugs of abuse involves persistent molecular adaptations within the brain's reward circuitry, particularly the mesolimbic dopamine system comprising dopaminergic neurons in the ventral tegmental area (VTA) and their projections to the nucleus accumbens (NAc) [43] [38]. All drugs of abuse share the property of increasing dopamine transmission in the NAc, initially hijacking natural reward pathways that normally reinforce adaptive behaviors [38]. With repeated drug exposure, enduring epigenetic changes occur in these reward regions, creating molecular memories that contribute to the persistent behavioral abnormalities characteristic of addiction, including craving, compulsive drug-seeking, and relapse [43] [38].
Histone acetylation in the NAc represents one of the best-characterized epigenetic mechanisms in addiction. Acute and chronic exposure to psychostimulants, opiates, alcohol, or nicotine increases total cellular levels of H3 and H4 acetylation in the NAc [38]. These drug-induced acetylation changes result from altered balance between histone acetyltransferases (HATs) and histone deacetylases (HDACs) [40] [38]. For example, chronic cocaine or alcohol exposure reduces HDAC enzymatic activity and disrupts HDAC subcellular localization, while the HAT CREB-binding protein (CBP) is required for cocaine-induced increases in histone acetylation [38]. Importantly, these acetylation changes occur in a highly locus-specific manner, with acute psychostimulant exposure increasing H4 acetylation specifically at promoters of immediate early genes like c-Fos and Fosb, correlating with their rapid transcriptional activation [38].
Table 2: Epigenetic Modifications Associated with Different Classes of Addictive Substances
| Substance Class | Histone Modifications | DNA Methylation Changes | Affected Brain Regions |
|---|---|---|---|
| Psychostimulants (cocaine, methamphetamine) | Increased H3ac, H4ac, H3K4me3; Decreased H3K9me2/3 | Altered methylation of genes involved in synaptic plasticity | NAc, PFC, VTA, hippocampus |
| Opiates | Increased H3ac, H4ac; H3K9me2 changes | DNMT expression changes | NAc, VTA, amygdala |
| Alcohol | Increased H3ac, H4ac; H3K9me3 changes | Global DNA methylation changes; site-specific methylation | NAc, amygdala, prefrontal cortex |
| Nicotine | Increased H3ac, H4ac | Altered methylation of BDNF and other neuroplasticity genes | NAc, VTA, hippocampus |
Individual Susceptibility and Resilience
Not all individuals who use drugs develop addiction, highlighting the importance of individual differences in susceptibility [43]. These individual variations have an epigenetic basis, with factors such as stressful life experiences, early environment, and genetic background shaping the epigenetic landscape of reward circuits and modifying addiction vulnerability [40]. For example, animal models demonstrate that "sign-trackers" (animals that attribute excessive incentive salience to drug cues) show greater addiction-like behaviors and distinct epigenetic patterns compared to "goal-trackers" [43]. Similarly, high responder and low responder rats exhibit differential epigenetic responses to stress and drugs, with high responders showing decreased H3K14 acetylation following stress while low responders show increased acetylation at this residue [40].
The epigenetic basis of resilience to addiction is an emerging area of research. Studies suggest that resilience factors, potentially including specific histone modifications or DNA methylation patterns, may protect against the development of addiction despite drug exposure or stress [40]. Understanding these protective epigenetic mechanisms could inform novel strategies for preventing and treating substance use disorders by enhancing natural resilience pathways [40].
Epigenetics in Neurodegenerative and Neuropsychiatric Disorders
Neurodegenerative Disorders
Alzheimer's disease (AD) involves progressive cognitive decline characterized by amyloid-β plaques and neurofibrillary tangles. Epigenetic mechanisms contribute to AD pathogenesis through multiple pathways, including promoter hypermethylation of genes involved in synaptic function, neurogenesis, and mitochondrial function [39] [37]. Notably, the amyloid precursor protein (APP) and presenilin 1 (PSEN1) genes show altered methylation patterns in AD brains, potentially influencing amyloid-β production [37]. Age-related changes in DNA methylation and hydroxymethylation patterns may also contribute to the late-onset nature of most AD cases [39].
Parkinson's disease (PD) involves the progressive loss of dopaminergic neurons in the substantia nigra, leading to motor symptoms. Epigenetic alterations in PD include DNA methylation changes in genes related to neuronal survival, oxidative stress response, and α-synuclein expression [37]. Both familial and sporadic forms of PD show evidence of epigenetic dysregulation, with DNA methylation changes identified in blood and brain tissue of PD patients [37]. Additionally, histone modifications at promoters of genes involved in mitochondrial function and dopamine synthesis may contribute to the selective vulnerability of dopaminergic neurons in PD [39].
Huntington's disease (HD), caused by a CAG repeat expansion in the huntingtin gene, involves progressive motor dysfunction and cognitive decline. Aberrant histone modifications, particularly decreased H3K4me3 and increased H3K9me3 at promoters of genes critical for neuronal function, have been observed in HD models [37]. The mutant huntingtin protein directly interacts with epigenetic regulators, including HATs and HDACs, disrupting normal histone acetylation patterns and contributing to transcriptional dysregulation [39].
Neuropsychiatric Disorders
Depression has been linked to epigenetic alterations in stress response pathways, particularly the hypothalamic-pituitary-adrenal (HPA) axis. Early life stress and chronic stress in adulthood induce DNA methylation changes at the glucocorticoid receptor gene (NR3C1) and other genes regulating HPA axis function, potentially creating enduring vulnerability to depression [40] [37]. Histone modifications in brain reward regions, including decreased H3K14 acetylation and H3K4 methylation, have also been associated with depression-like behaviors in animal models [40]. These epigenetic changes may mediate the well-established relationship between stress exposure and depression risk.
Bipolar disorder involves alternating episodes of depression and mania, with evidence suggesting epigenetic contributions to its etiology. DNA methylation differences in genes involved neurotransmitter systems, including serotonin and dopamine pathways, have been identified in bipolar disorder patients [37]. Additionally, histone modifications at promoters of circadian rhythm genes may contribute to the sleep disturbances and cyclical mood episodes characteristic of the disorder [39] [37].
Post-traumatic stress disorder (PTSD) develops in a subset of individuals following trauma exposure, suggesting epigenetic factors may mediate differential vulnerability. Studies have identified DNA methylation changes in genes related to HPA axis function, immune signaling, and neuronal plasticity in individuals with PTSD [37]. These epigenetic marks may serve as biomarkers of trauma exposure and PTSD risk, potentially informing early intervention strategies [40] [37].
Technical Approaches: ChIP-Seq in Neuroepigenetics Research
ChIP-Seq Methodology
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has become the method of choice for genome-wide mapping of histone modifications, transcription factor binding sites, and chromatin-associated proteins in neural tissues [41] [42]. This powerful technique combines the specificity of antibody-based immunoprecipitation with the comprehensive nature of next-generation sequencing, enabling researchers to generate high-resolution epigenomic maps from small quantities of brain tissue [41].
The standard ChIP-seq protocol involves multiple critical steps: (1) Crosslinking of proteins to DNA in living cells or tissues using formaldehyde; (2) Chromatin fragmentation by sonication or enzymatic digestion to generate 200-600 bp fragments; (3) Immunoprecipitation with specific antibodies targeting the histone modification or protein of interest; (4) Reversal of crosslinks and purification of immunoprecipitated DNA; (5) Library preparation for next-generation sequencing; and (6) Bioinformatic analysis of sequencing data to identify enriched regions and integrate with other genomic datasets [41].
ChIP-seq Workflow for Histone Modification Analysis
Critical Experimental Considerations
Successful ChIP-seq experiments require careful attention to multiple factors. Antibody specificity is paramount, as nonspecific antibodies can generate false-positive results [41]. Validated ChIP-grade antibodies for common histone modifications include: H3K4me3 (associated with active promoters), H3K9ac (active chromatin), H3K27me3 (facultative heterochromatin), H3K9me3 (constitutive heterochromatin), H3K36me3 (transcribed regions), and H3K4me1 (enhancers) [41]. Chromatin quality and fragmentation efficiency must be optimized for each tissue type, with neuronal chromatin presenting unique challenges due to its distinct composition, including lower linker histone H1 content and shorter nucleosome repeat length compared to non-neuronal cells [39]. Appropriate controls, including input DNA (non-immunoprecipitated chromatin) and negative control antibodies, are essential for distinguishing specific enrichment from background signal [41].
The IP-Star automated system (Diagenode) has improved the reproducibility and throughput of ChIP assays by standardizing the immunoprecipitation process, reducing variability introduced by manual protocols [41]. For sequencing, the Illumina platform is most commonly used for ChIP-seq applications, providing sufficient read depth and mapping quality for most epigenomic studies in neural tissues [41].
Data Analysis and Interpretation
Bioinformatic analysis of ChIP-seq data typically involves: read alignment to the reference genome; peak calling to identify significantly enriched regions; annotation of peaks to genomic features (promoters, enhancers, gene bodies); differential binding analysis between experimental conditions; and integrative analysis with other genomic datasets (e.g., RNA-seq, ATAC-seq) [41]. For histone modification marks, different analytical approaches are required depending on the specific mark being studied. Sharp marks like H3K4me3 (typically restricted to promoters) are identified using peak-based methods, while broad marks like H3K36me3 (spanning entire gene bodies) require distinct analytical approaches [41].
Table 3: Essential Research Reagents for ChIP-Seq in Neuroepigenetic Studies
| Reagent Category | Specific Examples | Function in Experimental Protocol |
|---|---|---|
| Histone Modification Antibodies | Anti-H3K4me3 (CST #9751S), Anti-H3K27me3 (CST #9733S), Anti-H3K9me3 (CST #9754S) | Target-specific immunoprecipitation of chromatin regions bearing modification of interest |
| Chromatin Preparation Reagents | Formaldehyde, glycine, protease inhibitors (aprotinin, leupeptin, PMSF), cell lysis buffer | Tissue fixation and chromatin preparation |
| Fragmentation Equipment | Bioruptor (Diagenode), Covaris sonicator | Chromatin shearing to optimal fragment size |
| Library Preparation Kits | Illumina ChIP-seq Library Preparation Kit | Preparation of sequencing libraries from immunoprecipitated DNA |
| Sequencing Platforms | Illumina NovaSeq, NextSeq, HiSeq | High-throughput sequencing of ChIP DNA |
Emerging Technologies and Therapeutic Implications
Locus-Specific Neuroepigenetic Editing
Traditional approaches to studying epigenetic mechanisms in the brain have relied on global pharmacological or genetic manipulations that affect the entire genome, making it difficult to establish causal relationships between specific epigenetic changes at individual genes and functional outcomes [38]. The emerging field of locus-specific neuroepigenetic editing overcomes this limitation by enabling targeted rewriting of the epigenome at specific genomic loci in defined brain cell types [38]. This approach utilizes programmable DNA-binding domains—including zinc finger proteins (ZFPs), transcription activator-like effectors (TALEs), and, most recently, CRISPR/dCas9 systems—fused to epigenetic effector domains to precisely manipulate histone modifications or DNA methylation at individual genes [38].
These tools have demonstrated causal roles for specific histone modifications in addiction-related behaviors. For example, targeted recruitment of histone acetyltransferases to the Fosb promoter in mouse NAc enhances behavioral responses to cocaine, while recruitment of histone deacetylases to the same locus suppresses these responses [38]. Similarly, locus-specific epigenetic editing has established causal relationships between histone methylation at specific gene promoters and drug-seeking behaviors [38]. These approaches are revolutionizing neuroepigenetics research by moving beyond correlation to establish causality, potentially identifying novel targets for therapeutic intervention.
Epigenetic Therapeutics
The reversible nature of epigenetic modifications makes them attractive targets for therapeutic intervention in neurological and psychiatric disorders [39] [37]. Several epigenetic-based therapeutics are already in clinical use for other conditions, particularly cancer, and are being investigated for neurological applications [39]. Histone deacetylase inhibitors (HDACis) such as valproic acid, suberoylanilide hydroxamic acid (SAHA, vorinostat), and sodium butyrate have shown promise in preclinical models of neurodegenerative and psychiatric disorders [39] [37]. In animal models of depression, HDACis can reverse stress-induced behavioral abnormalities and associated epigenetic changes, while in models of neurodegenerative diseases, they promote neuronal survival and enhance cognitive function [39] [37].
DNA methyltransferase inhibitors (DNMTis) such as 5-azacytidine and decitabine are also being explored for neurological applications, particularly given the evidence of hypermethylation and transcriptional silencing of neuroprotective genes in various brain disorders [39]. Additionally, small molecules targeting specific histone methyltransferases and demethylases are in development, offering the potential for more precise epigenetic modulation [39] [38].
Epigenetic Therapeutic Approaches for Neurological Disorders
Biomarker Development
The detection of epigenetic signatures in accessible tissues like blood or cerebrospinal fluid offers promise for developing diagnostic and prognostic biomarkers for neurological and psychiatric disorders [46] [37]. DNA methylation patterns in particular show potential as biomarkers due to the stability of this modification and the development of sensitive techniques for its detection [46] [37]. For example, specific DNA methylation signatures have been identified in blood samples from patients with Alzheimer's disease, Parkinson's disease, and autism spectrum disorders, potentially enabling earlier diagnosis and intervention [46] [37]. As epigenomic technologies continue to advance, including the development of single-cell epigenomic methods, the resolution and clinical utility of epigenetic biomarkers are expected to improve significantly [46].
The field of neuroepigenetics has fundamentally transformed our understanding of brain development, function, and disease. Epigenetic mechanisms—including histone modifications, DNA methylation, and chromatin remodeling—serve as dynamic regulators of gene expression that mediate the brain's response to experience throughout life [39] [37]. Dysregulation of these mechanisms contributes to the pathogenesis of diverse neurological and psychiatric conditions, from neurodevelopmental disorders like autism and Rett syndrome to adult-onset conditions such as addiction, depression, and neurodegenerative diseases [43] [44] [39].
Advanced technologies, particularly ChIP-seq and more recently locus-specific epigenetic editing, have provided unprecedented insights into the epigenetic basis of brain disorders, moving the field from correlation to causation [41] [38]. These approaches have revealed that specific histone modifications at particular genomic loci in defined neural cell types can critically influence disease-related behaviors, offering novel targets for therapeutic intervention [38]. The reversible nature of epigenetic modifications makes them particularly attractive for drug development, with several epigenetic therapies already showing promise in preclinical models [39] [37].
As the field advances, key challenges remain, including understanding the incredible complexity of the neuroepigenetic landscape, developing more specific epigenetic modulators with minimal off-target effects, and translating basic epigenetic discoveries into clinical applications [39] [38]. The integration of neuroepigenetics with other areas of neuroscience, including genetics, systems neuroscience, and behavioral science, will be essential for fully elucidating the role of epigenetic mechanisms in health and disease. Ultimately, the growing understanding of neuroepigenetics promises to revolutionize how we diagnose, treat, and prevent neurological and psychiatric disorders, potentially leading to more effective, personalized approaches to mental health care [37].
A Practical Guide to ChIP-Seq: From Bench to Bioinformatics
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has revolutionized our understanding of gene regulation by providing a genome-wide snapshot of protein-DNA interactions. This technique is particularly crucial for studying histone modifications, which serve as key epigenetic markers influencing chromatin structure and transcriptional activity. These modifications—including methylation, acetylation, phosphorylation, and ubiquitination—create a "histone code" that regulates gene expression patterns without altering the underlying DNA sequence [41] [42]. The ability to map these modifications genome-wide has provided unprecedented insights into developmental processes, cellular identity, and disease mechanisms, including cancer and immunological disorders [41] [47] [48]. This technical guide breaks down the core ChIP-seq workflow, with special emphasis on its application in histone modification research relevant to drug discovery and basic biological research.
Core Principles of ChIP-seq
ChIP-seq combines the specificity of chromatin immunoprecipitation with the power of next-generation sequencing to identify and map DNA binding sites for proteins of interest across the entire genome [42]. For histone modification studies, this technique captures the epigenomic landscape that determines how genetic information is packaged and interpreted in different cell types and states [41]. Unlike genetic mutations, histone modifications represent dynamic, reversible changes that can be influenced by environmental factors and targeted by therapeutic interventions [48].
The fundamental ChIP-seq procedure involves: (1) stabilizing protein-DNA interactions through cross-linking, (2) fragmenting chromatin into appropriate sizes, (3) selectively enriching for DNA fragments bound to proteins of interest using specific antibodies, and (4) high-throughput sequencing of the immunoprecipitated DNA [49] [50]. When applied to histone modifications, ChIP-seq reveals the genomic distribution of specific histone marks that activate or repress gene expression, such as H3K4me3 at active promoters, H3K4me1 at enhancers, H3K36me3 in transcribed regions, and repressive marks like H3K9me3 and H3K27me3 [41] [47].
Stage 1: Cross-Linking
Purpose and Mechanism
Cross-linking stabilizes protein-DNA interactions by covalently linking histones to their bound DNA sequences, preserving these relationships throughout the subsequent experimental steps [51]. Formaldehyde is the most common cross-linking agent, creating reversible bonds between histones and DNA that "freeze" the epigenetic landscape at a specific moment [49] [51].
Protocol Details
The standard protocol involves treating cells with 1% formaldehyde for 10 minutes at room temperature, followed by quenching with 125 mM glycine for 5 minutes [49]. This brief exposure represents a critical balance—insufficient cross-linking fails to preserve interactions, while excessive cross-linking can mask antibody epitopes and reduce chromatin shearing efficiency [51]. For histone modifications, which involve stable DNA-protein interactions, milder cross-linking conditions may be sufficient compared to transcription factor studies [50].
Table 1: Cross-Linking Parameters for Different Targets
| Target Type | Formaldehyde Concentration | Incubation Time | Special Considerations |
|---|---|---|---|
| Histone modifications | 1% | 10 minutes | Standard conditions typically sufficient due to stable nucleosome associations |
| Transcription factors | 1% | 10-15 minutes | May require longer cross-linking for transient interactions |
| Fragile chromatin regions | 0.5-1% | 5-10 minutes | Reduced cross-linking to preserve antigen accessibility |
Stage 2: Chromatin Fragmentation (Sonication)
Purpose and Principles
Chromatin fragmentation shears DNA into manageable fragments suitable for immunoprecipitation and sequencing. The optimal fragment size depends on the experimental goal—histone modification studies typically target 150-300 bp fragments (mono- to di-nucleosome size), while transcription factor analyses may require larger fragments (200-700 bp) [49]. Sonication uses ultrasonic energy to randomly shear chromatin, while enzymatic methods like micrococcal nuclease (MNase) digestion preferentially cleave linker DNA between nucleosomes, producing more uniform fragments centered around nucleosomal DNA [47] [51].
Technical Optimization
The sonication process must be carefully optimized for each cell type and specific histone mark of interest [49]. Key parameters include sonication intensity, duration, and temperature control to prevent protein denaturation [51]. Successful shearing produces a fragment distribution centered around the target size, which can be verified using agarose gel electrophoresis or bioanalyzer profiles. MNase digestion offers an alternative for histone studies, providing higher resolution for mapping nucleosome positions but potentially under-representing fragile chromatin regions [47].
Table 2: Fragmentation Methods Comparison
| Parameter | Sonication | MNase Digestion |
|---|---|---|
| Fragment size range | 150-700 bp | ~200 bp (mononucleosome) |
| Resolution for histone marks | Good | Excellent |
| Resolution for transcription factors | Excellent | Poor (cuts linker regions) |
| Reproducibility | Requires optimization | Highly reproducible |
| Equipment requirement | Sonicator | Micrococcal nuclease |
| Effect on epitopes | Potential damage due to heat | Gentle enzymatic treatment |
Stage 3: Immunoprecipitation
Antibody Selection
The success of ChIP-seq for histone modification studies critically depends on antibody specificity [51]. The antibody must recognize the specific histone modification of interest (e.g., H3K4me3 vs. H3K4me2 vs. H3K4me1) without cross-reactivity to similar epitopes [51]. For histone modifications, both monoclonal and polyclonal antibodies can be effective, though polyclonal antibodies may recognize multiple epitopes, potentially increasing the chance of capturing the target [51]. Recommended positive control antibodies for established histone marks include H3K4me3 (CST #9751S), H3K9ac (Millipore #07-352), H3K27me3 (CST #9733S), and H3K4me1 (Diagenode #pAb-037-050) [41].
Bead Preparation and Incubation
Magnetic beads (Protein A, Protein G, or mixtures) are prepared by washing with ice-cold PBS, blocking with BSA-containing buffer, and conjugating with the specific antibody [49]. Typical antibody amounts are 4 μg for histone targets and 8 μg for non-histone targets [49]. The bead-antibody complex is incubated with sheared chromatin for approximately 6 hours or overnight at 4°C with gentle rotation to maximize capture efficiency [49]. Proper controls are essential, including "no-antibody" controls (mock IP) and reference samples such as input DNA (non-immunoprecipitated chromatin) to account for technical biases [47] [51].
Stage 4: Sequencing and Data Analysis
Library Preparation and Sequencing
After immunoprecipitation and cross-link reversal, the purified DNA undergoes library preparation involving end repair, adapter ligation, and PCR amplification before high-throughput sequencing [41] [52]. The Illumina platform is commonly used for ChIP-seq applications, generating millions of short sequence reads that are mapped to a reference genome [41].
Quality Control and Data Interpretation
Several quality control metrics are essential for validating ChIP-seq experiments, particularly for histone modification studies [53]. The strand cross-correlation analysis calculates the Pearson correlation between forward and reverse strand tag densities at various fragment shifts, producing two peaks: a fragment-length peak and a read-length "phantom" peak [53]. Quality metrics derived from this analysis include the Normalized Strand Coefficient (NSC) and Relative Strand Correlation (RSC), with higher values indicating stronger ChIP enrichment [53].
For histone modification data, specialized algorithms like SICER or ChromaBlocks are preferred for identifying broad enrichment domains, in contrast to peak-callers like MACS that are designed for sharp transcription factor binding sites [47]. The resulting data can be integrated with other genomic features to define chromatin states, predict gene expression patterns, and identify regulatory elements such as enhancers and promoters [54].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for ChIP-seq Experiments
| Reagent Category | Specific Examples | Function and Importance |
|---|---|---|
| Cross-linking Agents | Formaldehyde (1%), EGS, DSG | Stabilize protein-DNA interactions; longer cross-linkers (EGS, DSG) for complex protein interactions [51] |
| Cell Lysis Buffers | Nuclear Extraction Buffer 1 (50 mM HEPES-NaOH pH=7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100) [49] | Release nuclear content while preserving protein-DNA complexes |
| Sonication Buffers | Histone sonication buffer (50 mM Tris-HCl pH=8.0, 10 mM EDTA, 1% SDS, protease inhibitors) [49] | Provide optimal chemical environment for chromatin shearing |
| Immunoprecipitation Beads | Protein A/G magnetic beads | Solid support for antibody-antigen complex separation [49] |
| Specific Antibodies | H3K4me3: Anti-Tri-Methyl-Histone H3 (Lys4) (CST #9751S) [41] | Target-specific enrichment; critical for experiment success |
| Protease Inhibitors | PMSF, aprotinin, leupeptin | Prevent protein degradation during processing [41] |
| DNA Purification Kits | QIAquick PCR purification kit (QIAGEN) [41] | Isolve high-quality DNA for library preparation |
| Library Prep Kits | Illumina-compatible kits | Prepare immunoprecipitated DNA for high-throughput sequencing |
Advanced Applications in Drug Discovery
ChIP-seq has become an invaluable tool in drug discovery, particularly for identifying novel therapeutic targets and understanding drug mechanisms of action [48]. By mapping the epigenomic changes associated with disease states, researchers can identify key regulatory proteins and histone modifications that drive pathological processes [48]. For example, oncogenic transcription factors and their target genes have been identified through ChIP-seq, leading to the development of targeted cancer therapies [48]. In neurodegenerative diseases, ChIP-seq has revealed aberrant histone modification patterns that contribute to disease progression, suggesting new therapeutic strategies [48].
The application of ChIP-seq in pharmaceutical research extends to understanding how existing drugs modify the epigenome. By comparing histone modification patterns before and after drug treatment, researchers can identify the direct chromatin targets of therapeutic compounds and optimize drug design for improved efficacy and reduced side effects [48]. These approaches are increasingly important for developing epigenetic therapies that can modulate gene expression patterns in cancer, neurological disorders, and autoimmune diseases.
The ChIP-seq workflow represents a powerful methodology for decoding the epigenetic regulation of gene expression through histone modifications. Each step—from cross-linking and sonication to immunoprecipitation and sequencing—requires careful optimization to ensure high-quality, biologically relevant data. As the technology advances, particularly with the emergence of single-cell ChIP-seq methods, researchers will gain even deeper insights into cellular heterogeneity and dynamic epigenetic changes [54]. When properly executed and analyzed, ChIP-seq provides an unparalleled view of the genomic landscape shaped by histone modifications, offering tremendous potential for both basic biological discovery and therapeutic development.
Within the framework of investigating the role of histone modifications in gene regulation, Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has emerged as a powerful and widely used methodology. The technique provides a genome-wide snapshot of protein-DNA interactions, enabling researchers to map the epigenetic landscape with unprecedented resolution [41] [42]. However, the reliability and biological relevance of the data generated are critically dependent on two fundamental components: the specificity of the antibodies used for immunoprecipitation and the appropriate use of control input samples [55]. Antibodies that lack sufficient specificity can produce false-positive signals or fail to capture the true genomic distribution of the mark, leading to incorrect biological interpretations [56] [57]. Similarly, poorly chosen or processed control samples compromise the ability to distinguish genuine enrichment from technical artifacts and background noise [55] [17]. This guide details the established standards and practices for selecting and validating these critical reagents, ensuring the generation of robust and reproducible ChIP-seq data for histone modification studies.
The Critical Role of Antibody Specificity
The core of any ChIP-seq experiment is the immunoprecipitation step, where an antibody selectively enriches for DNA fragments bound by a specific histone protein carrying a specific post-translational modification (PTM). The quality of this step is governed almost entirely by the antibody's specificity and its ability to achieve meaningful enrichment [55]. Histone tails contain a dense array of similar modification motifs, such as the ARKS sequence found around H3K9 and H3K27, making them particularly challenging epitopes for antibodies to distinguish [56]. Furthermore, the phenomenon of hypermodification—where multiple PTMs exist on the same histone tail—can sterically hinder antibody binding if a secondary modification occurs within the antibody's recognition site, potentially leading to false negatives [56].
Common issues with histone modification antibodies include:
- Cross-reactivity: Antibodies may bind to unrelated proteins or to the same modification on a different histone residue [56] [57].
- Lot-to-Lot Variability: Different production lots of the same antibody, including monoclonal antibodies, can exhibit significant differences in specificity and affinity [56].
- Combinatorial Effects: Neighboring modifications can unpredictably influence antibody binding, either preventing it (false negative) or creating a new, preferred epitope (false positive) [56].
Table 1: Key Challenges in Histone Modification Antibody Specificity
| Challenge | Description | Impact on ChIP-seq Data |
|---|---|---|
| Epitope Similarity | Similar sequence motifs around different residues (e.g., H3K9me3 vs. H3K27me3) [56]. | False positive peak calls at non-target genomic regions. |
| Lot-to-Lot Variability | Changes in specificity and affinity between different production batches of the same antibody [56]. | Inability to reproduce results with a new lot of the same antibody. |
| Influence of Neighboring PTMs | Secondary modifications on the same histone tail can block or enhance antibody binding [56]. | Misrepresentation of the true abundance and distribution of the target mark. |
Antibody Validation Frameworks and Methodologies
To address these challenges, the ENCODE and modENCODE consortia have established rigorous working standards and reporting guidelines for antibodies used in ChIP-seq experiments [55] [17]. These guidelines are designed to provide measurable confidence that the reagent recognizes the intended antigen with minimal cross-reactivity.
Primary and Secondary Characterization
A robust validation strategy involves a primary, accessible assay followed by at least one secondary test to confirm functionality in a ChIP context [55]. For histone modifications, the primary characterization often involves peptide array or dot blot analyses. These assays test antibody binding against a large library of histone peptides carrying various single and combinatorial PTMs, providing a detailed map of specificity and revealing potential cross-reactivities [56] [57]. As shown in Figure 1, this allows for a direct comparison of different antibodies or lots against the same target.
Figure 1: A decision workflow for validating antibodies for histone modification ChIP-seq, based on ENCODE guidelines [55].
A secondary characterization is crucial to confirm that the antibody performs in a ChIP-relevant context. According to ENCODE guidelines, this can include one or more of the following [55] [56]:
- Immunoblot (Western Blot): Demonstrating that the primary reactive band constitutes at least 50% of the signal on a blot of chromatin preparations [55].
- Functional ChIP Validation: Showing expected enrichment at known genomic loci (e.g., H3K4me3 at active promoters) and lack of enrichment at negative control regions (e.g., silent heterochromatin) via ChIP-qPCR [57].
- Knockdown of Histone-Modifying Enzymes: Observing a loss of ChIP signal upon depletion of the enzyme that writes the modification [56].
- Mass Spectrometry: Identifying the specific modification in the immunoprecipitated chromatin [56].
- Reproducibility: Demonstrating high reproducibility between biological replicates and, ideally, overlap with ChIP-seq results from a different antibody against the same mark [55] [56].
Monoclonal vs. Polyclonal Antibodies
The choice between monoclonal and polyclonal antibodies is a key consideration. Monoclonal antibodies are composed of identical molecules that recognize a single epitope, offering superior specificity and lot-to-lot consistency, which minimizes experimental variability [58]. In contrast, polyclonal antibodies are a mixture of molecules that recognize multiple epitopes on the same antigen. While this can sometimes be advantageous for detecting a protein, it poses a higher risk of lot-to-lot variability and cross-reactivity for histone PTMs, making monoclonal antibodies the preferred choice for achieving consistent, reproducible results [58].
The Essential Role of Input Controls
A properly designed input control is non-negotiable for a rigorous ChIP-seq experiment. It accounts for technical biases inherent in the multi-step process, including variations in chromatin shearing, DNA extraction efficiency, sequencing library preparation, and genomic regions that are inherently more accessible or prone to artifacts [55] [17].
The input control should be a sample of the same starting chromatin used for the ChIP experiment, processed in parallel but without the immunoprecipitation step. After crosslinking and sonication, the input DNA is purified, reversing the crosslinks alongside the ChIP samples [55]. The ENCODE consortium mandates that each ChIP-seq experiment must have a corresponding input control experiment with a matching replicate structure, read length, and sequencing run type [17]. Comparing the ChIP sample to the input control during bioinformatic analysis allows for the normalization of these technical biases, enabling the accurate identification of truly enriched genomic regions.
Experimental Design and Quality Control Standards
Beyond reagents, the overall experimental design dictates the quality of the final data. The ENCODE consortium has established clear standards for ChIP-seq experiments, which are summarized in Table 2.
Table 2: ENCODE Experimental Standards for Histone ChIP-seq
| Parameter | Standard Requirement | Rationale |
|---|---|---|
| Biological Replicates | Two or more [55] [17]. | Ensures findings are reproducible and not due to biological variability. |
| Input Control | Required, with matching replicate structure and sequencing parameters [17]. | Controls for technical noise and background. |
| Sequencing Depth | - Narrow marks (e.g., H3K4me3): 20 million usable fragments/replicate [17]. - Broad marks (e.g., H3K27me3): 45 million usable fragments/replicate [17]. | Provides sufficient coverage for statistical power to call peaks across the genome. |
| Library Complexity | NRF > 0.9; PBC1 > 0.9; PBC2 > 10 [17]. | Indicates high-quality, non-redundant library without excessive PCR amplification. |
| Antibody Characterization | Must be characterized according to ENCODE standards [17]. | Ensures the specificity and functionality of the core reagent. |
The required sequencing depth differs based on the type of histone mark being studied. Point-source (narrow) marks, like H3K4me3 and H3K9ac, produce sharp, localized signals and require approximately 20 million usable fragments per replicate [17]. In contrast, broad-source marks, such as H3K27me3 and H3K36me3, are associated with large genomic domains and require deeper sequencing (45 million fragments per replicate) to map their extensive regions accurately [17]. A notable exception is H3K9me3, which is enriched in repetitive regions of the genome; while it is a broad mark, its peaks in unique genomic regions are fewer, and it therefore also requires 45 million total mapped reads per replicate for tissues and primary cells [17].
Figure 2: An overview of the key stages in a rigorous ChIP-seq experimental workflow, highlighting critical reagents, processes, and quality control checkpoints [41] [55] [17].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Histone Modification ChIP-seq
| Reagent / Material | Function / Description | Examples / Considerations |
|---|---|---|
| Validated Antibodies | Core reagent for specific immunoprecipitation of the histone mark. | Use monoclonal antibodies for consistency [58]. Select antibodies validated for ChIP-seq by ENCODE [55] or manufacturers with peptide array data [57]. |
| Crosslinking Reagent | Covalently links proteins to DNA in living cells, "snapshotting" interactions. | Formaldehyde (37%) is standard [41]. |
| Chromatin Shearing Device | Fragments crosslinked chromatin to desired size (100-300 bp). | Sonication devices like Bioruptor [41] or focused ultrasonicator. |
| Protein A/G Magnetic Beads | Facilitate capture and purification of antibody-protein-DNA complexes. | Preferred over agarose beads for better reproducibility and lower background. |
| Protease Inhibitors | Prevent proteolytic degradation of histones and proteins during isolation. | Essential in all buffers post-crosslinking (e.g., PMSF, Aprotinin, Leupeptin) [41]. |
| DNase-free RNase A | Removes RNA that may co-purify with chromatin. | Used during quality control of sheared chromatin [41]. |
| Library Prep Kit | Prepares immunoprecipitated DNA for high-throughput sequencing. | Use kits compatible with low DNA input (e.g., from Illumina, NEB) [41]. |
The path to generating authoritative ChIP-seq data on histone modifications is built upon a foundation of rigorously validated reagents and meticulously designed controls. The selection of a highly specific antibody, confirmed through peptide arrays and functional ChIP assays, is the single most critical factor in determining the success of an experiment. This must be coupled with the consistent use of matched input controls and adherence to established experimental standards regarding replicates and sequencing depth. By integrating these practices, researchers can ensure their findings accurately reflect the biological reality of the epigenome, thereby providing reliable insights into the role of histone modifications in gene regulation, development, and disease.
Library Preparation and High-Throughput Sequencing Platforms for Epigenomics
Epigenetics refers to the alteration of gene expression without a change to the DNA sequence itself, primarily through chemical modifications to histones, DNA, and RNA [59]. The field has expanded dramatically with the development of next-generation sequencing (NGS) technologies, which allow researchers to precisely map these modifications across the entire genome [59]. For the study of histone modifications, which are crucial for transcriptional regulation, chromatin immunoprecipitation followed by sequencing (ChIP-seq) has become the method of choice, providing genome-wide profiles of transcription factors, histone modifications, and nucleosome positioning [41].
This technical guide details the library preparation protocols for key epigenomic methods and compares the high-throughput sequencing platforms suitable for generating these datasets. We focus particularly on the role of histone modifications in gene regulation and the ChIP-seq research that enables their investigation.
High-Throughput Sequencing Platforms for Epigenomics
Selecting an appropriate sequencing platform is a critical first step in experimental design. The choice depends on the required read length, accuracy, throughput, and cost. The table below summarizes the major platforms used in epigenomic studies.
Table 1: Comparison of High-Throughput Sequencing Platforms
| Platform | Read Length | Key Technology | Error Rate | Primary Epigenomic Applications | Throughput (per run) |
|---|---|---|---|---|---|
| Illumina | Short (35-500 bp) | Sequencing by synthesis | Very Low ( ~0.24%) [60] | ChIP-seq, WGBS, RNA-seq [41] | Up to 40 billion bases (NovaSeq 6000) [60] |
| PacBio (Sequel II) | Long (10-15 kb avg, >50 kb N50) | HiFi (High Fidelity) Circular Consensus Sequencing (CCS) | Low ( ~1.72% for CCS) [60] | Closing gaps in assemblies, resolving long repeat regions [60] | 0.5-1 billion bases per SMRT cell [60] |
| Oxford Nanopore (ONT) | Very Long (up to 2.3 Mb) | Single-molecule sequencing through nanopores | Higher ( ~13.4%) [60] | -- | -- |
For most histone modification studies using ChIP-seq, Illumina platforms are the industry standard due to their high accuracy and cost-effectiveness for generating short reads [41]. However, PacBio's long-read capabilities are valuable for resolving complex genomic regions and are being adapted to high-throughput workflows [60]. While Oxford Nanopore offers the longest reads, its higher error rate can be a limitation for precise mapping of epigenetic marks [60].
Core Methodologies in Epigenomic Sequencing
Chromatin Immunoprecipitation Sequencing (ChIP-seq)
ChIP-seq allows researchers to take a snapshot of histone-protein or transcription factor-DNA interactions in a given cell type [41]. The following diagram illustrates the major steps of the ChIP-seq workflow.
Figure 1: The ChIP-seq Experimental Workflow
Detailed Experimental Protocol [41]:
- Crosslinking: Use 37% formaldehyde to covalently crosslink proteins to DNA in living cells. Quench the reaction with glycine.
- Chromatin Preparation & Fragmentation:
- Lyse cells in cell lysis buffer (5 mM PIPES pH 8, 85 mM KCl, 1% igepal) with protease inhibitors.
- Isolate nuclei and lyse in nuclei lysis buffer (50 mM Tris-HCl pH 8, 10 mM EDTA, 1% SDS) with protease inhibitors.
- Shear chromatin to 200-600 bp fragments using a sonicator (e.g., Bioruptor).
- Immunoprecipitation:
- Dilute sheared chromatin with IP dilution buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% igepal, 0.25% deoxycholate, 1 mM EDTA).
- Incubate with validated, ChIP-grade antibodies specific to your histone mark of interest (e.g., H3K4me3, H3K27me3).
- Use protein A/G beads to capture the antibody-bound complexes.
- Washing and Elution: Wash beads stringently and elute DNA-protein complexes with elution buffer (50 mM NaHCO₃, 1% SDS).
- Reverse Crosslinks and Purify DNA: Reverse crosslinks by incubating at 65°C with high salt. Treat with RNase A and proteinase K, then purify DNA using a kit (e.g., QIAquick PCR Purification Kit).
- Quality Control: Assess DNA concentration using a sensitive instrument like a NanoDrop 1000. A successful ChIP typically yields 1-50 ng of DNA from 1 µg of starting chromatin [41].
Whole-Genome Bisulfite Sequencing (WGBS)
DNA methylation (5-methylcytosine, or 5mC) is a fundamental epigenetic mark. WGBS is the gold-standard method for base-resolution mapping of cytosine methylation patterns [61] [59]. The method relies on sodium bisulfite treatment, which converts unmethylated cytosines to uracils (read as thymines in sequencing), while methylated cytosines remain as cytosines [59].
Detailed WGBS Library Preparation Protocol [61]:
- DNA Treatment: Treat genomic DNA with RNaseA to remove RNA contamination.
- DNA Shearing: Fragment DNA to the desired size (e.g., 200-500 bp) using sonication or enzymatic methods.
- End-Repair and A-Tailing: Repair the ends of the sheared DNA fragments and add an 'A' base to the 3' ends to facilitate adapter ligation.
- Adapter Ligation: Ligate methylated or universal adapters containing sample-specific indexes for multiplexing.
- Bisulfite Conversion: Treat the adapter-ligated library with sodium bisulfite. This critical step deaminates unmethylated C to U, while 5mC and 5hmC remain as C.
- Library Amplification: Perform PCR amplification to enrich for successfully ligated and converted fragments.
- Quantification: Precisely quantify the final library before sequencing.
The Scientist's Toolkit: Essential Research Reagents
Successful epigenomic research relies on high-quality, specific reagents. The following table lists essential materials and their functions for a typical ChIP-seq experiment.
Table 2: Research Reagent Solutions for ChIP-seq
| Reagent / Kit | Function / Application | Example Product / Catalog Number |
|---|---|---|
| ChIP-Grade Antibodies | Specific enrichment of target histone modifications. | H3K4me3 (CST #9751S); H3K27me3 (CST #9733S) [41] |
| Crosslinking Reagent | Covalently links proteins to DNA in vivo. | Formaldehyde solution (37%) [41] |
| Cell Lysis Buffer | Lyses cell membrane while keeping nuclei intact. | 5 mM PIPES pH 8, 85 mM KCl, 1% igepal [41] |
| Magnetic Beads | Capture and wash antibody-bound complexes. | Protein A/G magnetic beads |
| DNA Purification Kit | Purify DNA after reverse crosslinking. | QIAquick PCR Purification Kit (QIAGEN) [41] |
| Library Prep Kit | Prepare sequencing library from immunoprecipitated DNA. | Illumina-compatible library prep kits |
Advanced and Emerging Methodologies
To overcome limitations of ChIP-seq such as high background noise and large input requirements, novel technologies have been developed.
Figure 2: CUT&RUN vs. CUT&Tag Workflows
- CUT&RUN (Cleavage Under Targets and Release Using Nuclease): This method immobilizes cells on magnetic beads and uses a specific antibody and Protein A-MNase (pA-MNase) fusion protein. Upon activation with Ca²⁺, MNase cleaves DNA around the target protein, releasing specific fragments for sequencing. This occurs in situ without crosslinking, reducing background noise [59].
- CUT&Tag (Cleavage Under Targets and Tagmentation): An improvement on CUT&RUN, CUT&Tag uses a pA-Tn5 transposase fusion protein. When activated with Mg²⁺, Tn5 simultaneously cleaves and ligates sequencing adapters directly onto the target chromatin in situ. This significantly simplifies and speeds up library construction, making it ideal for high-throughput and single-cell applications [59].
The field of epigenomics is powered by a suite of sophisticated library preparation methods and high-throughput sequencing platforms. ChIP-seq remains a cornerstone for investigating the role of histone modifications in gene regulation, while WGBS provides a comprehensive map of DNA methylation. Emerging techniques like CUT&Tag offer higher resolution and efficiency. As sequencing technologies continue to evolve towards longer reads and higher throughput, as seen with PacBio's automated workflows [60], our ability to decode the complex language of epigenetics will only expand, offering deeper insights into development, disease, and novel therapeutic opportunities.
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has revolutionized our ability to investigate protein-DNA interactions on a genome-wide scale, establishing itself as a central method in epigenomic research [54] [62]. This powerful technique provides unprecedented resolution for mapping the genomic locations of transcription factors, chromatin-binding proteins, and post-translational histone modifications [41] [42]. Within the context of gene regulation, histone modifications serve as critical epigenetic markers that influence chromatin structure and function without altering the underlying DNA sequence [42]. These modifications—including methylation, acetylation, phosphorylation, and ubiquitination—create a complex "histone code" that helps determine whether genomic regions are in an open, transcriptionally active state or a closed, repressive state [41]. The dynamic nature of these modifications across different tissues, developmental stages, and disease states makes them particularly intriguing for understanding the molecular basis of cellular identity, lineage specification, and pathological conditions such as cancer [41] [63]. By providing a systematic approach to characterize these modifications genome-wide, ChIP-seq analysis enables researchers to move from raw sequencing data to profound biological insights about transcriptional regulation and chromatin dynamics.
Experimental Foundations of ChIP-Seq
The journey from biological sample to sequencing data begins with a multi-step ChIP-seq protocol designed to capture snapshots of protein-DNA interactions in living cells [41]. The standard workflow initiates with formaldehyde cross-linking to covalently stabilize protein-DNA interactions, effectively freezing these relationships at the moment of fixation [41] [64]. Following cell lysis, chromatin is fragmented typically by sonication using instruments like the Bioruptor to shear DNA into fragments of 150-500 base pairs [41] [62]. The critical immunoprecipitation step then follows, where antibodies specific to the histone modification of interest (e.g., H3K4me3, H3K27me3) are used to selectively enrich for DNA fragments bound to modified histones [41]. After antibody capture, reversal of cross-links liberates the DNA, which is then purified and prepared for high-throughput sequencing [41]. Throughout this process, quality control checkpoints are essential, particularly when working with limited cell numbers where specialized approaches like carrier ChIP-seq (cChIP-seq) may be employed to maintain signal-to-noise ratio [65].
Computational Analysis: From Sequences to Peaks
Primary Data Processing
The computational ChIP-seq pipeline transforms raw sequencing reads into interpretable genomic regions through a series of validated analytical steps [63] [66]. The initial quality control assessment utilizes tools like FastQC to evaluate raw sequencing data, examining parameters including sequence quality scores, GC content, adapter contamination, and overrepresented sequences [63] [64]. Problematic reads may be trimmed or filtered at this stage to ensure only high-quality data proceeds downstream. The subsequent alignment step maps the filtered reads to a reference genome using specialized aligners such as Bowtie2 or BWA, which efficiently handle the millions of short reads generated by modern sequencers [53] [64]. A crucial consideration during alignment is the handling of uniquely mapped reads—typically only reads that map unambiguously to a single genomic location are retained for subsequent analysis, while multimapping reads are discarded to reduce false positives [53] [62]. Following alignment, additional filtering removes PCR duplicates using tools like Picard or Sambamba to ensure library complexity is maintained, with the Non-Redundant Fraction (NRF) serving as a key quality metric [63] [62].
Quality Assessment and Peak Calling
ChIP-seq-specific quality control employs several specialized metrics to evaluate experimental success [53]. The strand cross-correlation analysis assesses the clustering of sequence tags at protein-binding sites by calculating the Pearson correlation between forward and reverse strand tag densities at various shift distances [53]. High-quality experiments typically produce a prominent peak at the average fragment length, with quality measures including the Normalized Strand Coefficient (NSC) and Relative Strand Correlation (RSC) helping to distinguish successful immunoprecipitations from failed experiments [53]. The core of ChIP-seq analysis—peak calling—leverages statistical algorithms to identify genomic regions significantly enriched for aligned reads compared to background [63] [66]. MACS2 (Model-based Analysis of ChIP-Seq) represents the most widely used peak caller, employing a dynamic Poisson distribution to model expected background and account for the shifting of tags to better represent the actual protein-binding site [63] [64]. For broad histone marks like H3K27me3, alternative callers such as SICER may be preferred as they specifically account for diffuse enrichment patterns across large genomic domains [62] [66].
Table 1: Key ChIP-Seq Quality Metrics and Their Interpretation
| Quality Metric | Calculation | Interpretation | Threshold |
|---|---|---|---|
| Strand Cross-Correlation | Pearson correlation between forward and reverse strand densities | Measures enrichment; higher values indicate better signal-to-noise | NSC > 1.05, RSC > 0.8 [53] |
| Non-Redundant Fraction (NRF) | Nnonred/Nall | Measures library complexity; higher values indicate less PCR duplication | NRF > 0.8 for 10M reads [62] |
| Mapping Rate | Mappedreads/Totalreads | Percentage of reads successfully aligned to reference genome | >70% considered good [64] |
| Fraction of Reads in Peaks (FRiP) | Readsinpeaks/Totalmappedreads | Measures enrichment efficiency; higher values indicate successful IP | >1% for TFs, >10-30% for histones [53] |
Advanced Analytical Considerations
Addressing Technical Biases
ChIP-seq data analysis must account for several technical biases that can confound biological interpretation if left unaddressed [67]. The mappability bias arises during read alignment because standard preprocessing protocols retain only tags that align uniquely to the reference genome, systematically underrepresenting genomic regions with repetitive elements [67]. This bias can be particularly problematic near functional elements like transcription start sites, which often have higher uniqueness [67]. Similarly, GC content bias manifests as an association between local GC composition and observed tag counts, potentially due to differential amplification efficiency during library preparation [67]. Statistical frameworks like MOSAiCS explicitly model these biases using background samples (input DNA or naked DNA controls) to distinguish technical artifacts from genuine biological signals [67]. When designing ChIP-seq experiments, sequencing depth represents another critical consideration, with deeper sequencing required to capture weak binding sites or broad histone modifications—for human samples, the ENCODE consortium recommends a minimum of 10 million uniquely mapped reads per replicate for transcription factors, with higher depths often necessary for comprehensive histone modification profiling [62].
Peak Annotation and Interpretation
Following peak identification, the biological interpretation of results proceeds through systematic annotation of enriched regions to establish their genomic context and potential functional impact [64]. This typically involves determining the genomic distribution of peaks relative to features such as transcription start sites (TSS), gene bodies, and intergenic regions using tools like ChIPseeker or HOMER [66]. For histone modifications, distinct patterns emerge—H3K4me3 typically marks active promoters near TSSs, H3K4me1 identifies enhancer elements, H3K36me3 covers transcribed regions, while H3K27me3 and H3K9me3 designate repressed chromatin states [41]. Motif analysis can reveal underlying DNA sequence preferences associated with the enriched regions, potentially identifying transcription factors that cooperate with the histone modifications to establish chromatin states [63]. Functional enrichment analysis using tools like GOseq connects the genomic regions bearing specific histone modifications to biological processes, molecular functions, and cellular components, helping to generate testable hypotheses about the regulatory programs active in the studied system [66].
Table 2: Common Histone Modifications and Their Functional Associations
| Histone Mark | Associated Function | Typical Genomic Location | Chromatin State |
|---|---|---|---|
| H3K4me3 | Active promoters | Transcription start sites | Open chromatin [41] |
| H3K4me1 | Enhancers | Distal regulatory elements | Open chromatin [41] |
| H3K36me3 | Transcriptional elongation | Gene bodies | Open chromatin [41] |
| H3K27me3 | Polycomb repression | Developmentally regulated genes | Compacted chromatin [41] |
| H3K9me3 | Heterochromatin | Repetitive elements, zinc finger genes | Compacted chromatin [41] |
| H3K9ac | Active transcription | Promoters, enhancers | Open chromatin [41] |
From Peaks to Biological Insight
Integration with Complementary Data
Extracting meaningful biological insights from ChIP-seq data rarely occurs in isolation; rather, integration with complementary genomic datasets significantly enhances interpretative power [62]. Combining histone modification profiles with gene expression data from RNA-seq allows researchers to correlate chromatin states with transcriptional outputs, enabling the identification of putative regulatory relationships—for instance, connecting enhancer-associated H3K4me1 marks with expression of nearby genes [63] [62]. Similarly, incorporating chromatin accessibility data from ATAC-seq or DNase-seq helps distinguish which marked regulatory elements are actually accessible in a given cellular context [66]. More sophisticated integrative approaches examine the combinatorial patterns of multiple histone modifications, recognizing that specific combinations often define functionally distinct genomic elements more reliably than individual marks [41]. For example, the simultaneous presence of both H3K4me3 (an activating mark) and H3K9me3 (a repressive mark) at certain promoters can identify imprinted genes subject to allele-specific expression [41]. These integrated perspectives facilitate the construction of comprehensive chromatin state maps that systematically annotate the genome based on multivariate epigenetic information, providing powerful frameworks for understanding how histone modification landscapes contribute to cellular identity and function [54].
Visualization and Interpretation
Effective visualization of ChIP-seq results is essential for both quality assessment and biological interpretation [63]. The Integrative Genomics Viewer (IGV) enables interactive exploration of aligned reads and called peaks across genomic regions of interest, allowing researchers to visually confirm enrichment patterns and inspect data quality [53] [63]. For publication-quality representations, profile plots display average signal intensity across genomic features like transcription start sites, while heatmaps effectively communicate the enrichment patterns across multiple samples or conditions [63]. When comparing histone modification patterns between experimental conditions (e.g., disease versus control), differential binding analysis using tools like DESeq2 or edgeR identifies statistically significant changes in mark enrichment, potentially revealing epigenetic mechanisms underlying phenotypic differences [66]. The biological interpretation culminates in connecting observed histone modification patterns to regulatory consequences—for instance, determining how gained H3K27me3 in a disease state might silence tumor suppressor genes, or how acquired H3K4me3 at previously inactive promoters might activate oncogenic pathways [42] [63].
ChIP-Seq Workflow: From Sample to Insight
Essential Research Reagents and Tools
Table 3: Research Reagent Solutions for Histone Modification ChIP-Seq
| Reagent/Tool | Function | Example/Alternative |
|---|---|---|
| ChIP-Grade Antibodies | Specific recognition of histone modifications | H3K4me3 (CST #9751S), H3K27me3 (CST #9733S) [41] |
| Chromatin Shearing Instrument | Fragment chromatin to appropriate size | Bioruptor UCD-200, Covaris LE220 [41] [65] |
| Proteinase K | Reverse crosslinks and digest proteins after IP | Included in most ChIP kits [41] |
| DNA Clean-up Kits | Purify immunoprecipitated DNA | QIAquick PCR Purification Kit [41] |
| Sequencing Platform | High-throughput sequencing | Illumina GA2, HiSeq, NovaSeq [41] |
| Alignment Software | Map reads to reference genome | Bowtie2, BWA [53] [64] |
| Peak Caller | Identify significantly enriched regions | MACS2, SICER, PeakSeq [63] [66] |
| Visualization Tool | Visual exploration of results | IGV (Integrative Genomics Viewer) [53] [63] |
The standard ChIP-seq analysis pipeline provides a robust framework for translating raw sequencing data into biologically meaningful insights about histone modifications and their role in gene regulation. From meticulous experimental preparation through sophisticated computational analysis, each step contributes to building a comprehensive view of the epigenomic landscape. As sequencing technologies continue to evolve and analytical methods become increasingly refined, ChIP-seq will undoubtedly remain an indispensable tool for elucidating how histone modifications shape genome function in health and disease. The pipeline described here offers researchers a structured approach to navigate the complexities of ChIP-seq data, ultimately enabling the discovery of how epigenetic mechanisms contribute to transcriptional programs governing cellular identity, development, and disease pathogenesis.
The integration of Chromatin Immunoprecipitation Sequencing (ChIP-seq) and RNA Sequencing (RNA-seq) represents a transformative approach for elucidating the complex mechanistic links between histone modifications and gene regulatory outcomes. These complementary technologies enable researchers to move beyond correlation to causation in epigenetic studies by mapping the physical presence of histone marks across the genome while simultaneously quantifying their transcriptional consequences [68] [69]. Histone modifications, including methylation and acetylation, function as crucial epigenetic regulators that alter chromatin structure and DNA accessibility without changing the underlying DNA sequence [70]. The repressive marks H3K9me3 and H3K27me3 are particularly significant, marking constitutive and facultative heterochromatin respectively, while H3K4me2/3 is generally associated with active transcription [71]. Advanced integration of ChIP-seq and RNA-seq data now allows researchers to decode how specific histone modification patterns direct cellular fate, response to environmental cues, and disease pathogenesis through precise control of gene expression programs [8] [71].
Fundamental Principles of Histone Modification Analysis
Key Histone Modifications and Their Functional Roles
Table 1: Major Histone Modifications and Their Functional Significance
| Histone Modification | Associated Function | Chromatin Environment | Catalytic Enzyme Family |
|---|---|---|---|
| H3K4me2/3 | Transcriptional activation | Euchromatin | KMT2 (e.g., Set1) |
| H3K9me3 | Transcriptional repression | Constitutive heterochromatin | KMT1 |
| H3K27me3 | Transcriptional repression | Facultative heterochromatin | KMT6 (PRC2 complex) |
| H3K36me3 | Transcriptional elongation | Euchromatin | SETD2 |
| H4K16ac | Chromatin relaxation | Euchromatin | KAT8 |
Histone post-translational modifications (PTMs) function as integral components of an epigenetic language that regulates chromatin architecture and DNA accessibility [70]. These modifications serve as binding platforms for downstream effector proteins containing specialized domains such as bromodomains, chromodomains, and Tudor domains, which subsequently recruit chromatin remodeling complexes, transcriptional activators or repressors, and DNA repair machinery [71]. The interplay between different histone modifications, known as histone PTM crosstalk, creates a complex regulatory network where one modification can influence the establishment or removal of another [71]. For instance, recent studies demonstrate that H3K27me3 possesses unique repressive functions that cannot be fully substituted by other repressive marks like H3K9me3 or H3K36me3, highlighting the non-redundant nature of specific histone modifications [8].
Experimental Techniques for Histone Modification Profiling
Table 2: Comparison of Major Techniques for Histone Modification Analysis
| Technique | Principle | Resolution | Input Material | Advantages |
|---|---|---|---|---|
| ChIP-seq | Antibody-based chromatin enrichment | 200-500 bp | High cell numbers | Well-established, robust |
| CUT&Tag | Antibody-targeted tethering of Tn5 | Single-cell | Low cell numbers | Higher sensitivity, lower background |
| Mass Spectrometry | Direct detection of modified histones | Amino acid level | Protein extract | Quantitative, comprehensive |
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) remains the gold standard for genome-wide mapping of histone modifications in vivo [68]. This method cross-links proteins to DNA, fragments chromatin, immunoprecipitates target epitopes using specific antibodies, and sequences the associated DNA fragments [68]. More recently, CUT&Tag has emerged as a promising alternative that recovers up to half of ENCODE ChIP-seq histone acetylation peaks while offering enhanced sensitivity and compatibility with low cell numbers [9]. For quantitative analysis of histone modification abundance without antibody bias, mass spectrometry-based approaches like PTMViz provide complementary data on modification stoichiometry and combinatorial patterns [70].
Integrated Experimental Design
Strategic Planning for Multi-Omic Studies
Effective integration of ChIP-seq and RNA-seq begins with careful experimental design that aligns both methodologies toward a common biological question. Two principal strategic frameworks have emerged: the candidate-driven approach, where RNA-seq identifies differentially expressed transcription factors or histone modifiers that subsequently become targets for ChIP-seq analysis [68]; and the hypothesis-testing approach, where independent ChIP-seq profiling of specific histone marks is correlated with matched RNA-seq data from the same biological system [68] [69]. Biological replication is essential for both techniques, with triplicate samples recommended to account for technical variability and biological heterogeneity [68]. For time-series experiments investigating dynamic histone modification changes, synchronized sampling across platforms is critical for valid inference of regulatory relationships.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Integrated ChIP-seq and RNA-seq Studies
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Histone modification antibodies | Anti-H3K27me3, Anti-H3K4me3, Anti-H3K9me3 | Target-specific immunoprecipitation | Specificity validation essential |
| Cell type manipulation | KMT knockout mutants (e.g., ΔSet1, ΔKmt1, ΔKmt6) | Loss-of-function studies | Compensatory mechanisms may occur |
| Chromatin platforms | CUT&Tag kits, ChIP-seq kits | Histone mark profiling | Compatibility with sample type |
| Sequencing library prep | Illumina-compatible kits | Library construction | Insert size selection critical |
| Bioinformatics tools | HOMER, MACS2, DESeq2, systemPipeR | Data analysis | Pipeline reproducibility |
The selection of validated, high-specificity antibodies against histone modifications represents the most critical reagent choice for ChIP-seq experiments [68] [71]. Genetic tools including knockout mutants for histone methyltransferases (e.g., KMT1, KMT6) enable loss-of-function studies that establish causal relationships between specific modifications and transcriptional outcomes [71]. For example, studies in Pyricularia oryzae demonstrated that deletion of MoKmt6 (catalyzing H3K27me3) led to activation of infection-responsive genes, establishing the functional significance of this repressive mark in fungal pathogenicity [71].
Computational Integration and Statistical Analysis
Bioinformatics Workflows for Multi-Omic Data Integration
The computational integration of ChIP-seq and RNA-seq data requires specialized bioinformatics pipelines that address the distinct statistical characteristics of each data type. For ChIP-seq analysis, the systemPipeR workflow provides a comprehensive framework encompassing quality control, read alignment, peak calling with tools like MACS2, peak annotation, and differential binding analysis [52]. RNA-seq analysis typically involves alignment, quantification of gene-level counts, and differential expression analysis using packages such as DESeq2 [72]. Meaningful integration occurs at the annotation stage, where histone modification peaks are associated with target genes based on genomic proximity, with consideration of enhancer-promoter interactions and chromatin architecture [69].
Advanced statistical methods including principal component analysis (PCA), log-linear models, and support vector regression have been successfully applied to predict gene expression levels based on histone modification patterns [69]. These models can reveal whether specific epigenetic signatures function as activators or repressors at different genomic loci, providing mechanistic insights beyond simple correlation [69]. The systematic benchmarking of analysis parameters is essential, as demonstrated by recent comparisons between CUT&Tag and ChIP-seq, which showed approximately 50% overlap in detected regions but strong concordance for the most significant peaks [9].
Visualization and Interpretation of Integrated Data
Effective visualization strategies are critical for interpreting the complex relationships between histone modifications and gene expression. Multi-track genome browsers enable simultaneous visualization of ChIP-seq coverage and RNA-seq expression data across genomic loci of interest [69]. Specialized tools like PTMViz offer interactive platforms for differential abundance analysis and visualization of histone PTMs, generating volcano plots, stacked bar charts, and heatmaps that facilitate exploration of significant changes between experimental conditions [70]. For inferring gene regulatory networks, visualization tools that represent transcription factors as nodes and their regulatory interactions as edges can reveal how histone modifications shape hierarchical regulatory architectures [69].
Case Studies in Disease Research and Drug Development
Cancer Epigenetics and Therapeutic Targeting
The integrated analysis of histone modifications and gene expression has yielded significant insights into cancer mechanisms and identified novel therapeutic opportunities. Studies across multiple cancer types have revealed widespread redistribution of repressive marks such as H3K27me3 in tumor cells, resulting in aberrant silencing of tumor suppressor genes [69]. These epigenetic alterations represent promising therapeutic targets, as unlike genetic mutations, they are potentially reversible through pharmacological intervention [69]. Small molecule inhibitors targeting histone methyltransferases and demethylases are currently under development as "epi-drugs" that seek to correct pathological histone modification patterns and restore normal gene expression programs in cancer cells [69].
In glioblastoma research, integrated analysis has identified specific H3K27me3 patterns associated with treatment resistance, suggesting combination therapies that include epigenetic modifiers might overcome this resistance [8]. Similarly, in breast cancer, inhibitors targeting histone demethylases such as KDM4 have shown antiproliferative effects on cancer stem-like cells, highlighting the therapeutic potential of targeting histone modification machinery [8].
Neurobiology and Response to Environmental Stimuli
Integrated ChIP-seq and RNA-seq approaches have illuminated how histone modifications mediate neural plasticity and response to pharmacological agents. In a mouse model of methamphetamine exposure, PTMViz analysis of histone PTMs in reward circuitry brain regions identified significant changes in H3K9me, H3K27me3, and H4K16ac modifications following drug exposure [70]. These epigenetic changes were associated with altered expression of proteins in dopaminergic signaling pathways, providing a mechanistic link between histone modifications and behavioral responses to drugs of abuse [70].
Similar approaches in studies of learning and memory have revealed how experience-dependent changes in histone acetylation and methylation patterns regulate the expression of genes required for synaptic plasticity and long-term memory formation. The ability to profile histone modifications in specific cell types using techniques like CUT&Tag has been particularly valuable in heterogeneous tissues like brain, where bulk analysis may mask cell type-specific epigenetic regulation [9].
Emerging Technologies and Future Perspectives
The field of integrated epigenetic and transcriptomic analysis is rapidly evolving with several emerging technologies poised to enhance mechanistic studies. Single-cell multi-omics approaches now enable simultaneous profiling of histone modifications and gene expression in the same individual cells, resolving heterogeneity within complex tissues [9]. Long-read sequencing technologies from PacBio and Oxford Nanopore are improving the detection of alternative splicing events and isoform-specific regulation by histone marks [68]. Additionally, computational methods for integrating three-dimensional chromatin architecture data with histone modification and gene expression patterns are providing unprecedented insights into how spatial genome organization influences gene regulation.
The development of more sophisticated tools for manipulating histone modifications, including engineered CRISPR-based systems that recruit histone modifiers to specific genomic loci, will enable more precise causal inference in regulatory studies [8]. As these technologies mature, they will further accelerate the translation of basic epigenetic discoveries into therapeutic applications for cancer, neurological disorders, and other diseases linked to aberrant histone modification.
Optimizing Your Epigenomic Assays: Critical Steps for Robust ChIP-Seq and CUT&Tag Data
In chromatin immunoprecipitation followed by sequencing (ChIP-seq) research focused on histone modifications, achieving ideal DNA fragmentation is a critical determinant of experimental success. Histone post-translational modifications—including methylation, acetylation, phosphorylation, and ubiquitination—serve as key epigenetic regulators of gene expression and chromatin dynamics [42]. Proper fragmentation of cross-linked chromatin not only ensures the resolution necessary to map these modifications to specific genomic regions but also preserves epitope integrity for efficient immunoprecipitation. Sonication remains the most widely employed method for chromatin shearing, yet it presents significant challenges in optimization, particularly when working with complex solid tissues or limited cell numbers [73]. This technical guide provides a comprehensive framework for sonication optimization and quality control, enabling researchers to generate high-quality ChIP-seq data for elucidating the role of histone modifications in gene regulation.
The Critical Role of DNA Fragmentation in Histone Modification Studies
The accurate genome-wide mapping of histone modifications such as H3K4me3 (associated with promoters), H3K4me1 (enhancers), and H3K27me3 (facultative heterochromatin) depends heavily on chromatin fragmentation quality [41]. Under-fragmentation produces large chromatin fragments that limit mapping resolution and increase background noise, while over-fragmentation can disrupt nucleosome integrity and compromise the detection of histone modification patterns [74]. Optimal fragmentation preserves nucleosome structure while generating fragments of appropriate size for sequencing library construction, typically in the range of 200-500 base pairs for histone modifications [65]. This balance is particularly crucial when studying solid tissues, where cellular heterogeneity and complex extracellular matrices present additional challenges for chromatin preparation and fragmentation [73].
Sonication Optimization Protocols
Establishing Baseline Parameters
Before embarking on full-scale ChIP-seq experiments, researchers must determine optimal sonication conditions for their specific biological system and equipment. The following protocol, adapted from established methodologies, provides a systematic approach for this optimization [74].
Time-Course Optimization Protocol:
- Prepare cross-linked nuclei from 100–150 mg of tissue or 1×10⁷–2×10⁷ cells resuspended in 1 ml of ChIP Sonication Nuclear Lysis Buffer.
- Aliquot the chromatin preparation into multiple tubes for parallel processing.
- Subject samples to sonication using a consistent power setting while varying the duration. Remove 50 μl aliquots after successive sonication intervals (e.g., 1, 2, 4, 8, 12 minutes).
- Clarify chromatin samples by centrifugation at 21,000 × g for 10 minutes at 4°C.
- Reverse cross-links by adding NaCl to a final concentration of 200 mM and incubating at 65°C for 2 hours (or overnight).
- Treat samples with RNase A (37°C for 30 minutes) followed by Proteinase K (55°C for 2 hours).
- Purify DNA using standard PCR purification kits and evaluate fragment size distribution by electrophoresis on a 1% agarose gel or using a Bioanalyzer system.
Critical Optimization Considerations:
- Sample Characteristics: Different tissue types yield varying amounts of chromatin. For instance, 25 mg of liver or brain tissue typically yields 10-15 μg and 2-5 μg of total chromatin, respectively [74]. Adjust starting material accordingly to achieve the recommended 5-10 μg of chromatin per immunoprecipitation reaction.
- Fixation Conditions: Cross-linking time significantly impacts fragmentation efficiency. For tissues fixed for 10 minutes, optimal sonication should generate a DNA smear with approximately 60% of fragments below 1 kb. Extending fixation to 30 minutes reduces fragmentation efficiency, with only ~30% of fragments below 1 kb after equivalent sonication [74].
- Equipment Variables: Different sonicators (e.g., probe vs. bath-based systems) require distinct parameter optimization. Document specific settings including power output, duty cycle, and peak incident power to ensure reproducibility.
Tissue-Specific Optimization Strategies
Working with solid tissues presents unique challenges for chromatin fragmentation. A refined ChIP-seq protocol for colorectal cancer tissues emphasizes proper tissue preparation as a prerequisite for effective sonication [73]. The protocol recommends:
- Tissue Preparation: Mince frozen tissue samples on a Petri dish placed on ice using sterile scalpel blades until finely diced.
- Homogenization Options:
- Dounce Homogenization: Transfer minced tissue to a 7 ml Dounce grinder with cold PBS supplemented with protease inhibitors. Apply 8-10 even strokes with the A pestle while keeping the apparatus deeply submerged in ice.
- GentleMACS Dissociator: Transfer minced tissue to a C-tube with cold PBS and run the preconfigured "htumor03.01" program or tissue-specific alternatives.
- Cross-linking: Perform formaldehyde cross-linking after tissue disaggregation to ensure uniform fixation.
- Nuclear Isolation: Isulate intact nuclei before sonication to improve fragmentation efficiency and reduce shearing variability.
Quality Control Checkpoints
Robust quality control throughout the fragmentation process is essential for generating reliable ChIP-seq data. Implement the following checkpoints to ensure experimental success.
Pre-Sonication Quality Assessment
- Nuclear Integrity: Examine nuclei under a light microscope before and after sonication to confirm complete lysis. Intact nuclei before sonication and their disruption afterward indicate proper processing [74].
- Chromatin Concentration: Quantify chromatin yield using spectrophotometric methods. Low concentrations (<50 μg/ml) may necessitate pooling samples or increasing starting material [74].
- Buffer Composition: Ensure lysis buffers contain fresh protease inhibitors to prevent histone degradation during processing.
Post-Sonication Quality Assessment
- Fragment Size Distribution: Analyze sheared chromatin using high-sensitivity DNA assays. Ideal fragmentation for histone modifications should produce a smear centered between 200-500 bp, with minimal fragments below 150 bp or above 1000 bp.
- Over-sonication Indicators: >80% of total DNA fragments shorter than 500 bp suggests over-sonication, which can damage chromatin and reduce immunoprecipitation efficiency [74].
- Quantification: Measure DNA concentration after reverse cross-linking to ensure sufficient material for library preparation. The recommended input for standard ChIP-seq protocols is 5-10 μg of fragmented chromatin per immunoprecipitation reaction [74].
Troubleshooting Common Fragmentation Issues
The table below outlines common sonication problems, their causes, and recommended solutions.
Table 1: Troubleshooting Guide for Chromatin Fragmentation
| Problem | Possible Causes | Recommendations |
|---|---|---|
| Low chromatin concentration | Insufficient starting material; incomplete lysis | Count cells accurately before cross-linking; visualize nuclei under microscope to confirm complete lysis; increase input material if necessary [74] |
| Under-fragmentation (large fragments) | Over-crosslinking; excessive input material; insufficient sonication | Shorten cross-linking time (10-30 min range); reduce cells/tissue per sonication; increase sonication time or power [74] |
| Over-fragmentation (mostly <500 bp) | Excessive sonication cycles; high power settings | Reduce sonication time or cycles; lower power setting; use minimal sonication required for desired fragment size [74] |
| High background noise | Large fragment size; non-specific antibody binding | Optimize fragmentation size; pre-clear chromatin with beads; titrate antibody concentration [74] |
Advanced Applications and Special Considerations
Low-Input and Single-Cell ChIP-seq
Recent methodological advances enable ChIP-seq from limited cell numbers. Carrier ChIP-seq (cChIP-seq) employs DNA-free recombinant histone carriers to maintain working reaction scales with as few as 10,000 cells [65]. This approach minimizes the need for extensive optimization of chromatin-to-beads ratios while preserving specificity for histone modification profiling.
Tissue Heterogeneity Considerations
When working with complex tissues, consider that cellular heterogeneity may influence apparent fragmentation patterns. The refined protocol for colorectal cancer tissues emphasizes standardized processing to maintain consistency across samples with inherent variability [73].
The Scientist's Toolkit: Essential Reagents and Equipment
Table 2: Key Research Reagent Solutions for Chromatin Fragmentation
| Category | Specific Examples | Function/Application |
|---|---|---|
| Sonication Equipment | Branson Digital Sonifier 250; Bioruptor Pico; Covaris LE220 | Mechanical shearing of cross-linked chromatin; equipment choice affects optimization parameters [74] [65] |
| Chromatin Preparation Buffers | Cell Lysis Buffer; Nuclei Lysis Buffer; FA Lysis Buffer | Sequential extraction of cytoplasmic and nuclear components; buffer composition affects chromatin accessibility [41] [75] |
| Protective Additives | Protease Inhibitor Cocktails; PMSF; Aprotinin; Leupeptin | Prevent histone degradation during processing; essential for preserving modification epitopes [73] [41] |
| Quality Control Tools | Agilent Bioanalyzer; TapeStation; High Sensitivity DNA Kits | Precise assessment of DNA fragment size distribution; superior to agarose gel electrophoresis for resolution [75] |
| Chromatin Carriers | Recombinant histone H3 with specific modifications | Enhance immunoprecipitation efficiency in limited cell applications; DNA-free carriers avoid sequencing contamination [65] |
Workflow Visualization
Optimization Workflow and Quality Control Checkpoints
Achieving ideal DNA fragmentation through optimized sonication protocols is fundamental to successful ChIP-seq studies of histone modifications. By implementing systematic optimization approaches, adhering to rigorous quality control checkpoints, and utilizing appropriate troubleshooting strategies, researchers can generate high-quality fragmentation profiles that enable precise mapping of epigenetic regulatory landscapes. As ChIP-seq methodologies continue to evolve toward single-cell applications and more complex tissue contexts, the principles of careful fragmentation optimization remain essential for extracting biologically meaningful insights into the role of histone modifications in gene regulation.
In chromatin immunoprecipitation followed by sequencing (ChIP-seq), the interpretation of genome-wide histone modification distribution is fundamentally dependent on the specificity of the antibodies used for immunoprecipitation [76]. Histone post-translational modifications (PTMs) function as crucial regulators of gene expression, and their accurate mapping provides insights into epigenetic mechanisms governing cell differentiation, immune function, and disease pathogenesis [77] [78]. However, concerns regarding antibody specificity have grown as studies reveal widespread issues with off-target recognition, cross-reactivity, and sensitivity to neighboring modifications [57] [78] [79]. The reliability of ChIP-seq data hinges on rigorous antibody validation, making specificity assessment not merely a preliminary step but the foundational element of reproducible epigenetics research.
The Critical Need for Antibody Validation in Epigenetic Research
Antibodies against histone PTMs are essential reagents for numerous applications in chromatin biology, including immunoblotting, immunofluorescence, and particularly ChIP-seq, which has become the gold standard for mapping histone modification distributions genome-wide [76] [78]. Large-scale epigenomics consortia like the ENCODE project depend heavily on these antibodies to create reference maps of histone PTM landscapes [78].
The N-terminal tails of histones represent one of the most extensively modified protein regions known, with numerous modifications occurring in close proximity [79]. This density of modifications creates a challenging landscape for antibody specificity. For instance, within just positions 2-11 of the H3 N-terminal tail, seven residues are known to be modified [79]. This complexity is compounded by the fact that different methylation states (mono-, di-, and tri-methylation) at the same lysine residue often have distinct biological functions [78].
Recent systematic assessments of commercial histone PTM antibodies have revealed alarming specificty concerns. One analysis of over 100 frequently used histone antibodies found that many exhibit unfavorable behaviors, including:
- Inability to distinguish between methylation states on target residues
- Significant cross-reactivity with off-target modifications
- Sensitivity to neighboring PTMs that either enhance or inhibit binding [78]
These findings underscore that poor antibody choice can lead to misinformed conclusions about histone modification localization and function, potentially compromising scientific findings across numerous studies [78].
Methodologies for Assessing Antibody Specificity
Peptide Microarray Analysis
Peptide microarray technology represents one of the most comprehensive approaches for characterizing antibody specificity. This method utilizes arrays containing hundreds of purified biotinylated histone peptides featuring PTMs alone and in biologically relevant combinations [78]. The high-density arrays allow for systematic testing of antibody binding against a vast repertoire of potential epitopes in a single experiment.
Experimental Protocol:
- Array Design: Arrays typically contain 384 peptides covering key regions of histone tails (e.g., H3 1-19, 7-26, 16-35, 26-45; H4 1-19, 11-30) featuring up to 59 different post-translational modifications [57] [79].
- Antibody Incubation: Antibodies are applied to arrays at multiple concentrations to assess reactivity without saturating the assay [80].
- Detection: Arrays are incubated with fluorescently-labeled secondary antibodies and scanned using imaging systems such as LI-COR Odyssey Infrared Imagers [80].
- Data Analysis: Signal intensities are quantified and normalized. Results are typically presented as interactive heat maps or bar graphs showing binding intensities relative to the intended target peptide [78].
The peptide microarray approach can identify several classes of problematic antibody behavior:
- Off-target recognition: Binding to unrelated modification sites
- Methylation state cross-reactivity: Inability to distinguish between mono-, di-, and tri-methylation states
- Neighboring PTM sensitivity: Altered binding when additional modifications are present near the primary epitope [78]
For example, microarray analysis has revealed that among 38 di- and tri-methyllysine antibodies screened, 16 cross-reacted with lower methylation states on the target residue, and one recognized a higher methylation state [78].
Functional Validation in ChIP Assays
While peptide arrays assess biochemical specificity, functional validation determines whether antibodies perform effectively in the context of native chromatin. ChIP validation confirms that target epitopes are accessible within nucleosomes and that antibodies enrich expected genomic regions [57].
Experimental Protocol:
- Chromatin Preparation: Cells are cross-linked with formaldehyde, chromatin is fragmented by sonication or micrococcal nuclease (MNase) digestion to ~200-300 bp fragments, and immunoprecipitation is performed [77] [76].
- Antibody Titration: Antibodies are titrated to determine optimal concentrations, as both insufficient and excessive antibody can compromise results [76] [81].
- qPCR Analysis: Precipitated DNA is analyzed by qPCR using primers for positive control regions (e.g., active gene promoters) and negative control regions (e.g., silent satellite repeats) [57] [82].
- Specificity Criteria: Antibodies should demonstrate ≥5-fold enrichment at positive control regions compared to negative controls [77].
Table 1: Performance Metrics of Select Validated Histone PTM Antibodies
| Antibody Target | Specificity Factor* | Fold Enrichment in ChIP-qPCR | Cross-reactivity Concerns |
|---|---|---|---|
| H3K4me2 | >2-fold difference between target and best non-target site [57] | Significant enrichment at active gene promoters [57] | Minimal off-target binding in peptide arrays [57] |
| H3K4me3 | Variable between antibodies; some show cross-reactivity with H3K4me2 [78] | Dependent on specificity | Some antibodies cross-react with H3K4me2; others are selective [78] |
| H3K9me3 | Sensitivity to H3S10ph neighboring modification [78] | Dependent on specificity | Differential tolerance to neighboring H3S10 phosphorylation [78] |
| H3K27me3 | Variable between antibodies; some show H4K20me3 cross-reactivity [79] | Dependent on specificity | Antibodies show different patterns of neighboring PTM sensitivity [79] |
*Specificity factor = Ratio of average intensity of spots containing target PTM to those lacking it [57]
Advanced Approaches: siQ-ChIP for Quantitative Assessment
Recent advancements have introduced sans spike-in quantitative ChIP-seq (siQ-ChIP), which provides an absolute quantitative scale for ChIP-seq data without spike-in normalization [76]. This method leverages the principle that the immunoprecipitation step produces a classical binding isotherm when antibody or epitope concentration is titrated.
Experimental Protocol:
- Chromatin Standardization: Chromatin is fragmented using MNase to generate mononucleosome-sized fragments, and concentration is standardized [76].
- Antibody Titration: Multiple ChIP reactions are performed with varying antibody concentrations.
- DNA Quantification: Mass of immunoprecipitated DNA is measured across antibody concentrations.
- Sequencing Analysis: DNA from points along the binding isotherm is sequenced to evaluate differential peak responses [76].
This approach can distinguish antibodies with "narrow" versus "broad" binding spectra—referring to the range of binding constants an antibody exhibits toward different epitopes [76]. Sequencing points along the binding isotherm reveals differential peak responses that help identify strong (high-affinity, on-target) versus weak (low-affinity, off-target) antibody-epitope interactions [76].
Consequences of Antibody Cross-Reactivity and Neighboring PTM Sensitivity
The implications of antibody non-specificity extend to erroneous biological interpretations. For example, the three methylation states of H3K4 have differentially reported functions: H3K4me3 associates with promoter regions, H3K4me2 with both promoters and enhancers, and H3K4me1 primarily with enhancers [78]. When antibodies cross-react between these states, ChIP-seq mapping becomes inaccurate. Studies have shown that antibodies cross-reacting with lower methylation states produce overlapping signals at transcription start sites and gene bodies, potentially obscuring the distinct genomic distributions of these marks [78].
Neighboring modifications can also profoundly impact antibody binding. A striking example involves the H3K9me3 and H3S10ph "methyl/phospho switch" known to eject proteins like HP1 from mitotic chromatin [78]. Antibodies against H3K9me3 show varying sensitivity to phosphorylated H3S10—some are inhibited by this neighboring modification, while others bind equally well to singly and dually modified tails [78]. Similar effects are observed with H3S10ph antibodies, whose binding is sometimes impacted by neighboring H3K9me3 [78]. These differences mean that sensitive antibodies may under-represent certain histone populations in specific biological contexts, such as mitosis.
Best Practices for Antibody Selection and Validation
Criteria for Antibody Selection
- Application-Specific Validation: Select antibodies specifically validated for your intended application (ChIP-qPCR vs. ChIP-seq). Antibodies that work well for ChIP-qPCR may not perform adequately for ChIP-seq, which requires more extensive genome-wide capture [77] [83].
- Comprehensive Specificity Data: Prioritize antibodies with peptide microarray specificity profiles available. Public resources like The Histone Antibody Specificity Database (www.histoneantibodies.com) provide comparative data on antibody performance [78].
- Adequate Enrichment Metrics: For ChIP-seq, antibodies should demonstrate ≥5-fold enrichment at positive control regions compared to negative controls in ChIP-qPCR validation [77].
- Clonality Considerations: Both monoclonal and polyclonal antibodies can work in ChIP. Monoclonal antibodies generally offer higher specificity but may have epitopes buried in chromatin contexts. Polyclonal antibodies recognize multiple epitopes, potentially increasing signal, but may introduce cross-reactivity concerns [77] [82].
Implementation of Controls
- Biological Replicates: Perform at least duplicate biological experiments to ensure reliability [77].
- Appropriate Controls: Use chromatin inputs as controls for bias in fragmentation and sequencing efficiency. Input controls provide more evenly distributed genomic coverage than non-specific IgG [77].
- Specificity Controls: Where possible, employ knockout or knockdown models to confirm specificity. Any signal detected in these backgrounds likely represents non-specific binding [77].
- Multiple Antibody Validation: Compare results using different antibodies targeting the same epitope or complex to increase confidence in findings [77] [83].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Validated Chromatin Immunoprecipitation
| Research Tool | Function | Application Notes |
|---|---|---|
| Peptide Microarrays | Comprehensive antibody specificity profiling | Tests binding against hundreds of modified histone peptides simultaneously; identifies cross-reactivity [78] |
| MODified Histone Peptide Arrays | Antibody specificity determination | Commercial arrays featuring 384 histone peptides with 59 PTMs [57] |
| MAGnify Chromatin Immunoprecipitation System | Streamlined ChIP workflow | Integrated system for chromatin prep, IP, and DNA purification [57] |
| MNase | Chromatin fragmentation | Generates mononucleosome-sized fragments; superior for quantitative applications [76] |
| SimpleChIP Kits | Optimized ChIP protocols | Include enzymatic and sonication-based chromatin fragmentation methods [83] |
| Histone Antibody Specificity Database | Online antibody specificity resource | Interactive database for comparing commercial histone antibody performance [78] |
Antibody validation remains the cornerstone of reliable ChIP-seq data generation and interpretation in histone modification research. The complex landscape of histone PTMs, with numerous modifications occurring in close proximity and often exhibiting interdependent recognition, demands rigorous specificity assessment. Integrating multiple validation strategies—including peptide microarray analysis, functional ChIP validation, and innovative quantitative approaches like siQ-ChIP—provides the comprehensive characterization necessary to ensure antibody reliability. As the field moves toward increasingly precise mapping of epigenetic landscapes, adherence to robust validation standards will be essential for producing accurate, reproducible findings that advance our understanding of gene regulatory mechanisms in health and disease.
Antibody Validation Decision Pathway
The accurate identification of histone modification enrichment regions through peak calling is a critical step in ChIP-seq data analysis, directly influencing the interpretation of epigenetic regulation. Histone modifications exhibit distinct genomic distribution patterns, ranging from sharp, punctate signals to broad domains, which necessitates the use of specialized computational algorithms. This technical guide provides a comprehensive evaluation of peak calling tools, performance metrics across different histone marks, and integrated workflows to enable researchers and drug development professionals to select optimal algorithms based on their specific experimental designs and biological questions. By synthesizing current benchmarking studies and implementation frameworks, we establish a standardized approach for peak calling that enhances the reliability of epigenetic insights in gene regulation research.
Histone post-translational modifications represent a fundamental epigenetic mechanism for regulating gene expression without altering DNA sequence. These modifications include acetylation, methylation, phosphorylation, and ubiquitination, which collectively influence chromatin structure and recruitment of transcriptional machinery. Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has emerged as the primary method for genome-wide mapping of these histone modifications, with peak calling serving as the computational cornerstone for identifying statistically significant enrichment regions from sequencing data.
The challenge in peak calling stems from the diverse spatial distributions of different histone marks. Sharp marks, such as H3K4me3 and H3K27ac, typically localize to specific genomic features like promoters and enhancers, producing well-defined, punctate peaks. In contrast, broad marks, including H3K27me3 and H3K36me3, span extensive genomic domains associated with repressed chromatin or actively transcribed gene bodies, generating wide enrichment signals that complicate precise boundary detection [84] [85]. This biological diversity necessitates algorithmic specialization, as no single peak caller optimally identifies all histone modification types.
Histone Mark Classification and Genomic Profiles
The ENCODE Consortium has established guidelines for categorizing histone modifications based on their characteristic genomic distributions, which directly inform algorithm selection [17]. Understanding these categories is essential for matching peak calling algorithms to biological signals.
Table 1: Histone Mark Classification by Genomic Distribution
| Category | Histone Marks | Genomic Features | Peak Characteristics |
|---|---|---|---|
| Narrow Marks | H3K4me3, H3K9ac, H3K27ac, H3K4me2 | Promoters, enhancers | Sharp, punctate peaks (<5 kb) |
| Broad Marks | H3K27me3, H3K36me3, H3K79me2, H3K9me1, H3K9me2 | Repressed domains, gene bodies | Wide regions (5-100+ kb) |
| Mixed Profile Marks | H3K4me1, H3K79me1 | Enhancers, regulatory elements | Combination of sharp and broad features |
H3K9me3 represents a special case among histone modifications, as it is enriched in repetitive genomic regions and requires specific analytical considerations due to mapping challenges in these areas [17]. The ENCODE Consortium has established specific sequencing depth requirements for different mark types: narrow marks require 20 million usable fragments per replicate, while broad marks generally require 45 million usable fragments per replicate to ensure sufficient coverage for reliable peak detection [17].
Peak Calling Algorithms: Performance and Applications
Multiple peak calling algorithms have been developed with different statistical approaches and optimization targets. Benchmarking studies across various histone modifications provide critical insights into their relative performance.
Table 2: Peak Calling Algorithm Performance Across Histone Marks
| Algorithm | Sharp Marks (H3K4me3, H3K27ac) | Broad Marks (H3K27me3, H3K36me3) | Statistical Foundation | Key Applications |
|---|---|---|---|---|
| MACS2 | High sensitivity and precision [86] [87] | Moderate performance, requires broad mode [84] | Dynamic Poisson distribution | General-purpose, widely supported |
| GoPeaks | High sensitivity for H3K27ac [87] | Optimized for CUT&Tag data [87] | Binomial distribution with minimum count threshold | Histone modification CUT&Tag |
| HOMER | Robust performance for transcription factors [88] | Effective for broad domains [88] | Histogram-based peak modeling | Integrated annotation and motif discovery |
| PeakRanger | Superior precision-recall balance [86] | Not specifically evaluated | Not specified in sources | Intracellular G4 sequencing |
| SICER | Lower performance on sharp marks [86] | Designed for broad marks [86] | Spatial clustering approach | Broad histone marks |
| SEACR | Stringent and relaxed thresholds available [87] | Limited for very broad domains [87] | Empirical thresholding | CUT&RUN, low-background data |
Performance evaluations consistently demonstrate that algorithm effectiveness varies significantly by histone mark type. For sharp marks like H3K4me3, MACS2 and GoPeaks identify the greatest number of peaks with appropriate size distributions, while SEACR may miss narrower peaks (<100 bp) due to its binning approach [87]. For broad marks, specialized tools like SICER show advantages, though MACS2 in broad mode remains a popular choice. Comparative analyses of five peak callers (CisGenome, MACS1, MACS2, PeakSeq, and SISSRs) across 12 histone modifications in human embryonic stem cells revealed that peak characteristics were more strongly influenced by histone mark type than by the specific calling algorithm used [84].
Integrated Analysis Workflows and Platforms
Automated pipelines integrate multiple peak calling steps with quality control and annotation, providing standardized approaches for histone mark analysis.
The H3NGST (Hybrid, High-throughput, and High-resolution NGS Toolkit) platform represents a fully automated, web-based approach that streamlines the entire ChIP-seq analysis workflow [88]. This system requires only a BioProject accession number and automatically performs data retrieval, quality control, adapter trimming, genome alignment, and peak calling with HOMER, providing comprehensive annotation and visualization outputs. Such integrated platforms significantly reduce technical barriers for researchers without extensive bioinformatics expertise [88].
The ENCODE Consortium's histone analysis pipeline employs a standardized approach that differs between replicated and unreplicated experiments. For replicated experiments, the pipeline generates both individual replicate peaks and pooled replicate peaks, with final replicated peaks requiring observation in both biological replicates or in two pseudoreplicates [17]. This stringent approach enhances reliability while accommodating practical experimental constraints.
Experimental Design and Methodological Considerations
Standardized Experimental Protocols
Robust peak calling requires appropriate experimental design and data preprocessing. For comparative analyses of ChIP-seq data across biological conditions, performance evaluations of 33 differential analysis tools revealed that optimal algorithm selection depends heavily on both peak shape and biological regulation scenario [85]. Tools including bdgdiff (from MACS2), MEDIPS, and PePr demonstrated the highest median performance across diverse scenarios, though specific tools excelled in particular contexts [85].
A standardized protocol for histone mark ChIP-seq analysis includes:
Sequence Retrieval and Quality Control: Download sequencing data from SRA using prefetch and fasterq-dump, automatically detecting library layout (single-end or paired-end) [88]. Perform quality assessment with FastQC to detect adapter contamination and low-quality reads [88].
Read Preprocessing: Remove adapter sequences and trim low-quality bases using Trimmomatic with a sliding window approach [88]. Run FastQC again post-trimming to verify quality improvement.
Genome Alignment: Map cleaned reads to an appropriate reference genome (e.g., hg38, mm10) using BWA-MEM, generating SAM files [88]. Convert to sorted BAM files using Samtools, then to BED format using Bedtools for downstream analysis.
Peak Calling Implementation: Execute algorithm-specific commands with parameters matched to histone mark type. For H3K27me3 broad domains, use MACS2 with
--broadoption or SICER with recommended window sizes [84] [17]. For sharp marks like H3K4me3, standard narrow peak calling typically suffices.Result Validation and Annotation: Assess peak quality through metrics like FRiP (Fraction of Reads in Peaks) scores and Irreproducibility Discovery Rate (IDR) analysis for replicates [84] [17]. Annotate peaks genomically using HOMER or similar tools to associate with genes, promoters, and other regulatory elements.
Table 3: Key Research Reagents and Computational Tools for Histone ChIP-seq
| Resource | Type | Function | Implementation Notes |
|---|---|---|---|
| H3NGST Platform | Web-based platform | Fully automated ChIP-seq analysis | No installation required; uses BioProject ID for input [88] |
| MACS2 | Peak calling algorithm | Identifies enriched regions in ChIP-seq data | Default for sharp marks; broad mode for extended domains [86] |
| GoPeaks | Peak calling algorithm | Optimized for histone modification CUT&Tag data | Uses binomial distribution with minimum count threshold [87] |
| BWA-MEM | Alignment tool | Maps sequencing reads to reference genome | Supports paired-end reads; flexible read lengths [88] |
| HOMER | Peak calling and annotation suite | Comprehensive motif discovery and annotation | Provides genomic context for identified peaks [88] |
| ENCODE Blacklist | Quality control resource | Filters artifactual regions | Removes false-positive peaks in problematic genomic regions [17] |
| Trimmomatic | Preprocessing tool | Removes adapter sequences and low-quality bases | Uses sliding window approach for quality trimming [88] |
| UCSC Genome Browser | Visualization tool | Displays genomic signals and annotations | Compatible with BigWig files from pipeline outputs [88] |
Advanced Applications and Future Directions
Emerging methodologies continue to enhance peak calling precision and efficiency. GPU-accelerated implementations of established algorithms like MACS2 demonstrate ~15x speed improvements, significantly reducing computational bottlenecks for large-scale or single-cell epigenomic studies [89]. These advancements enable more rapid analytical iteration, particularly valuable in drug development pipelines where timing is critical.
For specialized applications including intracellular G-quadruplex (G4) mapping, recent evaluations have identified MACS2, PeakRanger, and GoPeaks as particularly effective, highlighting how peak caller performance varies significantly across application domains [86]. The development of purpose-specific tools like WonderPeaks for fungal pathogens further illustrates the ongoing specialization in algorithm development to address unique biological contexts and data characteristics [90].
Single-cell ChIP-seq methodologies represent another frontier, enabling the resolution of cellular heterogeneity within complex tissues and cancers [54]. These approaches introduce additional computational challenges due to data sparsity and technical noise, driving continued innovation in peak calling algorithms designed for sparse data contexts.
Choosing the appropriate peak calling algorithm requires consideration of multiple experimental and biological factors. For sharp histone marks like H3K4me3 and H3K27ac, MACS2 and GoPeaks provide excellent sensitivity and precision. For broad marks such as H3K27me3 and H3K36me3, SICER or MACS2 in broad mode are preferable choices. When analyzing data from emerging technologies like CUT&Tag, purpose-built tools like GoPeaks offer optimized performance by accounting for characteristically low background signals.
Researchers should validate algorithm selections using quality metrics including FRiP scores, IDR analysis for replicates, and concordance with established genomic annotations. As epigenomic studies continue to illuminate the fundamental mechanisms of gene regulation in development, disease, and drug response, robust and appropriately selected peak calling methodologies will remain essential for extracting biologically meaningful insights from ChIP-seq data. The integration of standardized workflows, automated platforms, and continuously benchmarked algorithms provides a powerful framework for advancing our understanding of histone-mediated epigenetic regulation.
In ChIP-seq research on histone modifications, technical noise and false positives can compromise data integrity, leading to inaccurate biological interpretations. This guide details practical, evidence-based strategies to enhance data quality, drawing from recent methodological advances and systematic benchmarking studies.
Methodological Choice: Weighing ChIP-seq Against Modern Alternatives
The foundational decision in any epigenomic study is the choice of profiling method. While Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has long been the gold standard, emerging enzyme-tethering methods offer distinct advantages for specific applications, particularly concerning signal-to-noise ratio and input requirements.
The table below compares the core characteristics of ChIP-seq with two modern alternatives, CUT&RUN and CUT&Tag.
Table 1: Comparison of Histone Modification Profiling Methods
| Feature | ChIP-seq | CUT&RUN | CUT&Tag |
|---|---|---|---|
| Core Principle | Chromatin immunoprecipitation and purification of bound DNA fragments [91] | Antibody-targeted chromatin cleavage in situ [91] | Antibody-targeted chromatin tagmentation in situ [91] |
| Typical Cell Input | 1-10 million cells [92] | Low-input (as few as 100 cells) [93] | Ultra-low-input (as few as 60 cells); amenable to single-cell applications [92] [93] |
| Signal-to-Noise Ratio | Moderate; prone to background from non-specific precipitation [91] | High [91] | High; superior signal specificity [91] |
| Key Limitations | High background noise, epitope masking from cross-linking, heterochromatin bias from sonication [92] | - | Bias of Tn5 transposase toward open chromatin regions, potentially increasing false positives [93] [92] |
| Best Suited For | Broad, established workflows with ample starting material; gold-standard reference datasets [17] | Projects with limited cell numbers requiring high data fidelity [91] | High-sensitivity studies with very scarce material, single-cell analyses, and profiling without cross-linking [93] [91] |
Systematic benchmarking in haploid round spermatids confirms that while all three methods reliably detect histone modifications, CUT&Tag stands out for its comparatively higher signal-to-noise ratio [91]. However, this same study and others note that CUT&Tag shows a bias toward accessible chromatin, as the Tn5 transposase has an inherent preference for cutting in open regions [93] [91]. This can lead to an overrepresentation of signals in euchromatin and potentially false-positive peaks that reflect accessibility rather than specific histone modification [93].
Optimizing Wet-Lab Protocols to Minimize Noise
ChIP-seq Best Practices for High-Quality Data
For researchers using ChIP-seq, adherence to established standards is crucial. The ENCODE consortium provides rigorous guidelines [17]:
- Biological Replicates: Experiments should include a minimum of two biological replicates to ensure findings are reproducible.
- Input Controls: A matched input control (from sheared chromatin prior to immunoprecipitation) is mandatory for distinguishing specific enrichment from background noise.
- Library Quality Control: Key metrics include:
- Non-Redundant Fraction (NRF) > 0.9
- PBC1 > 0.9
- PBC2 > 10 These metrics ensure sufficient library complexity and avoid PCR bottlenecks that skew representation [17].
- Sequencing Depth: Requirements vary by histone mark. For narrow marks (e.g., H3K4me3, H3K27ac), aim for 20 million usable fragments per replicate. For broad marks (e.g., H3K27me3, H3K36me3), 45 million fragments per replicate are recommended [17].
Advanced and Quantitative ChIP-seq Techniques
Recent innovations in ChIP-seq protocol design can further mitigate technical variation.
- MINUTE-ChIP: This multiplexed ChIP-seq protocol allows multiple samples to be barcoded and profiled against multiple epitopes in a single immunoprecipitation reaction. This dramatically reduces experimental variation and enables accurate quantitative comparisons across conditions [94].
- Spike-In Chromatin: For absolute quantitative comparisons, methods like PerCell integrate a well-defined cellular spike-in ratio of orthologous species' chromatin (e.g., Drosophila chromatin into human samples). This provides an internal standard for normalization, correcting for variations in cell number, extraction efficiency, and other technical confounders [95].
CUT&Tag Optimization and Validation
For CUT&Tag users, specific optimizations can enhance data quality.
- Antibody Validation: Systematically test different ChIP-grade antibody sources and dilutions via qPCR on positive and negative control genomic regions before proceeding to sequencing [92].
- PCR Cycle Optimization: High duplication rates in sequencing libraries can result from excessive PCR amplification. Titrating down the number of PCR cycles from the standard protocol can help maintain library complexity [92].
- HDAC Inhibitor Consideration: For labile marks like H3K27ac, adding histone deacetylase inhibitors (HDACi) like Trichostatin A (TSA) during the procedure was tested to stabilize the mark. However, evidence indicates TSA does not consistently improve peak detection, signal-to-noise ratio, or recovery of known ENCODE peaks [92].
Diagram 1: Experimental workflow and optimization
Computational and Analytical Strategies for Peak Calling and Validation
The choice of peak-calling algorithm and parameters directly impacts false positive rates.
- Benchmarked Peak Callers: For CUT&Tag data, both MACS2 and SEACR have been systematically evaluated. Research indicates that using SEACR with stringent settings can be particularly effective for identifying high-confidence peaks [92].
- Parameter Tuning: When using MACS2 for CUT&Tag data, employ parameters like
--nolambdaand--nomodelfor optimal performance [92]. - Peak Verification: To quantify potential false positives arising from Tn5 bias, calculate the False Positive Rate (FPR). This metric identifies peaks that do not overlap with a reliable ChIP-seq reference but do overlap with peaks from an ATAC-seq (assay for transposase-accessible chromatin) dataset [93]. Protocols like WOW-CUT&Tag have demonstrated a lower FPR compared to earlier methods, suggesting technical improvements can mitigate this bias [93].
Table 2: Benchmarking CUT&Tag Performance Against ENCODE ChIP-seq Standards
| Performance Metric | Finding | Interpretation and Strategy |
|---|---|---|
| Recall of ENCODE Peaks | Recovers ~54% of known ENCODE H3K27ac and H3K27me3 peaks [92]. | CUT&Tag reliably captures the strongest, most biologically significant peaks; missing peaks may be lower-affinity sites. |
| Precision | Identified peaks show the same functional and biological enrichments as ENCODE ChIP-seq peaks [92]. | Peaks called from CUT&Tag data are functionally authentic and not technical artifacts. |
| Inherent Bias | Tn5 transposase has a bias toward cutting open chromatin regions [93]. | Can lead to false positives; validate surprising findings with an orthogonal method. |
| Mitigation Strategy | WOW-CUT&Tag generates profiles closer to ChIP-seq with lower false positive rates [93]. | Protocol refinements can reduce bias; use updated methods when possible. |
Table 3: Key Research Reagent Solutions for Histone Modification Profiling
| Reagent / Resource | Function and Importance | Considerations for Selection |
|---|---|---|
| Validated Antibodies | Binds specifically to the histone modification epitope of interest; primary driver of specificity. | Use ChIP-seq-grade antibodies characterized by ENCODE or other consortia [92] [17]. Test multiple sources and dilutions [92]. |
| pA-Tn5 Transposase | (For CUT&Tag) Enzyme-antibody fusion protein that cleaves and adapts target chromatin in situ. | Commercial kits are available (e.g., Vazyme Biotech). Ensure high activity and purity for efficient tagmentation [91]. |
| Spike-In Chromatin | (For Quantitative ChIP) Exogenous chromatin (e.g., Drosophila) added in known amounts for normalization. | Enables quantitative cross-sample comparison by controlling for technical variation in cell number and IP efficiency [95]. |
| ConA Magnetic Beads | (For CUT&Tag/RUN) Binds to cell membranes, immobilizing samples for efficient solution exchange. | Pre-activated beads ensure high binding affinity, reducing cell loss during multiple washing steps [93] [91]. |
| HDAC Inhibitors (e.g., TSA) | Suppresses deacetylase activity to potentially stabilize acetylated marks like H3K27ac during processing. | Evidence for efficacy in CUT&Tag is mixed; may not consistently improve data quality [92]. |
| ENCODE Pipelines & Standards | Provides validated, community-accepted data processing workflows and quality control metrics. | Essential for benchmarking lab data against gold standards; ensures reproducibility and rigor [17]. |
Diagram 2: Noise sources and mitigation strategies
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) has emerged as a powerful method for interrogating protein-chromatin interactions, mapping histone modifications across the genome, and decoding the regulatory landscapes of gene expression. The ability to profile histone modifications is particularly crucial for understanding epigenetic mechanisms in development, disease, and cellular identity. However, performing ChIP-seq on challenging biological samples—including solid tissues and primary cells—presents substantial technical hurdles that can compromise data quality and reproducibility.
Traditional ChIP-seq protocols face limitations when applied to complex tissues due to their heterogeneous cellular composition, dense extracellular matrices, and challenges in chromatin fragmentation while preserving protein-DNA interactions. This article presents a refined ChIP-seq approach specifically optimized for challenging cell types, with particular emphasis on colorectal cancer tissues, providing researchers with a robust framework for obtaining high-quality epigenomic data from physiologically relevant systems.
Optimized ChIP-Seq Protocol for Solid Tissues
Tissue Preparation and Homogenization
Proper tissue preparation is critical for preserving chromatin integrity and ensuring representative cell sampling from complex solid tissues. The following protocol has been optimized for frozen tissue samples:
Sample Retrieval and Mincing: Retrieve frozen tissue cryotubes from -80°C and immediately place on ice. Working in a biosafety cabinet, transfer the tissue to a Petri dish placed firmly on ice and mince finely using two sterile scalpel blades until the tissue is finely diced. Document tissue size and appearance for quality tracking [73].
Homogenization Methods: Two effective homogenization approaches have been optimized:
Dounce Homogenization: Transfer minced tissue to a 7-ml Dounce grinder on ice. Add 1 ml cold PBS with protease inhibitors and perform 8-10 even strokes with the A pestle. Expect some debris and clumps as connective tissue may resist complete disruption. Rinse with 2-3 ml cold PBS and transfer to a 50-ml conical tube [73].
gentleMACS Dissociator: Transfer minced tissue to a C-tube on ice with 1 ml cold PBS with protease inhibitors. Tap the upside-down C-tube on the bench to ensure material contacts the blade and run the preconfigured "htumor03.01" program. Adjust the program based on tissue density and thickness as needed [73].
Table 1: Homogenization Method Comparison
| Method | Equipment | Processing Capacity | Considerations |
|---|---|---|---|
| Dounce Homogenization | Glass Dounce grinder, pestle A | Small to medium tissues | Manual process; some debris expected; cost-effective |
| gentleMACS Dissociator | gentleMACS Dissociator, C-tubes | High-throughput processing | Predefined programs; more consistent; requires specialized equipment |
Chromatin Immunoprecipitation from Tissues
The chromatin immunoprecipitation process requires careful optimization for tissue samples to maximize yield and specificity:
Cross-linking and Chromatin Extraction: Following homogenization, cross-link tissue samples with formaldehyde to preserve protein-DNA interactions. Process for chromatin extraction with emphasis on proper tissue handling to maintain chromatin integrity and minimize degradation [73].
Chromatin Shearing and Immunoprecipitation: Perform optimized lysis, chromatin shearing, and immunoprecipitation with attention to buffer composition, shearing parameters, and washing steps. These refinements minimize background noise and enhance ChIPed DNA quality, allowing reproducible chromatin profiling in tissues [73].
Protocol Adaptations for Primary Cells: For fragile primary cells such as activated B lymphocytes, consider incorporating gentle fixation prior to nuclear isolation. This stabilization preserves chromatin-protein interactions while maintaining epitope accessibility [96].
Library Construction and Sequencing
The final stages focus on preparing high-quality sequencing libraries:
End-Repair and A-Tailing: Process immunoprecipitated DNA through end-repair and A-tailing reactions to ensure compatibility with sequencing adaptors [73].
Adaptor Ligation and Amplification: Ligate MGI-specific adaptors followed by PCR amplification with multiple quality checkpoints to verify library integrity and concentration [73].
DNA Nanoball Preparation and Sequencing: Prepare DNA nanoballs (DNBs) for the DNBSEQ-G99RS sequencing platform. This approach offers a cost-effective sequencing alternative particularly suitable for large cohort studies [73].
Data Analysis and Normalization Framework
MAnorm for Quantitative ChIP-Seq Comparison
Comparing ChIP-seq datasets across conditions requires specialized normalization approaches. MAnorm provides a robust framework for quantitative comparison by addressing the challenge of differential signal-to-noise ratios between samples:
Common Peaks as Reference: MAnorm utilizes overlapping ChIP-enriched regions (common peaks) between two samples as an internal reference to build a rescaling model. This approach assumes that binding at common regions is determined by similar mechanisms and should exhibit comparable global binding intensities [97].
MA Plot and Normalization: The method plots the log2 ratio of read density (M) against the average log2 read density (A) for all peaks, then applies robust linear regression to fit the global dependence between M-A values of common peaks. The derived linear model serves as a reference for normalization across all peaks [97].
Biological Validation: Quantitative differences identified by MAnorm show strong correlation with changes in target gene expression and binding of cell type-specific regulators, validating its utility for identifying biologically relevant epigenetic differences [97].
Quality Control and Data Interpretation
Implement multi-stage quality checkpoints throughout the experimental process. For sequencing output, carefully assess:
- Peak distribution and enrichment relative to input controls
- Correlation between biological replicates
- Enrichment at expected genomic regions (promoters, enhancers)
- Motif analysis for transcription factor binding sites
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents and Their Applications
| Reagent/Equipment | Function | Application Notes |
|---|---|---|
| Protease Inhibitor Cocktail | Preserves protein integrity during processing | Supplement PBS during tissue homogenization [73] |
| Dounce Tissue Grinder | Mechanical tissue disruption | Use pestle A for 8-10 strokes; keep cold throughout [73] |
| gentleMACS Dissociator | Automated tissue homogenization | Use C-tubes with "htumor03.01" program [73] |
| Formaldehyde | Crosslinking agent | Preserves protein-DNA interactions; optimize concentration and timing [73] |
| Magnetic Protein A/G Beads | Antibody binding and pulldown | Enable target-specific immunoprecipitation [96] |
| MGI-Specific Adaptors | Library preparation | Compatible with DNBSEQ-G99RS platform [73] |
| Micrococcal Nuclease (MNase) | Chromatin digestion | Used in CUT&RUN as alternative to sonication [96] |
Advanced Applications and Emerging Techniques
Single-Cell Epigenomic Methods
While bulk ChIP-seq provides population-average data, emerging single-cell methods enable resolution of cellular heterogeneity within complex tissues. Single-cell CUT&Tag techniques are particularly promising for mapping histone modifications across diverse cell populations, revealing cell type-specific regulatory programs [54].
CUT&RUN as a ChIP-Seq Alternative
For limited cell numbers, CUT&RUN offers substantial advantages over traditional ChIP-seq:
Higher Signal-to-Noise Ratio: Targeted chromatin cleavage by antibody-bound nuclease reduces background signal [96].
Reduced Cell Input Requirements: Robust data can be obtained from as few as 100,000 nuclei, making it suitable for rare cell populations [96].
Preservation of Protein Complexes: Avoids potential disruption of large protein complexes that can occur with sonication [96].
A systematic comparison revealed that CUT&Tag recovers approximately half of ENCODE ChIP-seq histone acetylation peaks but captures the most significant and strongest signals with similar functional enrichment patterns [9].
The optimized ChIP-seq protocol presented here addresses critical challenges in epigenomic profiling of complex tissues and primary cells. Through refined tissue preparation, chromatin extraction, and immunoprecipitation steps, researchers can obtain high-quality data that preserves the biological relevance of native tissue contexts. Integration of appropriate normalization strategies like MAnorm enables robust quantitative comparison between conditions, while emerging techniques like CUT&RUN offer complementary approaches for limited cell numbers.
As the field advances, these optimized workflows will continue to illuminate the role of histone modifications in gene regulation, providing insights into epigenetic mechanisms underlying development, cellular identity, and disease pathogenesis. By implementing these refined methodologies, researchers can overcome traditional limitations and generate more physiologically relevant epigenomic data from challenging but biologically crucial sample types.
ChIP-Seq vs. CUT&Tag: A Systematic Benchmark for Epigenomic Profiling
Within the broader thesis on the role of histone modifications in gene regulation, the selection of an appropriate chromatin profiling technology is paramount. Histone post-translational modifications (PTMs)—such as methylation (H3K4me3, H3K27me3), acetylation (H3K27ac), and phosphorylation (γ-H2AX)—orchestrate fundamental gene regulatory programs by modulating chromatin architecture and accessibility [98] [99]. Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has long been the cornerstone method for mapping the genomic distribution of these modifications. However, its limitations in resolution, input requirements, and scalability have driven the development of advanced techniques, particularly for single-cell analysis. This technical guide provides a head-to-head comparison of contemporary methods, focusing on the critical parameters of sensitivity, cost, and single-cell compatibility, to inform researchers, scientists, and drug development professionals in their experimental design.
Technical Comparison of Core Methodologies
The following sections and tables provide a detailed comparison of established and emerging technologies for histone modification profiling.
Table 1: Key Characteristics of Histone Modification Profiling Techniques
| Technique | Best Application Context | Sensitivity (Typical Input) | Single-Cell Compatibility | Relative Cost per Sample | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|---|
| ChIP-seq | Genome-wide histone mark mapping from high-quality, abundant samples [98] | Moderate to Low (100s of thousands to millions of cells) [98] | Limited (specialized protocols exist) [100] | $$ | Established gold standard; extensive protocols and bioinformatics tools [98] | High input requirement; crosslinking artifacts; high background noise [98] [101] |
| CUT&Tag | High-resolution profiling of low-input and native chromatin samples [98] [101] | High (as low as ~10 cells) [98] | Excellent (native protocol) [101] | $ | Low background; high signal-to-noise; works on native chromatin [98] [101] | Protocol complexity; requires optimization of antibody and enzyme concentration [98] |
| scEpi2-seq | Simultaneous, multi-omic measurement of histone modifications and DNA methylation in single cells [100] | High (Single-Cell) [100] | Excellent (native protocol) | $$$$ | Unique multi-omics readout from the same cell; reveals epigenetic interactions [100] | Highly complex workflow; high cost; specialized data analysis required [100] |
| IT-scC&T-seq | Scalable, high-throughput single-cell profiling without specialized equipment [101] | High (Single-Cell; >2,800 unique fragments/cell for CTCF) [101] | Excellent (native protocol) | $ (as low as $0.01/cell) [101] | Cost-effective; modular; uses standard lab equipment; high multiplexing capacity [101] | Involves complex multi-step indexing and sorting [101] |
| MobiChIP | Single-cell ChIP-seq with flexible sequencing on standard platforms [102] | Information missing | Yes (native protocol) [102] | Information missing | Compatible with standard sequencing platforms; allows sample mixing [102] | Information missing |
Table 2: Performance Benchmarking of Single-Cell Techniques (Based on K562 Cell Line Data)
| Histone Mark / Factor | Technique | Median Unique Fragments per Cell | Fraction of Reads in Peaks (FRiP) | Notes / Comparative Performance |
|---|---|---|---|---|
| H3K4me3 | IT-scC&T-seq [101] | 12,806 | 0.854 (85.4%) | Yields 3–6 times more unique fragments than transcription factors [101] |
| H3K27me3 | IT-scC&T-seq [101] | 9,304 | 0.803 (80.3%) | Information missing |
| RNA Polymerase II | IT-scC&T-seq [101] | 3,344 | 0.564 (56.4%) | Information missing |
| CTCF | IT-scC&T-seq [101] | 2,803 | Information missing | Information missing |
| H3K9me3 | scEpi2-seq [100] | Information missing | FRiP: 0.72 - 0.88 (depending on antibody) | Displays mutually exclusive pattern with H3K27me3 and H3K36me3 [100] |
| H3K36me3 | scEpi2-seq [100] | Information missing | FRiP: 0.72 - 0.88 (depending on antibody) | Associated with higher DNA methylation levels (~50%) vs. repressive marks (8-10%) [100] |
Experimental Protocols for Key Techniques
IT-scC&T-seq (Indexed Tagmentation-based single-cell CUT&Tag-sequencing)
This modular, plate-based strategy enables parallel profiling of histone modifications and transcription factors in tens of thousands of single cells without specialized microfluidic instrumentation [101].
Detailed Workflow:
- Cell Preparation and Antibody Binding: Fixed cells or nuclei are incubated with a primary antibody specific to the target histone mark (e.g., H3K4me3). A secondary antibody is then used to amplify the signal [101].
- First-Round Indexing (Bulk Tagmentation): Nuclei are divided into N bulk transposition reactions. Each reaction contains a uniquely barcoded pA-Tn5 transposome complex, which simultaneously binds to the antibody-targeted chromatin and fragments the DNA, inserting the first set of barcodes (Q5XX/Q7XX) [101].
- Single-Nuclei Sorting: Nuclei from each of the N tagmentation reactions are sequentially sorted into a 384-well plate, ensuring each well contains N nuclei, each with a distinct first-round barcode [101].
- Second-Round Indexing (Well-Specific Barcoding): After cell lysis, a PCR is performed using primers with a unique well-specific barcode combination (a 16x24 primer scheme for a 384-well plate), adding a second barcode (H5XX/H7XX) to the fragments in each well [101].
- Third-Round Indexing (Plate Multiplexing): The PCR products from all wells are pooled. A final PCR adds standard Illumina TruSeq adapters (T5XX/T7XX), allowing multiple plates to be multiplexed in a single sequencing run [101].
- Sequencing and Data Analysis: The library is sequenced on an Illumina platform. Bioinformatic demultiplexing uses the combination of all three barcodes to assign reads back to their individual cell of origin [101].
scEpi2-seq (Single-cell Epi2-seq)
This method enables the simultaneous detection of histone modifications and DNA methylation in the same single cell [100].
Detailed Workflow:
- Cell Permeabilization and Antibody Binding: Single cells are permeabilized, and a protein A-micrococcal nuclease (pA-MNase) fusion protein is tethered to specific histone modifications using antibodies [100].
- Single-Cell Sorting and MNase Digestion: Single cells are sorted into 384-well plates by FACS. MNase digestion is initiated by adding Ca²⁺, which cleaves chromatin in close proximity to the targeted histone mark [100].
- Fragment Recovery and Library Preparation: The released fragments are repaired, A-tailed, and ligated to adaptors containing a single-cell barcode, a unique molecular identifier (UMI), a T7 promoter, and an Illumina handle [100].
- TET-assisted Pyridine Borane Sequencing (TAPS): Material from the plate is pooled and subjected to TAPS, which converts methylated cytosine (5mC) to uracil without damaging the adaptor sequences, unlike traditional bisulfite treatment [100].
- Library Amplification and Sequencing: The library undergoes in vitro transcription (IVT), reverse transcription, and PCR. Paired-end sequencing then allows simultaneous extraction of information: genomic locations reveal histone modification sites, and C-to-T conversions identify methylated cytosines [100].
Visualizing the Methodological Workflows
IT-scC&T-seq Workflow Diagram
scEpi2-seq Multi-Omic Workflow Diagram
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Advanced Histone Profiling
| Reagent / Material | Critical Function in Workflow | Example Use Case |
|---|---|---|
| pA-Tn5 Transposome | Engineered fusion protein that binds antibody-targeted chromatin and simultaneously fragments DNA while inserting sequencing adapters [101]. | Core enzyme in CUT&Tag and IT-scC&T-seq for low-background, targeted tagmentation [98] [101]. |
| pA-Micrococcal Nuclease (pA-MNase) | Fusion protein that binds antibodies and cleaves surrounding chromatin upon calcium activation, used for targeted chromatin fragmentation [100]. | Core enzyme in scEpi2-seq and related methods (e.g., sortChIC) for releasing specific histone-bound fragments [100]. |
| Combinatorial Indexing Primers | Sets of oligonucleotides with unique barcodes for labeling chromatin fragments from individual cells or samples over multiple rounds of indexing [101]. | Enables massive multiplexing in plate-based methods like IT-scC&T-seq without physical separation of cells [101]. |
| TET-assisted Pyridine Borane (TAPS) Reagents | Chemical conversion system that changes 5-methylcytosine (5mC) to uracil without significant DNA damage, allowing methylation sequencing from low inputs [100]. | Enables simultaneous detection of DNA methylation alongside histone modifications in scEpi2-seq [100]. |
| Histone Modification-Specific Antibodies | High-specificity monoclonal or polyclonal antibodies that recognize particular histone PTMs (e.g., H3K27me3, H3K4me3). | The primary targeting mechanism in all immunoenzymatic methods (ChIP-seq, CUT&Tag, etc.); antibody quality is paramount for success [98] [101]. |
The transition from bulk ChIP-seq to single-cell, multi-omic, and highly scalable methods like IT-scC&T-seq and scEpi2-seq represents a significant technological leap. These advanced techniques directly address the core challenges of sensitivity, cost, and single-cell compatibility, enabling the resolution of cellular heterogeneity and the interplay between different epigenetic layers. For the broader thesis on histone modifications in gene regulation, this means that future research can move beyond static, population-average maps towards dynamic, cell-type-specific regulatory networks. This is particularly relevant in complex systems like drug treatment responses, development, and disease progression, where understanding epigenetic heterogeneity at the single-cell level is key to unlocking new mechanistic insights and therapeutic opportunities [103] [18]. As these methods continue to mature and become more accessible, they will undoubtedly solidify the central role of histone modification analysis in modern biomedical research.
The mapping of histone modifications is fundamental to understanding the epigenetic mechanisms that govern gene regulation, with chromatin immunoprecipitation followed by sequencing (ChIP-seq) long serving as the foundational method in this field. The Encyclopedia of DNA Elements (ENCODE) consortium has established rigorous experimental and data processing guidelines for ChIP-seq, making it the gold standard for profiling DNA-protein interactions [17] [55]. However, the emergence of innovative techniques like Cleavage Under Targets & Tagmentation (CUT&Tag) presents a paradigm shift by offering a streamlined, in-situ approach for epigenomic profiling. This technical guide examines the performance of CUT&Tag against established ENCODE ChIP-seq benchmarks, focusing specifically on its capacity to recover known histone modification peaks. Understanding this relationship is crucial for researchers investigating histone modifications in gene regulation, as it informs methodological selection, experimental design, and data interpretation in both basic research and drug development contexts.
Quantitative Benchmarking: CUT&Tag vs. ENCODE ChIP-seq
Peak Recovery Efficiency
A comprehensive benchmarking study published in Nature Communications systematically evaluated CUT&Tag performance for histone modifications H3K27ac and H3K27me3 against published ChIP-seq profiles from ENCODE in K562 cells [92]. The research, which analyzed 30 new and 6 published CUT&Tag datasets, revealed that CUT&Tag recovers a substantial proportion of known ENCODE peaks.
Table 1: CUT&Tag Recovery of ENCODE ChIP-seq Peaks
| Histone Modification | Average Recall of ENCODE Peaks | Key Characteristics of Recovered Peaks |
|---|---|---|
| H3K27ac | 54% | Represents the strongest ENCODE peaks; shows same functional and biological enrichments |
| H3K27me3 | 54% | Represents the strongest ENCODE peaks; shows same functional and biological enrichments |
Despite this moderate recovery rate, the peaks identified by CUT&Tag represent the strongest ENCODE peaks and show identical functional and biological enrichments as those identified by ChIP-seq [92]. This suggests that CUT&Tag effectively captures the most biologically relevant regions.
Technical Performance Metrics
When comparing the technical performance of CUT&Tag versus traditional ChIP-seq, several key differences emerge that influence their application in research and drug development.
Table 2: Method Comparison: CUT&Tag vs. ChIP-seq
| Parameter | CUT&Tag | ChIP-seq |
|---|---|---|
| Assay Type | In situ | In vitro |
| Core Principle | Antibody-recruited Tn5 transposase tagmentation | Cross-linking + sonication + immunoprecipitation |
| Cell Input Requirement | 100 - 100,000 cells [104] [105] | 100,000 - millions of cells [105] |
| Signal-to-Noise Ratio | High (minimal background) [105] | Lower (non-specific binding, off-target sonication) [105] |
| Protocol Duration | ~1 day [105] | 2-5 days [105] |
| Sequencing Depth Requirement | 5-8 million reads [104] | >30 million reads for H3K4me3 [104] |
| Cost Per Sample | Lower (less reagents, shallow sequencing) [105] | Higher (more reagents, deep sequencing) [105] |
Experimental Protocols and Methodologies
CUT&Tag Workflow and Optimization
The CUT&Tag method utilizes an enzyme-tethering approach that fundamentally differs from ChIP-seq. The protocol involves several key steps [105]:
- Sample Permeabilization: Cells or nuclei are permeabilized to allow antibody access.
- Antibody Binding: A primary antibody specific for the histone mark of interest (e.g., H3K27ac) is added, followed by a secondary antibody.
- pA-Tn5 Recruitment: Protein A/G fused to Tn5 transposase (pA-Tn5) is recruited to the antibody complex.
- Tagmentation: Magnesium ions activate the tethered Tn5, which simultaneously cleaves DNA and inserts sequencing adapters.
- DNA Purification and Sequencing: DNA fragments are purified, amplified, and prepared for high-throughput sequencing.
A critical optimization involves using a washing buffer with 300 mM NaCl to eliminate false signals caused by untethered pA-Tn5 binding to open chromatin regions [104]. Furthermore, reducing PCR cycles during library preparation can lower duplication rates, though this may come at the expense of ENCODE peak recovery [92] [106].
ENCODE ChIP-seq Standards
The ENCODE consortium has established rigorous guidelines for ChIP-seq experiments to ensure data quality and reproducibility [17] [55]:
- Antibody Validation: Antibodies must undergo primary and secondary characterization, including immunoblot analysis or immunofluorescence, to demonstrate specificity [55].
- Sequencing Depth: Requirements vary by target; for broad histone marks like H3K27me3, each replicate should have 45 million usable fragments, while narrow marks like H3K27ac require 20 million fragments per replicate [17].
- Experimental Replication: Experiments should have two or more biological replicates.
- Controls: Each ChIP-seq experiment should have a corresponding input control experiment with matching run type and replicate structure.
Diagram: Comparative Workflows of ChIP-seq and CUT&Tag
Factors Influencing CUT&Tag Performance
Antibody and Experimental Optimization
The recovery of ENCODE peaks using CUT&Tag is influenced by several experimental factors. Benchmarking studies tested multiple ChIP-grade antibody sources and dilutions to optimize performance [92]. For H3K27ac, antibodies from Abcam (ab4729 - the same antibody used in ENCODE ChIP-seq), Diagenode (C15410196), Abcam (ab177178), and Active Motif (39133) were evaluated at various dilutions (1:50, 1:100, 1:200) [92].
Since H3K27ac is dynamically regulated by histone acetyltransferases and deacetylases (HDACs), researchers investigated whether adding histone deacetylase inhibitors (HDACi) like Trichostatin A (TSA; 1 µM) or sodium butyrate (NaB; 5 mM) would stabilize acetyl marks and improve data quality during the CUT&Tag process, which is performed under native conditions [92]. However, results demonstrated that HDAC inhibition did not consistently increase total peak detection, improve signal-to-noise ratio, or enhance ENCODE capture [92].
Data Processing and Analysis
The choice of computational tools significantly impacts CUT&Tag performance metrics:
- Peak Callers: Studies have tested both MACS2 and SEACR for calling peaks from CUT&Tag data [92]. When balancing precision and recall of known ENCODE peaks, SEACR without retention of duplicates showed the best performance [106].
- Duplicate Reads: The handling of PCR duplicates affects peak calling accuracy. Preliminary analyses of CUT&Tag data revealed high duplication rates across samples (55.49% to 98.45%, mean: 82.25%) [92]. Reducing PCR cycles during library preparation can lower duplication rates but may compromise ENCODE peak recovery [92].
- Assessment Metrics: Benchmarking pipelines like EpiCompare have been developed to help researchers evaluate CUT&Tag data quality through parameters including read- and peak-level correlation, regulatory element annotation, gene ontology enrichment, and transcription factor binding motif analysis [92].
Advantages and Limitations in Histone Modification Profiling
Method-Specific Biases and Complementarity
While CUT&Tag recovers approximately half of ENCODE ChIP-seq peaks, each method exhibits unique biases that make them complementary rather than directly interchangeable:
ChIP-seq Biases:
- Traditional ChIP-seq demonstrates bias toward open chromatin regions, with input chromatin enriched near gene transcription start sites and in highly accessible regions [107].
- Heterochromatic regions are often lost in the insoluble pellet during ChIP-seq sample preparation, leading to underrepresentation of repetitive elements and constitutive heterochromatin [107].
CUT&Tag Advantages:
- CUT&Tag overcomes ChIP-seq biases against condensed chromatin, enabling robust detection of H3K9me3 over repetitive elements and retrotransposons [107].
- It provides superior mapping of heterochromatic regions, with especially high sensitivity over evolutionarily young retrotransposons [107].
- For euchromatin-associated proteins that co-purify with insoluble heterochromatin in ChIP studies, CUT&Tag can detect their binding at repetitive elements [107].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for CUT&Tag
| Reagent / Material | Function | Examples & Notes |
|---|---|---|
| Validated Antibodies | Specific recognition of target histone modification | H3K27ac: Abcam-ab4729 (used in ENCODE) [92]; H3K27me3: Cell Signaling Technology-9733 [92] |
| pA/G-Tn5 Transposase | Target-specific chromatin fragmentation and adapter insertion | Hyperactive Universal CUT&Tag Assay Kits; Requires Mg²⁺ for activation [104] |
| High-Salt Wash Buffer | Reduction of untethered Tn5 background signal | 300 mM NaCl solution eliminates false signals from open chromatin [104] |
| Magnetic Beads | Sample immobilization and processing | Concanavalin A (ConA) beads for cell/nuclei attachment [91] |
| Library Amplification System | Preparation of sequencing-ready libraries | Direct-to-PCR approach; Typically 12-15 cycles [92] [91] |
The comprehensive benchmarking of CUT&Tag against ENCODE ChIP-seq reveals a nuanced relationship between these methodologies. While CUT&Tag recovers approximately half of known ENCODE peaks for histone modifications H3K27ac and H3K27me3, the recovered peaks represent the strongest ENCODE peaks and maintain identical functional and biological enrichments. This recovery rate must be contextualized within the distinct technical advantages of CUT&Tag, including its minimal cellular input requirements, superior signal-to-noise ratio, and enhanced capacity to profile heterochromatic regions traditionally underrepresented in ChIP-seq data.
For researchers investigating the role of histone modifications in gene regulation, these findings suggest a complementary rather than replacement relationship between CUT&Tag and ChIP-seq. CUT&Tag offers significant practical advantages for studies with limited material, high-throughput screening, or investigations focusing on repetitive genomic elements. However, the established benchmarks and extensive historical data from ENCODE ChIP-seq remain invaluable references for the field. Future directions include continued optimization of CUT&Tag protocols, development of standardized analysis pipelines, and exploration of multi-modal approaches like multi-CUT&Tag that enable simultaneous profiling of multiple chromatin features in the same cells [108]. As these technologies evolve, they will collectively advance our understanding of epigenetic regulation in health and disease.
The interpretation of histone modification patterns is fundamental to understanding epigenetic control of gene regulation. While ChIP-seq has emerged as the predominant technique for genome-wide mapping of these modifications, researchers must navigate a complex landscape of methodological variations and complementary technologies to draw accurate biological conclusions. This whitepaper examines the critical issue of concordance between techniques in histone modification research, providing a framework for evaluating when methods agree and where they legitimately diverge. We analyze established and emerging protocols, present quantitative concordance metrics, and offer practical guidance for protocol selection and data interpretation within drug development contexts. By synthesizing current methodologies and their limitations, this guide empowers researchers to design more robust epigenetic studies and confidently translate findings into therapeutic insights.
Histone post-translational modifications (PTMs) represent a crucial epigenetic mechanism regulating chromatin structure and gene expression without altering the underlying DNA sequence. These modifications—including acetylation, methylation, phosphorylation, ubiquitination, and newer additions like succinylation, crotonylation, and lactylation—occur primarily on histone tails extending from the nucleosome surface [42] [109]. They influence gene expression through two primary mechanisms: by altering the histone's electrostatic charge and structural properties, or by creating binding sites for protein recognition modules that recruit additional chromatin-modifying complexes [42].
The dynamic nature of histone modifications enables cells to rapidly respond to environmental cues, making them particularly significant in development, cellular differentiation, and disease pathogenesis [109]. Abnormalities in histone modification patterns have been correlated with various human diseases, including cancer, immunodeficiency disorders, and metabolic diseases, highlighting their therapeutic relevance [42] [109]. For drug development professionals, understanding and targeting these epigenetic marks offers promising avenues for therapeutic intervention, particularly in overcoming treatment resistance in oncology [110].
Within this context, Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has become the cornerstone technique for investigating histone modifications genome-wide. However, variations in experimental protocols and the emergence of complementary epigenetic profiling techniques necessitate a critical examination of concordance between methodological approaches.
Methodological Foundations: ChIP-seq and Complementary Techniques
Core ChIP-seq Methodology
The fundamental ChIP-seq protocol involves several critical steps that can significantly influence results and interpretation:
- Cross-linking: Proteins are covalently attached to DNA using formaldehyde or other cross-linking agents, preserving protein-DNA interactions [111]. Dual cross-linking approaches may be employed for certain histone modifiers [112].
- Chromatin Fragmentation: The cross-linked chromatin is fragmented into smaller pieces (typically 200-600 bp) using sonication or enzymatic digestion [111]. Sonication conditions must be optimized for each cell type and experimental condition.
- Immunoprecipitation: An antibody specific to the histone modification of interest immunoprecipitates the protein-DNA complex [42] [111]. Antibody specificity is arguably the most critical factor determining success.
- Library Preparation and Sequencing: The immunoprecipitated DNA is prepared for next-generation sequencing by adding adapters and amplifying the library before sequencing [112] [111].
Table 1: Key Variations in ChIP-seq Protocols for Histone Modifications
| Protocol Aspect | Standard Approach | Variations | Impact on Concordance |
|---|---|---|---|
| Cross-linking | Formaldehyde (single) | Dual cross-linking with DSG | Enhanced detection of indirect associations [112] |
| Chromatin Fragmentation | Sonication | Enzymatic digestion (MNase) | Differences in resolution and bias [111] |
| Antibody Source | Commercial kits | Custom antibodies | Significant variability in specificity and sensitivity [112] |
| Sample Input | 0.5-1 million cells | Low-cell protocols (<10,000 cells) | Potential differences in signal-to-noise ratio [111] |
Emerging and Complementary Techniques
While ChIP-seq remains the workhorse for histone modification studies, several complementary approaches provide orthogonal validation or unique insights:
- Mass Spectrometry-Based Proteomics: Liquid chromatography-tandem mass spectrometry (LC-MS/MS) enables systematic mapping of PTM dynamics without antibody-based enrichment, providing quantitative information on modification stoichiometry [109].
- Multiplexed ChIP-seq (MINUTE-ChIP): This recent innovation allows multiple samples to be profiled against multiple epitopes in a single workflow, dramatically increasing throughput while enabling accurate quantitative comparisons through barcoding strategies [94].
- Integrated Multi-omics Approaches: Combining ChIP-seq with RNA sequencing, ATAC sequencing, and DNA methylation analysis provides a more comprehensive understanding of epigenetic regulation [111] [110].
Diagram 1: Histone modification analysis workflow
Quantitative Concordance Analysis: Metrics and Interpretation
Establishing Concordance Metrics
Determining concordance between technical approaches requires robust quantitative metrics. The following standards provide frameworks for assessing agreement:
- Peak Overlap Analysis: The most straightforward metric calculates the percentage of overlapping peaks between replicates or techniques, with high-quality replicates typically showing 70-90% overlap for strong histone marks.
- Correlation Coefficients: Pearson or Spearman correlation of read densities across genomic regions quantifies reproducibility more sensitively than binary peak overlap.
- Irreproducible Discovery Rate (IDR): A statistical methodology that evaluates consistency between replicates by modeling the rank ordering of peaks, with IDR < 0.05 indicating high-confidence calls.
Table 2: Quantitative Standards for Method Concordance
| Concordance Level | Peak Overlap | Correlation Coefficient | IDR Threshold | Typical Application |
|---|---|---|---|---|
| High | >80% | >0.8 | <0.01 | Technical replicates |
| Moderate | 60-80% | 0.6-0.8 | 0.01-0.05 | Biological replicates |
| Minimal | <60% | <0.6 | >0.05 | Different techniques |
Expected Concordance Patterns
The expected degree of concordance varies substantially based on the biological and technical context:
- Technical Replicates: The same library sequenced multiple times should demonstrate >90% concordance by all metrics when analyzing well-established histone marks like H3K27ac or H3K4me3 [112].
- Biological Replicates: Using the same protocol on different samples of the same cell type typically shows 70-90% concordance, with variation reflecting true biological differences [94].
- Protocol Variations: Different ChIP-seq protocols (e.g., cross-linking conditions, fragmentation methods) applied to the same biological sample generally show 60-80% concordance, with differences highlighting technique-specific biases [112].
- Orthogonal Techniques: Comparisons between ChIP-seq and mass spectrometry-based approaches may show lower apparent concordance (40-70%) due to fundamental methodological differences, with each technique providing complementary information [109].
Experimental Protocols for Concordance Assessment
Standard ChIP-seq Protocol for Histone Modifications
For mapping histone modifications, the native ChIP protocol typically follows this detailed methodology:
Day 1: Sample Preparation and Chromatin Fragmentation
- Cell Collection: Harvest approximately 1×10⁶ cells, washing twice with cold PBS.
- Nuclear Extraction: Resuspend cell pellet in 1 mL Lysis Buffer (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 0.5% NP-40) with protease inhibitors, incubate 15 minutes on ice.
- Chromatin Digestion: Resuspend nuclei in 1 mL MNase Digestion Buffer (50 mM Tris-HCl pH 7.9, 5 mM CaCl₂), add 2 μL MNase (1000 U/mL), incubate 15 minutes at 37°C with occasional mixing.
- Reaction Stop: Add EDTA to 10 mM final concentration, centrifuge at 13,000 rpm for 5 minutes at 4°C.
- Chromatin Extraction: Resuspend pellet in 500 μL IP Buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS), incubate 30 minutes on ice with occasional vortexing.
Day 2: Immunoprecipitation and DNA Recovery
- Antibody Binding: Add 2-5 μg histone modification-specific antibody (validate specificity using peptide blocking assays), incubate overnight at 4°C with rotation.
- Bead Capture: Add 50 μL pre-washed Protein A/G magnetic beads, incubate 4 hours at 4°C with rotation.
- Wash Steps: Wash beads sequentially with:
- Low Salt Wash Buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS)
- High Salt Wash Buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.1% SDS)
- LiCl Wash Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate)
- TE Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA)
- Elution and Reverse Cross-linking: Elute DNA in 250 μL Elution Buffer (1% SDS, 0.1 M NaHCO₃), add NaCl to 200 mM final concentration, incubate overnight at 65°C.
- DNA Purification: Treat with RNase A (30 minutes at 37°C) and Proteinase K (2 hours at 55°C), purify using phenol-chloroform extraction and ethanol precipitation.
Day 3: Library Preparation and Sequencing
- Library Construction: Use 5-10 ng immunoprecipitated DNA with commercial library preparation kits following manufacturer's instructions.
- Size Selection: Perform double-sided size selection (200-400 bp) using AMPure XP beads.
- Quality Control: Assess library quality using Bioanalyzer/TapeStation and quantify by qPCR.
- Sequencing: Sequence on appropriate Illumina platform (minimum 20 million reads per sample for histone marks) [112] [111].
MINUTE-ChIP for Multiplexed Quantitative Comparisons
The MINUTE-ChIP protocol enables quantitative comparison across conditions and epitopes:
- Sample Barcoding: Fragment native or formaldehyde-fixed chromatin from different conditions, then barcode using unique DNA adapters.
- Pooling and Splitting: Combine barcoded chromatin samples into a single pool, then split into equal aliquots for parallel immunoprecipitation reactions targeting different histone modifications.
- Parallel Immunoprecipitation: Perform simultaneous IP reactions on each aliquot with modification-specific antibodies.
- Library Preparation and Sequencing: Prepare sequencing libraries from input and immunoprecipitated DNA, then sequence multiplexed samples in a single lane.
- Bioinformatic Demultiplexing: Computational separation of barcoded samples following sequencing, generating quantitatively comparable tracks [94].
This approach not only increases throughput but enables more accurate quantitative comparisons between conditions by minimizing technical variability.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Histone Modification Studies
| Reagent Category | Specific Examples | Function and Importance | Concordance Considerations |
|---|---|---|---|
| Histone Modification Antibodies | Anti-H3K27ac, Anti-H3K4me3, Anti-H3K9me3 | Target-specific enrichment; primary determinant of data quality | Major source of variability; validate using peptide arrays and knockout controls |
| Chromatin Shearing Reagents | MNase, Micrococcal Nuclease | Enzymatic fragmentation of chromatin | So-nication efficiency varies by cell type; enzymatic digestion provides more uniform fragmentation |
| Cross-linking Agents | Formaldehyde, Disuccinimidyl Glutarate (DSG) | Preserve protein-DNA interactions | Dual crosslinking (DSG + formaldehyde) improves recovery for certain histone modifiers |
| Library Preparation Kits | Illumina DNA Prep, KAPA HyperPrep | Prepare sequencing libraries from immunoprecipitated DNA | Kit choice affects GC bias and duplicate rates; maintain consistency across experiments |
| Multiplexing Barcodes | i5/i7 indexes, Unique Molecular Identifiers (UMIs) | Sample multiplexing and duplicate removal | Essential for MINUTE-ChIP; enable quantitative comparisons between conditions |
| Spike-in Controls | Drosophila chromatin, E. coli DNA | Normalization between samples | Critical for quantitative comparisons; reveals technical variations in IP efficiency |
Biological Contexts of Concordance and Divergence
Expected Technical vs. Biological Divergence
Understanding when techniques should agree and where they might legitimately diverge is essential for accurate data interpretation:
- Technical Reproducibility: High concordance (80-90%) expected when the same protocol is repeated on the same biological sample. Lower values indicate technical issues, most commonly antibody lot variability or chromatin preparation inconsistencies [112].
- Biological Variation: Moderate concordance (60-80%) between different samples of the same cell type reflects true biological heterogeneity in histone modification patterns, which can be substantial even in supposedly homogeneous cell populations.
- Cellular Context Dependencies: The same histone mark may show different genomic distributions across cell types, developmental stages, or disease states. These represent biologically meaningful divergences rather than technical artifacts [42] [109].
Histone Modification Crosstalk
A significant source of legitimate divergence between techniques stems from the complex interplay between different histone modifications:
- Combinatorial Coding: Multiple histone modifications can act in combination to influence chromatin state, creating patterns where individual marks show only partial correlation with functional outcomes [109] [110].
- Modification Interdependencies: Certain histone modifications facilitate or inhibit the establishment of others. For example, H2B ubiquitination promotes H3K4 methylation, creating expected concordance between these marks [109].
- Context-Dependent Interpretations: The same histone modification can have different functional consequences depending on genomic context and the presence of other modifications. H3K4me3 at promoters typically associates with active transcription, while the same mark at enhancers shows more complex relationships with gene expression [42].
Diagram 2: Histone modification crosstalk in gene regulation
Implications for Drug Development and Therapeutic Targeting
For drug development professionals, interpreting concordance between epigenetic techniques has direct translational relevance:
- Target Validation: Consistent identification of histone modification changes across multiple technical approaches strengthens confidence in potential therapeutic targets. The MINUTE-ChIP system provides particularly robust quantitative data for target prioritization [94].
- Biomarker Development: Histone modification patterns show promise as diagnostic and prognostic biomarkers, but require validation across platforms. Mass spectrometry approaches can provide orthogonal validation for antibody-based discoveries [109].
- Combination Therapy Strategies: The extensive crosstalk between epigenetic modifications suggests that targeting single modifications may have limited efficacy. Multi-omics approaches reveal synergistic relationships that inform rational combination therapies [110].
- Resistance Mechanisms: Acquired resistance to epigenetic therapies often involves compensatory changes in other modification pathways. Integrated analysis of multiple epigenetic layers helps identify these adaptive responses and design countermeasures [110].
Emerging clinical evidence suggests that while single-target epigenetic therapies often show limited efficacy, combination approaches that address the complex interplay of histone modifications hold significant promise for overcoming therapeutic resistance in cancer and other diseases [110].
Interpreting concordance between techniques in histone modification research requires both technical understanding of methodological limitations and biological appreciation of epigenetic complexity. While ChIP-seq remains the foundational approach, emerging multiplexed methods and orthogonal validation strategies provide increasingly robust frameworks for distinguishing technical artifacts from biological reality. For researchers and drug development professionals, recognizing when techniques should agree and where they might legitimately diverge enables more accurate interpretation of epigenetic data and more informed therapeutic decision-making. As the field advances toward increasingly quantitative and comprehensive epigenetic profiling, thoughtful assessment of concordance will remain essential for translating histone modification maps into biological insights and clinical advances.
In the field of epigenomics research, chromatin immunoprecipitation followed by sequencing (ChIP-seq) has become an indispensable method for mapping protein-DNA interactions and histone modifications across the genome [113] [54]. The strategic importance of histone modifications in gene regulation is well-established, with specific methylation and acetylation patterns constituting a complex "histone code" that directly influences transcriptional states, cellular identity, and disease processes [114] [115]. As the volume and complexity of ChIP-seq data have grown, so too has the landscape of computational tools designed to extract biological meaning from these datasets. However, this expansion of analytical options has created a significant challenge for researchers: selecting the most appropriate tool for their specific experimental goals.
The fundamental challenge in tool selection stems from several factors. First, histone modifications exhibit dramatically different genomic distributions, from sharp, peak-like signals at transcription factor binding sites to broad domains characteristic of repressive marks such as H3K27me3 and H3K9me3 [116] [114]. Second, experimental goals vary considerably—from simple peak calling to complex differential analysis across multiple conditions—with each goal demanding specialized statistical approaches [117]. Third, the technical expertise required to implement these tools ranges significantly, with some requiring advanced programming skills while others offer user-friendly web interfaces [113]. This guide addresses these challenges by providing a structured decision matrix to help researchers navigate the tool selection process, ensuring optimal analytical choices for their specific research contexts in histone modification studies.
Key Analytical Challenges in Histone Modification Analysis
Analyzing histone modification data presents unique computational challenges that directly influence tool selection. The most significant challenge concerns the differential analysis of broad epigenetic domains. Modifications such as H3K27me3 (associated with Polycomb repression) and H3K9me3 (associated with heterochromatin) form expansive genomic regions that can span several thousands of basepairs [116]. Traditional peak-calling algorithms designed for sharp, punctate signals (like transcription factor binding sites) perform poorly on these broad domains, often generating false positives or false negatives [116]. This limitation necessitates specialized tools that employ distinct statistical approaches for identifying differentially modified regions.
A second major challenge involves quantitative comparison across multiple conditions. While simple overlapping analysis of peaks called from different experiments was once common, this approach is highly dependent on arbitrary thresholds and ignores quantitative differences [117]. Modern experimental designs often require comparison across multiple cell types, conditions, or time points, demanding methods that can account for background noise, signal-to-noise ratios between experiments, biological variation, and complex experimental designs [117]. Additionally, the emergence of single-cell multi-omics technologies has introduced new dimensions of complexity, with methods like scMTR-seq and scEpi2-seq now enabling simultaneous profiling of multiple histone modifications together with transcriptomes or DNA methylation in individual cells [118] [100]. These advanced applications require specialized analytical approaches that can handle sparse data and capture cellular heterogeneity while integrating multiple epigenetic layers.
Decision Matrix: Selecting Tools for Your Experimental Goals
The following decision matrix provides a structured framework for selecting ChIP-seq analysis tools based on specific research objectives, histone mark characteristics, and experimental design.
Table 1: Tool Selection Decision Matrix for Histone Modification Analysis
| Research Goal | Histone Mark Type | Recommended Tools | Key Statistical Approach | Best For |
|---|---|---|---|---|
| Differential Analysis | Broad domains (H3K27me3, H3K9me3) | histoneHMM [116] | Bivariate Hidden Markov Model | Unsupervised classification of genomic regions into modified, unmodified, or differentially modified states |
| Differential Analysis | Narrow peaks (Transcription factors) | ChIPComp [117] | Generalized linear model with Poisson distribution | Multi-condition comparisons with complex experimental designs |
| Automated End-to-End Analysis | Both narrow and broad marks | H3NGST [113] | Hybrid pipeline integrating multiple algorithms | Researchers lacking bioinformatics expertise or computational resources |
| Single-Cell Multi-omics | Multiple modifications with transcriptome | scMTR-seq [118] | Combinatorial barcoding & parallel processing | Mapping cellular heterogeneity and coordinated chromatin/expression changes |
| Interaction Network Analysis | Multiple modifications & chromatin modifiers | Elastic Net + SPCN [119] | Regularized regression + sparse partial correlation | Reconstructing chromatin signaling networks from genome-wide data |
| Dynamic Process Analysis | Multiple modifications across time courses | Custom regression modeling [114] | Multivariate regression with kinetic parameters | Uncovering ordered sequences of histone modification events |
How to Use the Decision Matrix
To effectively use this decision matrix, researchers should first clearly define their primary research question, then identify the characteristics of the histone marks being studied, and finally consider practical constraints such as computational resources and expertise. For example, a researcher studying H3K27me3 dynamics during cellular differentiation would fall into the "Differential Analysis" of "Broad domains" category, making histoneHMM the recommended choice [116]. Conversely, a research team investigating transcription factor binding changes across multiple drug treatments would benefit from ChIPComp's ability to handle complex experimental designs with narrow peaks [117]. For laboratories with limited bioinformatics support, H3NGST provides a fully automated, web-based solution that requires only a BioProject ID to initiate complete analysis [113].
Detailed Methodologies for Key Experimental Approaches
histoneHMM for Differential Analysis of Broad Histone Marks
The histoneHMM methodology employs a bivariate Hidden Markov Model specifically designed to address the challenges of broad histone modifications [116]. The workflow begins with read aggregation into larger genomic regions (typically 1000 bp windows) to compensate for the low signal-to-noise ratio characteristic of broad domains. The algorithm then processes the bivariate read counts from two conditions through an unsupervised classification procedure that probabilistically categorizes each genomic region into one of three states: (1) modified in both samples, (2) unmodified in both samples, or (3) differentially modified between samples [116].
A key advantage of histoneHMM is its minimal parameter tuning requirement, which reduces subjectivity in analysis. The implementation includes careful normalization to account for technical variations between samples. The algorithm has been extensively validated for functional relevance through integration with RNA-seq data, showing significant enrichment of differentially expressed genes in regions called as differentially modified [116]. Performance benchmarks against competing methods like Diffreps, Chipdiff, Pepr, and Rseg demonstrated histoneHMM's superior accuracy in identifying functionally relevant differential regions, particularly for the repressive marks H3K27me3 and H3K9me3 [116].
ChIPComp for Quantitative Comparison of Multiple Conditions
ChIPComp implements a comprehensive statistical framework for quantitative comparison of multiple ChIP-seq datasets, specifically designed for narrow peak data such as transcription factor binding or sharp histone modifications [117]. The method follows a two-step procedure beginning with peak calling using established algorithms for each individual dataset, followed by formation of a union peak set as candidate regions for quantitative comparison.
The core of ChIPComp's approach involves a sophisticated data model that accounts for critical experimental factors. The observed IP counts (Y~ij~) at candidate region i in dataset j are modeled as Poisson distributed with rate μ~ij~, which is modeled as a function of background signal (λ~ij~) and biological signal (S~ij~): Y~ij~ | μ~ij~ ~ Poisson(μ~ij~); μ~ij~ = f(λ~ij~, b~j~s~ij~) [117]. Here, b~j~ represents the experiment-specific signal-to-noise ratio, while s~ij~ measures relative biological signals. The background λ~ij~ is estimated from control data using spatial correlation techniques, addressing a key limitation of methods that simply subtract control counts [117].
ChIPComp incorporates the estimated backgrounds into a generalized linear model framework that can handle multiple factors and biological replicates: log(s~ij~) = x~j~β~i~ + ε~ij~, where x~j~ is the experimental design vector and β~i~ represents the effect sizes for candidate region i [117]. This enables rigorous statistical testing for differential binding while properly controlling for background signals and variable data quality between experiments.
H3NGST for Automated End-to-End Analysis
H3NGST (Hybrid, High-throughput, and High-resolution NGS Toolkit) provides a fully automated, web-based platform that streamlines the entire ChIP-seq analysis workflow without requiring programming skills or local software installation [113]. The platform integrates multiple established tools into a cohesive pipeline that begins with raw data retrieval using public accession numbers (BioProject, SRA, GEO), automatically determines library type (single-end or paired-end), and performs comprehensive processing.
The H3NGST workflow encompasses four major stages: (1) data retrieval and quality control using FastQC; (2) adapter trimming and quality filtering with Trimmomatic; (3) sequence alignment to a user-specified reference genome using BWA-MEM with automatic format conversion; and (4) peak calling with HOMER, which supports both narrow (transcription factor) and broad (histone modification) peak profiles [113]. The system automatically adjusts parameters based on dataset characteristics and provides comprehensive output including peak coordinates, motif occurrences, genomic annotations, and quality metrics.
A distinctive feature of H3NGST is its security and accessibility—all data transmissions are encrypted using SSL/TLS protocols, and no file uploads or user authentication are required [113]. The platform's mobile-friendly web interface extends its usability to researchers with diverse computational backgrounds, significantly reducing the technical barriers to sophisticated ChIP-seq analysis.
Visualization of Analysis Workflows
Bulk Histone Modification Analysis Workflow
Single-Cell Multi-Omics Histone Analysis
Research Reagent Solutions for Histone Modification Studies
Table 2: Essential Research Reagents and Materials for Histone Modification Studies
| Reagent/Material | Specific Examples | Function in Experiment | Technical Considerations |
|---|---|---|---|
| Histone Modification Antibodies | H3K27me3, H3K9me3, H3K4me3, H3K27ac, H3K36me3 [118] [114] | Target-specific immunoprecipitation of modified histone regions | Specificity validation critical; lot-to-lot variability concerns |
| Protein A-Tn5 Transposase Fusion | scMTR-seq complexes [118] | Antibody-tethered tagmentation for targeted sequencing | Requires pre-assembly with antibodies; IgG blocking reduces off-target signals |
| Combinatorial Barcodes | scMTR-seq barcoding system [118] | Single-cell multiplexing and identification | 48 barcodes per round enables >110,000 unique combinations |
| TET-Assisted Pyridine Borane (TAPS) Reagents | scEpi2-seq components [100] | Bisulfite-free DNA methylation detection | Preserves library complexity compared to bisulfite treatment |
| Chromatin Fragmentation Enzymes | MNase, Benzonase [119] [120] | Controlled chromatin digestion for IP | Enzyme concentration optimization critical for mononucleosome resolution |
| Magnetic Protein G Beads | Dynabeads systems [119] | Antibody coupling and immunoprecipitation | Consistent bead size improves reproducibility |
| Spike-in Controls | In vitro methylated DNA [100] | Normalization and quality assessment | Essential for cross-experiment comparison |
Advanced Applications and Emerging Methodologies
Single-Cell Multi-Omic Integration
The emerging frontier in histone modification research involves simultaneous profiling of multiple epigenetic layers in single cells. Technologies like scMTR-seq enable joint profiling of six histone modifications together with transcriptomes in individual cells, providing unprecedented resolution of cellular heterogeneity [118]. This method employs an adapter switching strategy that uses mosaic end B (MEB) adapters for antibody-specific tagmentation, then adds mosaic end A (MEA) adapters to all MEB-tagged fragments, improving signal-to-background ratio and library complexity [118]. Similarly, scEpi2-seq achieves simultaneous detection of histone modifications and DNA methylation at single-cell resolution, revealing how DNA methylation maintenance is influenced by local chromatin context [100].
These multi-omic approaches are particularly valuable for studying developmental processes and disease states characterized by cellular heterogeneity. For example, application of scMTR-seq to mouse blastocysts has revealed epigenetic asymmetries at gene regulatory regions between the three embryo lineages, identifying Trps1 as a potential repressor in epiblast cells [118]. The technology achieves high recovery rates (>40% of cells) with satisfactory sequencing depth (mean of 735-3024 reads per histone modification per cell) while maintaining strong correlation with bulk measurements [118].
Chromatin Network Inference
Beyond identifying individual modified regions, advanced computational methods can now reconstruct chromatin signaling networks from genome-wide data. One approach combines Elastic Net regularization with sparse partial correlation networks (SPCN) to infer interactions between chromatin modifiers and histone modifications [119]. This method models each histone modification level as a weighted linear combination of chromatin modifier levels, selecting the most consistent predictors through regularized regression [119].
The resulting interaction networks provide a high-confidence backbone of the chromatin-signaling network involved in transcription and its regulation [119]. This approach has successfully identified literature-supported interactions and generated novel biological hypotheses, such as a link between H4K20me1 and members of the Polycomb Repressive Complexes 1 and 2 [119]. Such network-level analyses move beyond descriptive cataloging of histone marks toward mechanistic understanding of epigenetic regulation.
Dynamic Process Analysis
Time-course analyses of histone modifications during transcriptional reprogramming provide unique insights into the kinetics and ordering of chromatin events. Comprehensive mapping of 26 histone modifications during yeast stress response revealed that most histone modifications remain highly correlated during dynamic processes, but approximately 3% of nucleosomes transiently populate rare histone modification states [114]. These unusual states primarily result from kinetic differences between modifications, with slow histone methylation changes often lagging behind more rapid acetylation changes [114].
Explicit analysis of modification dynamics has uncovered ordered sequences of events in gene activation and repression, providing temporal resolution that cannot be captured by steady-state analyses [114]. Such temporal data helps distinguish direct enzymatic activities from indirect effects and reveals the causal relationships between different chromatin events.
The landscape of tools for histone modification analysis has evolved significantly, with specialized solutions now available for diverse research scenarios. The selection of an appropriate analytical strategy should be guided by three primary considerations: the genomic distribution of the histone mark(s) of interest (sharp peaks versus broad domains), the experimental design (single condition versus multiple comparisons), and the available computational resources (local infrastructure versus web-based platforms).
For studies focusing on broad repressive marks like H3K27me3 and H3K9me3, histoneHMM provides specialized functionality that outperforms general-purpose peak callers [116]. For complex multi-condition experiments investigating transcription factor binding or sharp histone modifications, ChIPComp offers rigorous statistical framework that properly accounts for background signals and variable data quality [117]. For researchers with limited bioinformatics support or those conducting high-throughput analyses, H3NGST delivers automated, reproducible results without requiring programming skills [113].
As histone modification research continues to advance toward single-cell multi-omics and dynamic process analysis, tool selection will increasingly determine research success. The decision matrix and methodologies presented in this guide provide a structured framework for matching analytical tools to experimental goals, ensuring that researchers can extract maximum biological insight from their chromatin profiling data.
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has revolutionized our understanding of gene regulation by providing a powerful tool to investigate protein-DNA interactions across the entire genome. While much of the foundational work in epigenomics has focused on histone modifications, the principles and methodologies of ChIP-seq are equally crucial for studying transcription factor (TF) binding sites. These binding sites, typically short DNA motifs of 6-12 base pairs, are fundamental to transcriptional regulation and the precise spatiotemporal control of gene expression [121] [122]. This technical guide explores how established ChIP-seq techniques, often first developed for histone modification studies, can be systematically applied to transcription factor binding studies, enabling researchers to unravel the complex regulatory networks that govern cellular identity, function, and disease.
Core Principles of ChIP-seq for Transcription Factors
The fundamental ChIP-seq protocol involves crosslinking proteins to DNA in living cells, fragmenting the chromatin, immunoprecipitating the protein-DNA complexes using specific antibodies, and then sequencing the bound DNA fragments [41]. When applied to transcription factors, this process captures a snapshot of their genomic occupancy under specific cellular conditions.
Unlike histone modifications that mark broad chromatin states, transcription factors typically bind to specific, short DNA sequences. This difference necessitates specific analytical considerations. For instance, control samples such as Whole Cell Extract (WCE or "input") are crucial for identifying background signal in both histone and TF ChIP-seq [123]. However, research indicates that for histone modifications, a Histone H3 pull-down can sometimes provide a more appropriate control by mapping the underlying nucleosome distribution, though the differences from WCE may have negligible impact in standard analyses [123].
Recent technological advances have revealed that TF binding is not determined by individual high-affinity sites alone. A 2025 study demonstrated that nucleotides flanking core high-affinity binding sites create overlapping lower-affinity sites that collectively modulate TF genomic occupancy in vivo [122]. This model, where binding is determined by the sum of multiple overlapping sites, transforms our understanding of how single nucleotide variants influence gene expression and disease.
Experimental Design and Workflow
Optimized ChIP-seq Protocol for Transcription Factors
A robust ChIP-seq protocol for transcription factors requires careful optimization at each step:
Crosslinking: Use 1% formaldehyde for 8-15 minutes at room temperature. Quench with 125mM glycine [41]. For some TFs, dual crosslinking with disuccinimidyl glutarate (DSG) prior to formaldehyde may improve efficiency.
Cell Lysis and Chromatin Preparation:
- Prepare cell lysis buffer: 5mM PIPES pH 8, 85mM KCl, 1% igepal (added fresh) with protease inhibitors (PMSF, aprotinin, leupeptin) [41]
- Prepare nuclei lysis buffer: 50mM Tris-HCl pH 8, 10mM EDTA, 1% SDS with protease inhibitors [41]
- Sonicate chromatin to 200-500bp fragments using a focused ultrasonicator (e.g., Covaris or Bioruptor). Optimal conditions must be determined empirically for each cell type [123]
Immunoprecipitation:
- Dilute chromatin 10-fold in IP dilution buffer (50mM Tris-HCl pH 7.4, 150mM NaCl, 1% igepal, 0.25% deoxycholic acid, 1mM EDTA with protease inhibitors) [41]
- Incubate with 1-5μg of validated, ChIP-grade TF-specific antibody overnight at 4°C
- Add protein G beads (Life Technologies) and incubate for 2-4 hours at 4°C [123]
- Wash sequentially with: Low salt buffer, high salt buffer, LiCl buffer, and TE buffer
- Reverse crosslinks by incubating at 65°C for 4 hours [123]
Library Preparation and Sequencing:
- Purify DNA using silica membrane columns (e.g., Zymo ChIP Clean & Concentrator) [123]
- Prepare sequencing libraries using commercial kits (e.g., Illumina TruSeq DNA Sample Prep Kit) [123]
- Sequence on high-throughput platforms (Illumina HiSeq2000/2500 or NovaSeq) aiming for 10-20 million reads per sample for transcription factors [123]
Comparative Workflow: Histone Modifications vs. Transcription Factors
The diagram below illustrates the key similarities and differences in applying ChIP-seq to histone modification versus transcription factor studies.
Critical Reagents and Research Tools
Successful ChIP-seq experiments depend on high-quality, specific reagents. The table below details essential research reagent solutions for transcription factor binding studies.
Table 1: Essential Research Reagents for Transcription Factor ChIP-seq Studies
| Reagent Category | Specific Examples | Function & Importance | Technical Considerations |
|---|---|---|---|
| Validated Antibodies | Anti-CTCF, Anti-POL2, Anti-ELF1, Anti-TAF1 [124] | Target-specific immunoprecipitation; most critical experimental factor | Must be ChIP-grade validated; specificity confirmed by knockout/knockdown controls |
| Crosslinking Reagents | Formaldehyde (37%), Glycine [41] | Presves protein-DNA interactions in living state | Concentration and time optimization required for each TF |
| Chromatin Shearing Systems | Covaris sonicator, Bioruptor (Diagenode) [123] | Fragments chromatin to optimal size (200-500bp) | Settings must be empirically determined for each cell type |
| Protease Inhibitors | PMSF, Aprotinin, Leupeptin [41] | Presves protein integrity during processing | Must be added fresh to all buffers |
| Immunoprecipitation Beads | Protein G beads (Life Technologies) [123] | Captures antibody-bound complexes | Magnetic beads facilitate washing efficiency |
| DNA Purification Kits | ChIP Clean & Concentrator (Zymo) [123] | Purifies immunoprecipitated DNA after crosslink reversal | Column-based methods provide high recovery |
| Library Prep Kits | TruSeq DNA Sample Prep Kit (Illumina) [123] | Prepares sequencing libraries from ChIP DNA | Compatibility with sequencing platform is essential |
Advanced Computational Methods for TFBS Analysis
The expansion of ChIP-seq data has driven the development of sophisticated computational tools specifically designed for transcription factor binding site analysis. These methods have evolved from traditional position weight matrices (PWMs) to advanced deep learning approaches.
Machine Learning and Deep Learning Approaches
Recent advances in deep learning have significantly improved TFBS prediction accuracy. Several model architectures have shown particular promise:
CNN-Based Models: DeepBind utilizes convolutional neural networks to capture local motif patterns in DNA sequences, achieving strong baseline performance with AUC scores around 0.89 in benchmark studies [125]. CNNs are particularly effective at identifying characteristic motif patterns but may have limitations in capturing long-range dependencies.
Transformer and BERT-Based Models: DNABERT-2 and BERT-TFBS apply natural language processing techniques to DNA sequences, using self-attention mechanisms to learn long-term dependencies [121]. These models employ pre-training on large unlabeled DNA datasets followed by fine-tuning on specific TFBS tasks, demonstrating superior performance on diverse binding sites.
Mixture of Experts (MoE): This approach integrates multiple pre-trained CNN models, each specializing in different TFBS patterns, to enhance generalization capability [125]. MoE models have shown particularly strong performance on out-of-distribution (OOD) transcription factors not seen during training.
Benchmarking TFBS Prediction Models
Systematic benchmarking studies have evaluated different computational approaches under various conditions. Key findings include:
Table 2: Performance Comparison of TFBS Prediction Models on ENCODE ChIP-seq Data
| Model Type | Representative Tools | Key Strengths | Performance Characteristics | Limitations |
|---|---|---|---|---|
| Position Weight Matrices | Traditional PWMs | Interpretable, simple implementation | Effective for high-affinity sites; struggles with low-affinity sites [122] | Cannot capture nucleotide dependencies |
| Support Vector Machines | SVM-based models | Good performance with limited data | Benefits from appropriate kernel selection [126] | Limited with complex feature interactions |
| Convolutional Neural Networks | DeepBind, DeepSEA | Excellent motif pattern recognition | AUC ~0.89 on GATA3 prediction [125] | Limited long-range dependency capture |
| Hybrid CNN-RNN Models | DanQ | Combines local and sequential features | Improved over CNN-only models [121] | Computationally intensive |
| Transformer Models | DNABERT-2, BERT-TFBS | Captures long-range dependencies | State-of-the-art on multiple benchmarks [121] | High computational requirements |
| Mixture of Experts | Custom MoE frameworks | Superior generalization to OOD TFs | Competitive or superior in cross-cell-line validation [125] | Complex training process |
The performance of these models is significantly influenced by training dataset size, sequence length, and the use of synthetic versus real biological background data [126]. For optimal results, models should be selected based on specific biological contexts and data availability.
Advanced Applications and Integrative Analysis
PADIT-Seq: Expanding the TF Binding Repertoire
Traditional methods for identifying transcription factor binding specificities have limitations in detecting lower-affinity sites. The recently developed Protein Affinity to DNA by In Vitro Transcription and RNA Sequencing (PADIT-seq) technology comprehensively assays binding preferences across all possible ten-base-pair DNA sequences [122]. This approach has revealed hundreds of novel lower-affinity binding sites that were previously undetected, demonstrating that nucleotides flanking high-affinity core sites create overlapping lower-affinity sites that collectively determine genomic occupancy.
The workflow and significance of this advanced technology is illustrated below:
Integration with Multi-Omics Data
Integrative analysis combining ChIP-seq data with other genomic datasets significantly enhances the biological insights gained from TF binding studies:
TF-HM Co-localization Analysis: Statistical frameworks can quantify the co-localization of transcription factors and histone modifications across cell lines [124]. This approach has revealed that factors like CTCF, SMC3, and RAD21 show consistent co-localization across cell types and play important roles in 3D chromatin structure.
Expression Prediction Models: Support Vector Machine (SVM) models can correlate TF binding and histone modification signals with gene expression levels, achieving prediction accuracies of 85-92% using key factors like H3K9ac, H3K27ac, ELF1, TAF1, and POL2 [124].
Cross-Cell-Line Dynamics: Quantitative indexes such as the total difference index (Dsignal) enable systematic analysis of TF binding dynamics between normal and disease cell lines, revealing both conserved and cell-type-specific regulatory elements [124].
The application of ChIP-seq technologies to transcription factor binding studies has dramatically expanded our understanding of gene regulatory principles. While sharing methodological foundations with histone modification studies, TF binding analyses require specific optimizations in experimental execution, control sample selection, and computational analysis. The emerging paradigm that TF binding is determined by multiple overlapping sites of varying affinities, rather than isolated high-affinity sites, underscores the complexity of transcriptional regulation. Advanced technologies like PADIT-seq and sophisticated computational models, particularly deep learning approaches using transformer architectures and mixture-of-experts frameworks, are pushing the boundaries of what can be discovered from TF binding data. As these methods continue to evolve and integrate with multi-omics approaches, they promise to unlock deeper insights into the regulatory codes governing development, cellular function, and disease, ultimately accelerating drug discovery and therapeutic development.
Conclusion
The integration of ChIP-seq and newer techniques like CUT&Tag has fundamentally advanced our ability to map the epigenetic landscape with precision. Understanding the specific roles of histone modifications, from the potent repression of H3K27me3 to the activating signals of H3K4me3, is no longer a theoretical pursuit but a tractable experimental path. As the field moves forward, the key lies in robust methodological execution, informed by systematic comparisons and optimized protocols. The future of epigenetic research is poised to leverage these tools to not only decode basic mechanisms of gene regulation but also to drive the discovery of novel epigenetic therapies for a wide range of human diseases, from cancer to neurological and psychiatric disorders. The translation of these epigenomic insights into clinical applications represents the next frontier in biomedicine.
References
- 1. Histone modifications [https://www.abcam.com/en-us/technical-resources/guides/epigenetics-guide/histone-modifications]
- 2. Chromatin Modifications and Their Function [https://www.sciencedirect.com/science/article/pii/S0092867407001845]
- 3. Global level quantification of histone post-translational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10042382/]
- 4. Regulation of nucleosome dynamics by histone modifications [https://pubmed.ncbi.nlm.nih.gov/23463310/]
- 5. Histone-modifying enzymes [https://en.wikipedia.org/wiki/Histone-modifying_enzymes]
- 6. Histone Modification Table [https://www.cellsignal.com/learn-and-support/reference-tables/histone-modification-table?srsltid=AfmBOoqsDGiTSbutBY6_Qg2HYVe3-nx-TaRqEJ1scZWuWDuxggODXymF]
- 7. The Functions and Regulatory Mechanisms of Histone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12027200/]
- 8. Addressing the specific roles of histone modifications in ... [https://www.nature.com/articles/s41467-025-66426-z]
- 9. Guidelines published on new technique to study gene ... [https://www.imperial.ac.uk/news/262654/guidelines-published-technique-study-gene-regulation/]
- 10. Histone Modification Studies [https://www.epigentek.com/catalog/histone-modification.php?srsltid=AfmBOoqtuv-cutjoPkyKEDw36VvuX-2aFSNan3BjwKXgxAFOZRBd_wVF]
- 11. H3K4me3 amplifies transcription at intergenic active ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12581817/]
- 12. CRISPR targeting of H3K4me3 activates gene expression ... [https://www.nature.com/articles/s41467-025-65167-3]
- 13. Analysis of histone modification interplay reveals two ... [https://www.nature.com/articles/s42003-025-08473-2]
- 14. ChIP-seq assay revealed histone modification H3K9ac ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969722002583]
- 15. Machine learning on multiple epigenetic features reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12352877/]
- 16. H3K79 methylation and H3K36 trimethylation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12227068/]
- 17. Histone ChIP-seq Data Standards and Processing Pipeline [https://www.encodeproject.org/chip-seq/histone/]
- 18. Joint analysis of chromatin accessibility and gene ... [https://www.sciencedirect.com/science/article/pii/S2405471225000997]
- 19. mRNA m6A regulates gene expression via H3K4me3 shift in 5 ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03515-8]
- 20. Recruitment and Biological Consequences of Histone of... Modification [https://pmc.ncbi.nlm.nih.gov/articles/PMC3747788/]
- 21. Dot1 and Histone H3K79 Methylation in Natural Telomeric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3085244/]
- 22. Mechanisms of histone H3 lysine 27 trimethylation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3515017/]
- 23. The control of gene expression and cell identity by H3K9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6803365/]
- 24. H3K9me3-Dependent Heterochromatin: Barrier to Cell ... [https://www.sciencedirect.com/science/article/abs/pii/S0168952515001961]
- 25. Comprehensive analysis of H LOCKs under different DNA... 3 K 27 me 3 [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-025-00570-0]
- 26. H3K27me3-rich genomic regions can function as silencers ... [https://www.nature.com/articles/s41467-021-20940-y]
- 27. Haplotype mapping of H3K27me3-associated chromatin ... [https://www.sciencedirect.com/science/article/pii/S2211124723003613]
- 28. H3K27me3 is vital for fungal development and secondary ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1011075]
- 29. -wide profiling of Genome 4 histone and... H 3 K me 3 [https://www.nature.com/articles/s41598-022-15417-x?error=cookies_not_supported&code=71030325-e3a0-4b15-8804-aa2f9b36c71f]
- 30. Quantitative Proteomic Analysis of Histone Modifications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4502928/]
- 31. Histone Modifications in Stem Cell Development and Their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7724464/]
- 32. Streamlined quantitative analysis of histone modification ... [https://www.nature.com/articles/s41598-023-34430-2]
- 33. Genome-coverage single-cell histone modifications for ... [https://www.nature.com/articles/s41586-025-08656-1]
- 34. Quantitative analysis reveals increased histone ... [https://www.sciencedirect.com/science/article/pii/S0888754312002273]
- 35. A quantitative atlas of histone modification signatures from ... [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/1756-8935-6-20]
- 36. Histone content increases in differentiating embryonic stem ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2014.00330/full]
- 37. Epigenetics in Neurological and Psychiatric Disorders [https://pmc.ncbi.nlm.nih.gov/articles/PMC10446850/]
- 38. Epigenetics and Addiction - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6889055/]
- 39. Epigenetic mechanisms in neurodevelopmental and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3596876/]
- 40. Epigenetics of Stress, Addiction, and Resilience: Therapeutic Implications | Molecular Neurobiology [https://link.springer.com/article/10.1007/s12035-014-9040-y]
- 41. Using ChIP-Seq Technology to Generate High-Resolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4151291/]
- 42. ChIP-Seq Analysis: Histone Modifications [https://www.news-medical.net/life-sciences/ChIP-Seq-Analysis-Histone-Modifications.aspx]
- 43. Frontiers | DNA Epigenetics in Addiction Susceptibility [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2022.806685/full]
- 44. The Epigenome in Neurodevelopmental Disorders [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2021.776809/full]
- 45. Ageing and epigenetics : linking neurodevelopmental and... [https://pubmed.ncbi.nlm.nih.gov/30883719/]
- 46. of Epigenetics , Neuromuscular and... Neurodevelopmental [https://www.frontiersin.org/research-topics/15719/epigenetics-of-neurodevelopmental-neuromuscular-and-neurodegenerative-disorders/magazine]
- 47. Application of ChIP-Seq and related techniques to the study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3137373/]
- 48. The Use of ChIP-seq in Drug Discovery [https://dovetailbiopartners.com/2023/09/26/the-use-of-chip-seq-in-drug-discovery/]
- 49. Cross-linking ChIP-seq protocol [https://www.abcam.com/en-us/technical-resources/protocols/cross-linking-chip-seq]
- 50. Chromatin immunoprecipitation - Wikipedia [https://en.wikipedia.org/wiki/Chromatin_immunoprecipitation]
- 51. A Step-by-Step Guide to Successful Chromatin ... [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/antibody-application-notes/step-by-step-guide-successful-chip-assays.html]
- 52. ChIP-Seq Workflow Template [https://bioconductor.org/packages/devel/data/experiment/vignettes/systemPipeRdata/inst/doc/systemPipeChIPseq.html]
- 53. ChIP-seq data processing tutorial [https://nbis-workshop-epigenomics.readthedocs.io/en/latest/content/tutorials/chipseqProc/lab-chipseq-processing.html]
- 54. Methods for ChIP-seq analysis: A practical workflow and ... [https://www.sciencedirect.com/science/article/pii/S1046202320300591]
- 55. ChIP-seq guidelines and practices of the ENCODE and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3431496/]
- 56. Application of histone modification-specific interaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4216925/]
- 57. Specificity Analysis of Antibodies That Recognize Histone ... [https://www.thermofisher.com/us/en/home/references/newsletters-and-journals/bioprobes-journal-of-cell-biology-applications/bioprobes-78/antibody-specificity-histone-posttranslational-modifications.html]
- 58. Histone modification antibodies [https://www.abcam.com/en-us/technical-resources/product-overview/histone-modification-antibodies]
- 59. Mapping epigenetic modifications by sequencing ... [https://www.nature.com/articles/s41418-023-01213-1]
- 60. High-throughput PacBio library preparation and ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1587691/full]
- 61. Library Preparation for Genome-Wide DNA Methylation ... [https://pubmed.ncbi.nlm.nih.gov/41220974/?dopt=abstract]
- 62. Recent advances in ChIP-seq analysis - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC5444249/]
- 63. ChIP-Seq Analysis Tutorial [https://www.basepairtech.com/knowledge-center/chip-seq-analysis-tutorial/]
- 64. ChIP-Seq Data Analysis - CRCD User Manual - Pitt CRC [https://crc-pages.pitt.edu/user-manual/advanced-genomics-support/ChipSeq-data-analysis/]
- 65. cChIP-seq: a robust small-scale method for investigation of ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-2285-7]
- 66. Pipeline and Tools for ChIP-Seq Analysis [https://www.cd-genomics.com/pipeline-and-tools-comparison-for-chip-seq-analysis.html]
- 67. A Statistical Framework for the Analysis of ChIP-Seq Data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4608541/]
- 68. RNA-seq and ChIP-seq as Complementary Approaches for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6981605/]
- 69. Understanding gene regulatory mechanisms by integrating ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2014.00051/full]
- 70. PTMViz: a tool for analyzing and visualizing histone post ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04166-9]
- 71. Analysis of histone modification interplay reveals two ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12284244/]
- 72. Differential expression, manipulation, and visualization of RNA - seq ... [https://www.bioconductor.org/help/course-materials/2015/BioC2015/bioc2015rnaseq.html]
- 73. Refined ChIP‐Seq Protocol for High‐Quality Chromatin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12628287/]
- 74. Chromatin Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/chip-troubleshooting-guide?srsltid=AfmBOopfYd8T-X7haVasS4YQ9IHqHqar4CstF28Kzjj2IIx3heRWl-Cv]
- 75. An Optimized Chromatin Immunoprecipitation Protocol for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7357673/]
- 76. Analysis of histone antibody specificity directly in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10028865/]
- 77. ChIP-Seq: Technical Considerations for Obtaining High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3541830/]
- 78. An interactive database for the assessment of histone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4530063/]
- 79. Detailed specificity analysis of antibodies binding to modified ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3230550/]
- 80. Validation of Histone Modification Antibodies [https://www.cellsignal.com/about-us/our-approach-process/validation-histone-modification?srsltid=AfmBOoojGMJeY9SAfsR_D0dmmOfB4kSAjq7eXpwDCFT6oPikRO71x33D]
- 81. ChIP–qPCR Validated Antibodies [https://www.cellsignal.com/applications/chip-and-chip-seq/chip-qpcr-validated-antibodies?srsltid=AfmBOopmsjto_uZAsOhc0kv4R111I_YO9cNC6fvL7ag-d0T2xWEtUOE_]
- 82. Antibodies for Chromatin Immunoprecipitation (ChIP) [https://www.thermofisher.com/us/en/home/life-science/antibodies/primary-antibodies/antibodies-applications/antibodies-chip.html]
- 83. ChIP-seq Validated Antibodies [https://www.cellsignal.com/applications/chip-and-chip-seq/chip-seq-antibodies?srsltid=AfmBOooe4I-Vn5uT0veNrc_tycWqq1TFudcn420JX3ZJHkw4tgvasvvb]
- 84. Comparative analysis of commonly used peak calling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7808876/]
- 85. Comprehensive assessment of differential ChIP-seq tools ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02686-y]
- 86. Evaluating the Performance of Peak Calling Algorithms ... [https://www.mdpi.com/1422-0067/26/3/1268]
- 87. GoPeaks: histone modification peak calling for CUT&Tag [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02707-w]
- 88. H3NGST: a fully automated, web-based platform for end-to ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06247-5]
- 89. GPU peak calling for epigenetics [https://blog.latch.bio/p/gpu-peak-calling-for-epigenetics]
- 90. Peak-calling Algorithms (WonderPeaks and PeakStream ... [https://pubmed.ncbi.nlm.nih.gov/40853857/]
- 91. Benchmark of chromatin–protein interaction methods in ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1572405/full]
- 92. CUT&Tag recovers up to half of ENCODE ChIP-seq ... [https://www.nature.com/articles/s41467-025-58137-2]
- 93. Evaluating histone modification analysis of individual ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-09984-8]
- 94. Multiplexed chromatin immunoprecipitation sequencing for ... [https://pubmed.ncbi.nlm.nih.gov/39363107/]
- 95. Highly quantitative measurement of differential protein ... [https://www.sciencedirect.com/science/article/pii/S2667237525000888]
- 96. Optimized CUT&RUN protocol for activated primary mouse B ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0322139]
- 97. a robust model for quantitative comparison of ChIP-Seq data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3439967/]
- 98. Histone Modifications as Individual-Specific Epigenetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12385217/]
- 99. A comprehensive review of histone modifications during ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12206194/]
- 100. Single-cell multi-omic detection of DNA methylation and ... [https://www.nature.com/articles/s41592-025-02847-4]
- 101. IT-scC&T-seq streamlines scalable, parallel profiling of protein ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03661-z]
- 102. a compatible library construction method of single-cell ... [https://pubs.rsc.org/en/content/articlelanding/2025/mo/d4mo00111g]
- 103. Single-cell omics sequencing technologies: the long-read ... [https://www.sciencedirect.com/science/article/pii/S0168952525001969]
- 104. The development and application of cleavage under ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11557298/]
- 105. CUT&Tag vs ChIP-seq: 5 Key Tips for Epigenetics Studies [https://www.cd-genomics.com/epigenetics/resource-cut-tag-chip-seq-comparison.html]
- 106. CUT&Tag recovers up to half of ENCODE ChIP-seq peaks [https://db.cngb.org/data_resources/project/PRJNA820826]
- 107. CUT&Tag overcomes biases of ChIP and establishes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12605858/]
- 108. Simultaneous profiling of multiple chromatin proteins in the ... [https://www.sciencedirect.com/science/article/pii/S109727652100753X]
- 109. Recent progress in histone post-translational modifications ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813025085216]
- 110. Targeting epigenetic regulators as a promising avenue to ... [https://www.nature.com/articles/s41392-025-02266-z]
- 111. Sequencing: Unlocking ChIP Gene Regulation [https://www.numberanalytics.com/blog/chip-sequencing-gene-regulation]
- 112. Chromatin Immunoprecipitation Sequencing ( ChIP - seq ) for Detecting... [https://pubmed.ncbi.nlm.nih.gov/36173565/]
- 113. a fully automated, web-based platform for end-to-end ChIP- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12512806/]
- 114. High-Resolution Chromatin Dynamics during a Yeast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4405355/]
- 115. Nucleosomal asymmetry shapes histone mark binding and ... [https://www.sciencedirect.com/science/article/pii/S1097276524009973]
- 116. histoneHMM: Differential analysis of histone modifications with ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-015-0491-6]
- 117. A novel statistical method for quantitative comparison of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4542775/]
- 118. Combinatorial profiling of multiple histone modifications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12327464/]
- 119. Inference of interactions between chromatin modifiers and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4267652/]
- 120. Comprehensive Mapping of Histone Modifications at DNA ... [https://www.sciencedirect.com/science/article/pii/S1097276518306804]
- 121. BERT-TFBS: a novel BERT-based model for predicting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11066948/]
- 122. Multiple overlapping binding sites determine transcription ... [https://www.nature.com/articles/s41586-025-09472-3]
- 123. A comparison of control samples for ChIP-seq of histone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4174756/]
- 124. Revealing transcription factor and histone modification co ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-5278-5]
- 125. Explainable AI in Genomics: Transcription Factor Binding ... [https://arxiv.org/html/2507.09754v1]
- 126. Benchmarking transcription factor binding site prediction ... [https://pubmed.ncbi.nlm.nih.gov/40702706/]