Creating Interactive Gene Expression Heatmaps with heatmaply: A Complete Guide for Biomedical Researchers
This comprehensive guide provides researchers, scientists, and drug development professionals with both theoretical foundations and practical implementation strategies for creating interactive gene expression heatmaps using the R package heatmaply.
Creating Interactive Gene Expression Heatmaps with heatmaply: A Complete Guide for Biomedical Researchers
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with both theoretical foundations and practical implementation strategies for creating interactive gene expression heatmaps using the R package heatmaply. Covering everything from basic installation to advanced customization, the article explores how interactive heatmaps can reveal patterns in high-dimensional biological data, facilitate cluster analysis, and serve as diagnostic tools in sequencing experiments. Readers will learn to transform raw count data into publication-quality visualizations, troubleshoot common issues, and understand how heatmaply compares to alternative heatmap tools in the R ecosystem for effective data communication in biomedical research.
Understanding Interactive Heatmaps: Why Gene Expression Visualization Matters
What Are Heatmaps and Dendrograms? Core Concepts for Biological Data
A heatmap is a graphical representation of data where individual values contained in a matrix are represented as colors [1]. This technique makes it easy to visualize complex data at a glance, as color is often easier to interpret and distinguish than raw numerical values [1]. In biological research, heatmaps are extensively used to visualize data such as gene expression across samples, correlation matrices, and disease case distributions [1] [2].
A dendrogram, or tree diagram, is a network structure used to visualize hierarchy or clustering in data [1]. These tree-like diagrams illustrate the arrangement of clusters produced by hierarchical clustering analysis, showing the relationships between similar data points [3]. When combined with heatmaps, dendrograms reveal natural groupings in the data that might not be immediately apparent through other analytical methods [3].
Clustered Heat Maps (CHMs) represent the integration of these two techniques, combining heat mapping with hierarchical clustering to reveal patterns and relationships in complex datasets [3]. This powerful visualization approach has become indispensable in biological research, particularly for analyzing high-dimensional data generated by modern molecular biology techniques such as RNA sequencing, metabolomics, and proteomics [3].
Key Concepts and Components
Fundamental Elements of Clustered Heatmaps
- Heat Map Matrix: The main grid where each cell's color represents data values, with rows typically representing observations (e.g., genes) and columns representing features or samples (e.g., experimental conditions) [3].
- Dendrogram: Tree-like structures showing hierarchical clustering of rows and columns, displaying the relationships and similarities between data points [3].
- Row and Column Labels: Identifiers for data points, such as gene names or sample IDs, which are essential for interpreting the biological significance of patterns [1] [3].
- Color Legend: A scale that maps color intensities to data values, allowing quantitative interpretation of the color representations in the heatmap [1] [4].
Distance, Clustering, and Scaling
The construction of meaningful clustered heatmaps relies on three critical computational parameters [1]:
- Distance Calculation: The method for calculating similarity or dissimilarity between data points. Common metrics include Euclidean distance (straight-line distance between points) and Pearson correlation (measures linear relationship between variables) [1] [3]. The choice of distance metric significantly influences the clustering results [3].
- Clustering Algorithm: The method for grouping similar objects together based on the calculated distances. Hierarchical clustering is most commonly used, which can be agglomerative (bottom-up, merging small clusters) or divisive (top-down, splitting large clusters) [1] [3]. The linkage method (e.g., complete, average, or single linkage) determines how distances between clusters are calculated [1].
- Data Scaling: The process of standardizing data to ensure comparability, particularly when variables have different units or scales. Z-score normalization is frequently employed, which transforms values based on their relationship to the mean and standard deviation of the dataset [1]. This prevents variables with large values from disproportionately influencing the analysis and ensures patterns in variables with lower values remain visible [1].
Table 1: Common Distance Metrics and Clustering Methods
| Category | Method | Description | Typical Use Case |
|---|---|---|---|
| Distance Metrics | Euclidean Distance | Straight-line distance between points in multidimensional space | General purpose clustering |
| Pearson Correlation | Measures linear relationship between variables | Gene expression patterns | |
| Manhattan Distance | Sum of absolute differences between coordinates | High-dimensional data | |
| Clustering Algorithms | Hierarchical Clustering | Creates a tree of clusters using linkage methods | Most biological applications |
| k-means Clustering | Partitioning method that requires pre-specified k | Large datasets with known clusters | |
| Linkage Methods | Complete Linkage | Distance between clusters = farthest neighbor distance | Compact, evenly sized clusters |
| Average Linkage | Distance between clusters = average of all pairwise distances | Balanced approach | |
| Single Linkage | Distance between clusters = closest neighbor distance | Elongated, chain-like clusters |
Experimental Protocol: Creating an Interactive Gene Expression Heatmap with Heatmaply
Materials and Software Requirements
Table 2: Research Reagent Solutions and Computational Tools
| Item | Function/Description | Example/Note |
|---|---|---|
| Normalized Gene Expression Data | Matrix of expression values (e.g., log2 CPM, TPM) | Typically from RNA-seq or microarray experiments |
| R Statistical Software | Programming environment for data analysis | Version 4.0.0 or higher recommended |
| heatmaply R Package | Generates interactive cluster heatmaps | Enables zooming and value inspection via hovering |
| pheatmap R Package | Produces publication-quality static heatmaps | Highly customizable with automatic scaling |
| ggplot2 R Package | Grammar of graphics for data visualization | Used for additional plot customization |
| ColorBrewer Palettes | Color-blind friendly color schemes | Accessed through RColorBrewer package |
Step-by-Step Protocol
Step 1: Software Environment Preparation
Begin by installing and loading the required R packages. Execute the following code in your R environment:
Step 2: Data Import and Preprocessing
Import your gene expression data, typically stored in a comma-separated values (CSV) file. Ensure the first column contains gene identifiers and subsequent columns represent samples:
Step 3: Data Scaling and Normalization
Apply Z-score standardization to normalize expression values across genes, enabling meaningful comparison of expression patterns:
Step 4: Color Scheme Selection
Select appropriate color palettes for your data type. For gene expression data with positive and negative values (e.g., Z-scores), use a diverging color scale. For strictly positive values (e.g., expression counts), use a sequential scale [4]:
Step 5: Generate Interactive Heatmap with Heatmaply
Create an interactive clustered heatmap with customizable clustering parameters:
Step 6: Generate Publication-Quality Static Heatmap
Create a high-resolution static version for publications using pheatmap:
Step 7: Interpretation and Analysis
Interpret the resulting visualization by examining:
- Sample Clustering: Check if samples from the same experimental conditions or phenotypes cluster together in the column dendrogram.
- Gene Clustering: Identify groups of genes with similar expression patterns across samples in the row dendrogram.
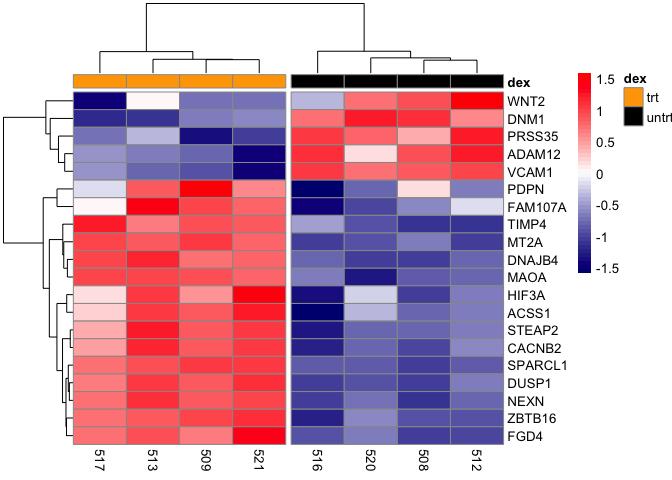
- Expression Patterns: Look for coordinated up-regulation (red shades) or down-regulation (blue shades) of gene clusters in specific sample groups.
- Cluster Validation: Use statistical methods (e.g., silhouette width) or biological validation to confirm the significance of identified clusters.
Applications in Biological Research and Drug Development
Clustered heatmaps with dendrograms have become fundamental tools across multiple domains of biological research and pharmaceutical development [3]:
- Gene Expression Studies: Identification of co-expressed gene clusters across different conditions, tissues, or disease states, enabling discovery of biomarker signatures and potential drug targets [3]. For example, The Cancer Genome Atlas (TCGA) projects extensively use heatmaps to classify cancer subtypes based on molecular profiles [3].
- Patient Stratification: Classification of patients into molecularly distinct subgroups based on genomic, transcriptomic, or proteomic profiles, facilitating personalized treatment approaches and clinical trial design [3].
- Compound Screening Analysis: Visualization of high-throughput drug screening results, clustering compounds by their activity profiles across multiple cell lines or targets to identify promising lead compounds and mechanism of action [2].
- Toxicogenomics: Assessment of drug-induced toxicity patterns by clustering gene expression responses to various compounds, predicting potential adverse effects early in drug development [3] [2].
- Pathway Analysis: Integration of clustered expression data with pathway databases to identify activated or suppressed biological processes in response to treatments or disease states [3].
Best Practices and Troubleshooting
Color Scale Selection Guidelines
Table 3: Color Scale Recommendations for Different Data Types
| Data Type | Recommended Scale | Rationale | Example Applications |
|---|---|---|---|
| Expression Z-scores | Diverging (Blue-White-Red) | Clear visualization of up/down regulation | Differential expression analysis |
| Raw Expression Values | Sequential Single-Hue | Intuitive for low-to-high progression | Expression level comparisons |
| Correlation Coefficients | Diverging (Blue-White-Red) | Natural inflection at zero | Correlation matrices |
| Statistical Significance | Sequential (White to Dark) | Emphasizes strength of effect | p-value or enrichment displays |
Optimization Strategies
- Color Selection: Avoid rainbow color scales and red-green combinations that are problematic for color-blind users [4]. Instead, use color-blind friendly palettes like blue-orange or blue-red [4].
- Data Scaling: Always scale your data appropriately before heatmap generation. For gene expression data, row-wise scaling (by gene) is typically most biologically meaningful as it highlights relative expression patterns across samples [1].
- Cluster Validation: Remember that clusters identified in a heatmap represent patterns of similarity but do not necessarily imply biological relevance or causation [3]. Always validate clusters using statistical methods or experimental approaches.
- Handling Large Datasets: For datasets with numerous genes or samples, consider filtering to the most variable features prior to heatmap generation to reduce visual clutter and computational demands.
- Interactive Features: Leverage the interactive capabilities of heatmaply, including zooming for detailed inspection of specific regions and hovering to display exact values, gene names, and sample information [5] [6].
Common Issues and Solutions
- Poor Cluster Separation: Try alternative distance metrics (e.g., correlation instead of Euclidean distance) or clustering methods (e.g., average linkage instead of complete linkage).
- Color Interpretation Difficulties: Ensure your color legend clearly indicates the value range and check that extreme values are not saturated.
- Uninformative Clustering: Pre-filter genes by variance or significance to remove uninformative features that may obscure meaningful patterns.
- Large Dataset Performance: For datasets with thousands of genes, use specialized packages like ComplexHeatmap that optimize handling of large matrices [7].
In the field of genomics and drug development, the ability to visually interrogate complex datasets is paramount. Gene expression studies, which simultaneously measure the activity of thousands of genes across multiple experimental conditions, present a particular challenge for data visualization and interpretation. Static heatmaps have long served as a fundamental tool for representing this high-dimensional data as grids of colored cells, where expression levels are encoded by color intensity [8]. However, these traditional visualizations suffer from inherent limitations—fixed resolution obscures fine details, and the underlying numerical values remain hidden from immediate view.
The introduction of interactive heatmaps represents a transformative advancement in biological data exploration. Tools like the heatmaply R package have empowered researchers to move beyond passive observation to active investigation through three critical interactive features: hover for instant value inspection, zoom for focused region analysis, and dynamic exploration for pattern discovery [5] [9]. These capabilities are particularly valuable in gene expression analysis, where identifying subtle expression patterns, verifying specific gene behaviors, and communicating findings to collaborative teams can directly impact research outcomes and therapeutic development decisions.
This protocol details the implementation of interactive heatmaps for gene expression analysis using the heatmaply package, providing researchers with a structured methodology to enhance their data exploration processes and extract more meaningful insights from their experimental data.
Research Reagent Solutions
Table 1: Essential computational tools and their functions for creating interactive heatmaps.
| Tool Name | Category | Primary Function |
|---|---|---|
| heatmaply R Package | Main Software | Generates interactive cluster heatmaps with hover and zoom functionality [5] |
| ggplot2 | Visualization Engine | Provides foundational plotting system for heatmap construction [5] |
| plotly.js | Interactive Graphics | Enables client-side interactivity including hovering and zooming [5] |
| dendextend | Dendrogram Manipulation | Customizes clustering trees with branch coloring and rotation [9] |
| seriation | Matrix Ordering | Optimizes row/column arrangement to highlight patterns [9] |
| RColorBrewer | Color Schemes | Provides colorblind-friendly palettes for data representation [9] |
| viridis | Color Palettes | Offers perceptually uniform color scales [8] |
The following diagram illustrates the comprehensive workflow for creating and analyzing interactive gene expression heatmaps, from data preparation through interactive exploration and interpretation.
Equipment and Software Setup
Computational Environment Specifications
A standard desktop or laptop computer with the following specifications is sufficient for most gene expression datasets:
- Operating System: Windows 10+, macOS 10.14+, or Linux Ubuntu 18.04+
- Memory: Minimum 8 GB RAM (16+ GB recommended for large transcriptomic datasets)
- R Version: R 4.0.0 or newer
- RStudio: Version 1.4 or newer (recommended for integrated viewing)
Software Installation Protocol
Install Core Dependencies: Begin by installing fundamental statistical and graphical packages required for data manipulation and visualization:
Install heatmaply Package: Install the main interactive heatmap package from CRAN:
Verify Installation: Confirm successful installation and load the package:
Install Supplementary Packages (Optional): For specialized genomic analyses, additional bioconductor packages may be required:
Data Preparation Methods
Data Transformation Procedures
Gene expression data requires appropriate transformation to ensure meaningful visual comparisons across genes with potentially different expression ranges. The choice of transformation method depends on the biological question and data characteristics.
Table 2: Data transformation methods for gene expression analysis.
| Method | Formula | Application Context | Advantages |
|---|---|---|---|
| Scaling | ( X_{\text{scaled}} = \frac{X - \mu}{\sigma} ) | Normally distributed data | Standardizes to Z-scores; comparable units |
| Normalization | ( X{\text{norm}} = \frac{X - X{\min}}{X{\max} - X{\min}} ) | Non-normal distributions | Preserves distribution shape; [0,1] range |
| Percentile | ( X_{\text{percentile}} = \frac{\text{rank}(X)}{n} ) | Ordinal data or ties | Robust to outliers; intuitive interpretation |
| Square Root | ( X_{\text{sqrt}} = \sqrt{X} ) | Count-based data (RNA-seq) | Stabilizes variance of count data |
Protocol: Data Preprocessing for RNA-seq Expression Data
Load Expression Matrix: Import your gene expression data, typically as a data frame or matrix with genes as rows and samples as columns:
Apply Appropriate Transformation: Select and apply the most suitable transformation method based on your data characteristics. For RNA-seq count data:
Filter Low-Expressed Genes: Remove genes with minimal expression across samples to reduce noise:
Verify Data Integrity: Check the dimensions and range of the processed data:
Interactive Heatmap Generation
Core Visualization Protocol
Basic Interactive Heatmap Generation: Create a fundamental interactive heatmap with default parameters:
Advanced Configuration with Customization: Implement a highly customized heatmap with controlled clustering and coloring:
Correlation Heatmap for Sample Relationships: Visualize correlations between samples rather than direct expression values:
Hover Text Customization Protocol
Enhance the interactive hover functionality by adding contextual biological information:
Prepare Annotation Data: Create a matrix of hover text with the same dimensions as your expression matrix:
Generate Heatmap with Custom Hover Information: Incorporate the hover text into the visualization:
Interactive Exploration Framework
Hover Inspection Protocol
The hover functionality transforms static visualization into an interactive query system, enabling researchers to extract precise numerical values and annotations directly from the heatmap.
- Position the cursor over any cell in the heatmap to activate the hover tooltip
- Observe the displayed information which includes:
- Row (gene) and column (sample) identifiers
- Exact numerical value of the cell
- Additional biological annotations (if configured)
- Compare multiple values by hovering across different conditions for the same gene
- Identify expression trends by hovering along a row (gene) or column (sample)
Zoom and Pan Navigation Protocol
Large gene expression matrices often contain more information than can be effectively displayed at once. The zoom functionality enables focused analysis of specific gene sets or sample groups.
Region Selection Zoom:
- Click and drag to draw a rectangle around the region of interest
- The display will automatically zoom to the selected area
- Use this to focus on clusters of co-expressed genes or sample subgroups
Dendrogram-Based Zoom:
- Click on dendrogram branches to zoom to specific clusters
- Particularly useful when combined with branch coloring (krow/kcol parameters)
Navigation Controls:
- Use the plotly toolbar to reset zoom, pan across the visualization, or adjust display options
- Return to the full view using the "Reset axes" option in the toolbar
Pattern Discovery Protocol
Cluster Identification:
- Observe dendrogram branching patterns to identify natural groupings in the data
- Note the height of branch points as an indicator of cluster stability
- Use branch coloring (via krow/kcol parameters) to emphasize predefined clusters
Expression Pattern Recognition:
- Scan for consistent color patterns across sample groups
- Identify genes with similar expression profiles by examining row patterns
- Detect sample outliers by examining column patterns that deviate from group norms
Cross-Validation with Biological Context:
- Correlate expression patterns with known biological pathways
- Validate cluster compositions using gene ontology information
- Compare with experimental metadata (e.g., treatment groups, time points)
Advanced Applications
Missing Data Visualization Protocol
Identify and visualize patterns in missing data, which is particularly common in large-scale genomic studies:
Publication-Quality Export Protocol
Generate shareable, publication-ready outputs from interactive heatmaps:
HTML Export for Supplementary Materials:
Static Image Export for Manuscripts:
Embedding in R Markdown Documents:
Interactive Heatmap for Time-Series Expression Data
Specialized protocol for visualizing temporal expression patterns:
Troubleshooting and Optimization
Performance Optimization for Large Datasets
Visualization of genome-scale datasets (e.g., >10,000 genes) requires optimization strategies:
Data Subsetting Approaches:
- Filter to top variable genes:
top_genes <- names(sort(apply(expression_data, 1, sd), decreasing = TRUE)[1:2000]) - Focus on statistically significant genes from differential expression analysis
- Pre-cluster data and select cluster representatives
- Filter to top variable genes:
Computational Efficiency Settings:
Common Issues and Solutions
Table 3: Troubleshooting guide for interactive heatmap generation.
| Problem | Potential Cause | Solution |
|---|---|---|
| Blank heatmap output | Missing or infinite values | Apply data[is.infinite(data)] <- NA and filter complete cases |
| Poor color contrast | Unsuitable color palette | Use colors = viridis(256) for perceptually uniform scaling |
| Uninformative clustering | Inappropriate distance metric | Test dist_method = "euclidean", "correlation", or "manhattan" |
| Slow rendering | Large matrix size | Implement data subsetting or use plot_method = "plotly" |
| Overlapping labels | Too many rows/columns | Set showticklabels = c(FALSE, FALSE) or use label grouping |
Interactive heatmaps represent a significant advancement over traditional static visualizations for gene expression analysis. The implementation of hover, zoom, and exploration capabilities transforms the analytical process from passive observation to active investigation, enabling researchers to uncover subtle patterns, verify specific gene behaviors, and generate more reliable biological insights.
The protocols outlined in this document provide a comprehensive framework for implementing interactive heatmaps in genomic research and drug development contexts. By following these standardized methodologies, research teams can enhance their analytical capabilities, improve reproducibility, and accelerate the translation of genomic data into biological understanding and therapeutic applications.
The true power of these interactive approaches emerges not merely from the individual technical capabilities, but from their integrated implementation within a coherent analytical workflow—enabling researchers to ask more nuanced questions of their data and to discover meaningful biological stories that might otherwise remain hidden in numerical matrices.
Application Note: Interactive Gene Expression Visualization
Purpose and Scope
Interactive heatmaps are indispensable tools in biomedical research for visualizing high-dimensional gene expression data. These visualizations transform complex expression matrices into colored grids where rows typically represent genes and columns represent samples or experimental conditions, enabling researchers to quickly identify patterns of co-expression, sample clustering, and potential outliers [10]. The heatmaply R package extends these capabilities by creating interactive visualizations that allow direct inspection of values via mouse hover and zooming into specific regions, facilitating deeper exploration of large datasets common in genomics and transcriptomics [11] [10].
Key Applications in Biomedical Research
- Gene Expression Analysis: Visualize expression levels across multiple samples or experimental conditions to identify upregulated and downregulated genes [10].
- Sample Correlation: Assess biological replicates and experimental consistency through correlation patterns between samples.
- Cluster Diagnostics: Validate clustering results and identify potential misclassifications or novel subgroups through dendrogram inspection.
- Spatial Transcriptomics: Explore spatial gene expression patterns in tissue sections, complementing emerging technologies in spatial biology [12].
- Quality Control: Identify technical artifacts, batch effects, or outlier samples that may affect downstream analysis.
Quantitative Performance Metrics
Table 1: Key performance metrics for evaluating gene expression patterns in biomedical research
| Metric Category | Specific Metrics | Application Context | Typical Values |
|---|---|---|---|
| Predictive Performance | Pearson Correlation Coefficient (PCC) | Gene expression prediction accuracy | 0.2-0.5 [12] |
| Mutual Information (MI) | Information content in predicted patterns | ~0.06 [12] | |
| Structural Similarity Index (SSIM) | Spatial pattern preservation | 0.2-0.65 [12] | |
| Biological Relevance | Highly Variable Genes (HVG) | Identification of biologically relevant genes | p<0.05 [12] |
| Spatially Variable Genes (SVG) | Detection of spatial expression patterns | p<0.05 [12] | |
| Visual Quality | Color Contrast Ratio | Accessibility compliance | ≥3:1 [13] |
Experimental Protocols
Protocol 1: Basic Interactive Heatmap Creation with Heatmaply
Materials and Reagents
Table 2: Essential computational tools and packages
| Tool/Package | Function | Installation Command |
|---|---|---|
| R Statistical Environment | Base computational platform | https://cran.r-project.org/ |
| heatmaply package | Interactive heatmap creation | install.packages('heatmaply') |
| ggplot2 | Underlying graphics system | install.packages('ggplot2') |
| plotly | Interactive visualization engine | install.packages('plotly') |
Step-by-Step Procedure
Environment Setup: Install and load required packages in R:
Data Preparation: Load and preprocess gene expression data (e.g., RNA-seq count data, microarray intensities). Normalize data using appropriate methods (e.g., TPM for RNA-seq, RMA for microarrays).
Basic Heatmap Generation: Create an interactive heatmap with default parameters:
Enhanced Clustering: Generate a heatmap with predefined cluster numbers:
Output Saving: Export the interactive visualization as an HTML file:
Quality Control Checks
- Verify that the color scale adequately represents expression ranges
- Confirm that dendrograms appropriately reflect sample relationships
- Ensure interactive features (hover, zoom) function correctly in output file
- Check that all samples and genes are properly labeled
Protocol 2: Advanced Cluster Diagnostics and Validation
Procedure
Data Clustering: Perform hierarchical clustering on both rows (genes) and columns (samples) using Euclidean distance and complete linkage.
Cluster Determination: Utilize the Gap Statistic or silhouette width to determine optimal cluster numbers.
Visual Validation: Inspect cluster stability through interactive dendrogram manipulation in the heatmaply output.
Biological Interpretation: Correlate identified clusters with known biological annotations (e.g., pathway enrichment, disease subtypes).
Interpretation Guidelines
- Strong Clusters: Tightly grouped branches with high bootstrap values
- Marginal Groupings: Weakly supported branches that may represent transitional states
- Outliers: Single elements distant from main clusters that may represent technical artifacts or biologically distinct entities
Visualization Workflows
Heatmap Creation and Validation Workflow
Sample Correlation Analysis Workflow
Research Reagent Solutions
Table 3: Essential computational reagents for interactive heatmap analysis
| Reagent Type | Specific Tool/Package | Function in Analysis |
|---|---|---|
| Programming Environment | R Statistical Software | Base platform for statistical computing and graphics |
| RStudio IDE | Integrated development environment for R | |
| Visualization Packages | heatmaply | Primary interactive heatmap generation [10] |
| ggplot2 | Underlying graphics system for static plots | |
| plotly | Interactive visualization engine | |
| Data Manipulation | dplyr | Data wrangling and transformation |
| tibble | Modern data frame implementation | |
| Specialized Analysis | dendextend | Dendrogram manipulation and visualization |
| ComplexHeatmap | Advanced static heatmap creation | |
| Biological Annotation | biomaRt | Genomic data annotation retrieval |
| clusterProfiler | Functional enrichment analysis |
Heatmaps are a fundamental tool in scientific research for visualizing complex, high-dimensional data. They function by encoding a matrix of numerical values as a grid of colored cells, allowing for immediate visual identification of patterns, clusters, and outliers [8] [9]. In fields such as genomics and drug development, they are indispensable for tasks ranging from interpreting gene expression levels to examining correlations among variables [4] [8].
The evolution of heatmaps has progressed from static graphics to interactive visualizations. Static heatmaps, often published as PNG or PDF images, provide a fixed view of the data. In contrast, interactive heatmaps, enabled by modern web technologies, allow researchers to engage directly with the data through operations like hovering to inspect precise values, zooming into specific regions, and dynamically reordering clusters [8] [9]. This article examines the heatmaply R package as a premier tool for creating interactive heatmaps and provides a structured framework for selecting the appropriate visualization type for your research needs, with a specific focus on gene expression analysis.
Critical Visualization Concepts and Data Preprocessing
Data Transformation and Scaling Protocols
The first and most critical step in creating a meaningful heatmap is the appropriate transformation and scaling of the raw data. This ensures that the visual output is a true and comparable representation of the underlying biology. The choice of method depends entirely on the data's structure and distribution.
Table: Data Transformation Methods for Gene Expression Analysis
| Method | Best Use Case | Protocol Formula | Effect on Data |
|---|---|---|---|
Z-Score Standardization (scale) |
Normally distributed data; comparing deviations from mean [8]. | ( X_{\text{scaled}} = \frac{X - \mu}{\sigma} ) | Centers to mean=0, scales to SD=1. |
Min-Max Normalization (normalize) |
Non-normal distributions; bringing all variables to a 0-1 range while preserving shape [9]. | ( X{\text{norm}} = \frac{X - X{\text{min}}}{X{\text{max}} - X{\text{min}}} ) | Bounds data between 0 and 1. |
Percentile Transformation (percentize) |
Non-parametric data; interpreting values as empirical percentiles [9]. | ( X_{\text{perc}} = \frac{\text{rank}(X)}{N} ) | Represents % of observations ≤ value. |
| Square Root/Variance Stabilizing | Count data (e.g., TPM, raw reads) with right-tailed distribution [8]. | ( X_{\text{trans}} = \sqrt{X} ) | Reduces skew from extreme observations. |
For gene expression data, if the matrix contains raw counts or TPMs, a variance-stabilizing transformation like the square root is often recommended as a first step to prevent a few highly expressed genes from dominating the color scale [8]. When comparing genes measured on different scales, the normalize or percentize functions are more robust alternatives to standard Z-score scaling, especially when dealing with binary or categorical variables mixed with continuous data [9].
Color Palette Selection for Scientific Clarity
The choice of color palette is not merely an aesthetic concern; it is a critical factor in accurate data interpretation and accessibility for color-blind readers [4].
- Sequential Palettes: Use a single hue (e.g., light yellow to dark red) or a sequence of related hues (e.g., the Viridis scale) to represent a progression from low to high values [4] [8]. These are ideal for representing non-negative data, such as raw gene expression TPM values [4].
- Diverging Palettes: Use two contrasting hues that meet at a neutral central color (e.g., blue-white-red) [4] [14]. These are essential for highlighting deviations from a critical reference point, such as zero in a correlation matrix or the mean in standardized gene expression data (e.g., up-regulated and down-regulated genes) [4] [8].
- Accessibility and Pitfalls: It is essential to choose color-blind-friendly combinations. Avoid the common but problematic red-green palette [4]. Instead, opt for proven accessible combinations like blue & orange or blue & red [4]. Furthermore, avoid the "rainbow" scale, as it creates perceptual illusions with abrupt changes between hues that can misrepresent smooth data gradients [4].
Diagram 1: A workflow for selecting an appropriate and accessible color palette for a scientific heatmap.
The Heatmaply R Package: A Detailed Protocol
heatmaply is an R package designed to create interactive, publication-quality cluster heatmaps that can be shared as standalone HTML files [8] [9]. Its synergy with the plotly.js engine allows it to handle large matrices efficiently, a common requirement in genomics [5].
Core Functionality and Basic Protocol
The following is a basic protocol for generating an interactive heatmap using heatmaply:
- Installation: Install the package from CRAN using
install.packages('heatmaply')or the development version from GitHub [9] [5]. - Basic Function Call: The simplest heatmap can be generated with
heatmaply(data_matrix)[9]. This will perform hierarchical clustering on rows and columns and display the result with a default Viridis color palette. - Saving Output: To save the interactive heatmap as a shareable HTML file, use:
heatmaply(data_matrix, file = "my_heatmap.html")[8]. A static image (PNG/JPEG/PDF) can also be generated using the same function, which requires thewebshotpackage [5].
Advanced Customization for Gene Expression Analysis
The true power of heatmaply lies in its extensive customization options that cater to complex analytical needs.
- Dendrogram Customization: Control the clustering of rows and columns. You can use the
dendrogramargument to show dendrograms on both sides, one side, or none. TheRowvandColvparameters allow you to supply custom dendrogram objects, providing full control over the clustering structure [8] [9]. - Optimal Leaf Ordering: The
seriationargument controls the ordering of leaves in the dendrogram. The default"OLO"(Optimal Leaf Ordering) rearranges branches to minimize the sum of distances between adjacent leaves, often revealing clearer patterns [9]. - Cluster Identification: The
k_rowandk_colarguments can be set to a specific integer (e.g.,k_row = 3) to cut the dendrogram and visually highlight a predefined number of clusters. Setting these toNAinstructs the function to automatically find the number of clusters (from 2 to 10) that yields the highest average silhouette coefficient [8].
Specialized Functions for Common Analyses
heatmaply includes wrappers optimized for specific analytical tasks:
- Correlation Heatmaps: The
heatmaply_corfunction is specifically designed for correlation matrices. It automatically uses a diverging color palette (e.g.,RdBu) and sets sensible limits from -1 to 1 [9]. Advanced versions can encode p-values into the point size of each correlation cell [9]. - Missing Data Visualization: The
heatmaply_nafunction (or usingis.na10with the main function) is highly effective for visualizing patterns of missing data in a dataset, which is a common step in quality control [9].
Interactive vs. Static Heatmaps: A Decision Framework
The choice between an interactive and a static heatmap is dictated by the goals of the research phase, the nature of the data, and the intended audience for the visualization.
Table: Decision Matrix: Interactive vs. Static Heatmaps
| Criterion | Interactive Heatmaps (e.g., heatmaply) | Static Heatmaps (e.g., base R heatmap, pheatmap) |
|---|---|---|
| Primary Use Case | Data exploration, hypothesis generation, supplementary online material [8]. | Final publication figures, reports, presentations. |
| Data Size | Suitable for larger matrices; allows zooming [5]. | Better for smaller matrices to avoid overplotting. |
| Key Advantage | Tooltip values, zooming, dynamic manipulation [8] [9]. | Simplicity, universal compatibility (PDF/PNG). |
| Audience | Co-investigators, reviewers (as supplementary), interactive dashboards. | Journal readers, conference audiences, broad dissemination. |
| Clustering | Dynamic reordering and cluster exploration is possible. | Fixed clustering based on final parameters. |
Diagram 2: A decision tree to guide researchers in choosing between interactive and static heatmaps for their specific task.
Essential Toolkit for Heatmap Creation in R
A suite of R packages complements and enhances the functionality of heatmaply, forming a comprehensive toolkit for modern biological data visualization.
Table: Research Reagent Solutions for Heatmap Creation in R
| Package/Function | Category | Function in Analysis |
|---|---|---|
heatmaply |
Core Visualization | Creates interactive cluster heatmaps for online sharing and exploration [8] [9]. |
dendextend |
Dendrogram Manipulation | Enables visualizing, adjusting, comparing, and coloring dendrogram branches [8] [9]. |
seriation |
Optimal Ordering | Provides algorithms for finding an optimal ordering of rows and columns to reveal patterns [9]. |
viridis / RColorBrewer |
Color Palettes | Supplies perceptually uniform and color-blind-friendly sequential and diverging color palettes [8] [9]. |
normalize / percentize |
Data Transformation | Transforms data to a 0-1 scale for comparable visualization, preserving distribution shape [9]. |
The dichotomy between interactive and static heatmaps is not a question of which is superior, but of which is more appropriate for a given scientific context. For the dynamic, data-rich world of gene expression research and drug development, heatmaply offers a powerful solution for the exploratory phase, enabling deep, interactive interrogation of complex datasets. Its capacity to create shareable HTML files makes it an ideal tool for collaborative science and for providing rich supplementary material alongside traditional static figures. By mastering the protocols for data preprocessing, color selection, and tool customization outlined here, researchers can leverage the full potential of interactive visualizations to generate more impactful biological insights.
In the field of genomic research, effective data visualization begins with proper data preparation. The quality and structure of your expression matrix directly determine the clarity, accuracy, and biological insights you can derive from interactive heatmaps generated with the heatmaply package in R. This protocol details the essential steps for structuring expression data to optimize visualization outcomes, specifically framed within the context of creating interactive gene expression heatmaps for research and drug development applications.
Properly structured expression data enables researchers to visualize complex gene expression patterns across samples, identify potential biomarkers, and communicate findings effectively to interdisciplinary teams. The heatmaply package builds upon ggplot2 and plotly to create interactive cluster heatmaps that allow inspection of specific values by hovering over cells and zooming into regions of interest [9] [15]. However, these advanced visualization capabilities depend entirely on receiving properly formatted input data.
## Expression Matrix Fundamentals
An expression matrix is a structured data representation where rows typically correspond to features (genes, transcripts) and columns represent samples or experimental conditions. Each cell contains the expression value of a particular feature in a specific sample. This matrix structure serves as the direct input for the heatmaply() function and determines how effectively the package can visualize patterns, calculate distances, and perform clustering.
### Core Structural Requirements
- Dimensional Consistency: All samples must measure the same set of features without missing values in the matrix structure
- Appropriate Data Types: Numeric values for expression measurements with proper row and column identifiers
- Metadata Integration: Sample groups, experimental conditions, and feature annotations must be integrable with the core expression data
The matrix structure enables heatmaply to perform its key analytical operations, including distance calculation between samples or features, hierarchical clustering, and color mapping of expression values [1].
## Data Collection and Experimental Design
### Sample Considerations
When designing experiments for expression heatmap visualization, several factors directly impact data quality:
- Biological Replicates: Essential for assessing experimental variability and ensuring statistical robustness
- Sample Size: Balance between statistical power and computational limitations of visualization tools
- Control Samples: Critical for interpreting expression changes in experimental conditions
In a typical gene expression study, such as research investigating influenza virus effects on human plasmacytoid dendritic cells, experimental design includes both infected and control cells to enable meaningful comparisons [16].
### Expression Quantification Methods
Table 1: Common Expression Measurement Technologies
| Technology | Output Type | Data Structure | Preprocessing Needs |
|---|---|---|---|
| RNA Sequencing | Count Data | Integer Values | Normalization, Transformation |
| Microarrays | Fluorescence Intensity | Continuous Values | Background Correction, Normalization |
| qPCR | Cycle Threshold | Continuous Values | Delta-CT Calculation |
For RNA-seq data, expression values typically begin as raw counts that require normalization to account for sequencing depth and other technical variables [17]. The heatmaply package can visualize various normalized expression measures, including counts per million (CPM), fragments per kilobase million (FPKM), and transcripts per million (TPM).
## Data Transformation Protocols
### Protocol 1: Reshaping to Tidy Data Format
The heatmaply package integrates with ggplot2, which operates most effectively with data in "tidy" format [16]. This protocol transforms a wide-format expression matrix into a long-format structure suitable for visualization.
Materials:
- R statistical environment (v4.0 or higher)
- tidyverse package suite
- Expression matrix in wide format
Procedure:
- Import your expression matrix where rows are genes and columns are samples
- Preserve gene identifiers as row names during import
- Convert the matrix to a data frame with gene identifiers as a proper column
- Use
pivot_longer()to transform sample columns to key-value pairs
Example Implementation:
Validation:
- Confirm all expression values are numeric
- Verify no gene identifiers were lost during transformation
- Ensure sample names are properly recorded
### Protocol 2: Expression Value Normalization
Raw expression values often require transformation to improve visualization effectiveness, particularly when dealing with RNA-seq count data that exhibits mean-variance relationship.
Materials:
- Normalized expression values (e.g., CPM, TPM, or FPKM for RNA-seq)
- R environment with preprocessed expression data
Procedure:
- Log Transformation: Apply log2 transformation to count data to reduce dynamic range and stabilize variance
- Z-Score Standardization: Scale expression values for each gene to have mean = 0 and standard deviation = 1
- Alternative Approaches: Consider using the
normalize()orpercentize()functions from the heatmaply package for specific applications [9]
Example Implementation:
Validation:
- Confirm transformation has preserved biological signal
- Check for infinite values introduced during transformation
- Verify that the matrix retains proper dimensions
## Expression Matrix Quality Assessment
### Quality Control Metrics
Before proceeding to visualization, assess your expression matrix using these essential quality metrics:
Table 2: Expression Matrix Quality Control Checklist
| QC Metric | Optimal Range | Assessment Method | Corrective Action |
|---|---|---|---|
| Missing Values | <5% of cells | sum(is.na(matrix)) |
Imputation or filtering |
| Value Distribution | Approximates expected | Histogram, Q-Q plot | Transformation |
| Feature Variance | Sufficient for clustering | Variance calculation | Filter low-variance genes |
| Sample Correlation | Replicates >0.8 | Correlation heatmap | Investigate outliers |
### Protocol 3: Handling Missing Data
Missing values in expression matrices can disrupt distance calculations and clustering in heatmap visualizations.
Materials:
- Expression matrix with identified missing values
- R environment with impute package
Procedure:
- Identify missing values using
is.na()function - Assess pattern of missingness (random vs. systematic)
- Apply appropriate imputation method:
- K-nearest neighbors (KNN) imputation for random missingness
- Minimum value imputation for detection limit missingness
- Document imputation method for reproducibility
Example Implementation:
Validation:
- Confirm no remaining missing values
- Verify imputation hasn't introduced artificial patterns
- Document method in analysis records
## Matrix Optimization for Heatmap Visualization
### Feature Selection Strategies
Large expression matrices with thousands of genes can produce cluttered, uninterpretable heatmaps. Strategic feature selection improves visualization clarity.
Approaches:
- High-Variance Genes: Select genes with the highest expression variance across samples
- Differentially Expressed Genes: Identify statistically significant genes between experimental conditions
- Pathway Representatives: Select genes from biologically relevant pathways
- Custom Gene Sets: Use predefined gene sets based on prior knowledge
Example Implementation:
### Protocol 4: Integration with Metadata
Incorporating sample metadata (e.g., treatment groups, time points, patient characteristics) enhances heatmap interpretability.
Materials:
- Expression matrix (features × samples)
- Sample metadata table (samples × attributes)
- R environment with heatmaply package
Procedure:
- Prepare metadata data frame with samples as rows and attributes as columns
- Ensure perfect alignment between metadata samples and expression matrix columns
- Code categorical variables as factors with intuitive level ordering
- Incorporate metadata using the
side_colorparameters in heatmaply
Example Implementation:
## Visualization Workflow Integration
### Data Flow from Matrix to Interactive Heatmap
The following diagram illustrates the complete workflow for transforming raw expression data into an optimized matrix for interactive heatmap visualization:
### Advanced Matrix Preparation for Specialized Applications
#### Correlation Matrices
For visualizing correlation patterns rather than direct expression values, heatmaply provides the heatmaply_cor() function with presets optimized for correlation matrices [9] [15].
Procedure:
- Compute correlation matrix using
cor()function - Apply
heatmaply_cor()with diverging color palette - Set appropriate limits (-1 to 1) for proper color scaling
Example Implementation:
#### Missing Data Visualization
The heatmaply_na() function provides a specialized approach for visualizing patterns in missing data [9].
Procedure:
- Convert expression matrix to binary missingness indicator
- Apply
heatmaply_na()with appropriate grid formatting - Interpret missingness patterns for potential technical artifacts
## Troubleshooting Common Matrix Issues
### Dimension Mismatch Errors
Problem: Row or column counts between expression matrix and metadata don't match.
Solution: Verify alignment using rownames() and colnames() functions, then reorder using index matching.
### Color Scaling Artifacts
Problem: Heatmap colors don't adequately represent expression patterns due to extreme outliers. Solution: Apply Winsorization (capping extreme values) or use non-linear color scales.
### Memory Limitations
Problem: Large expression matrices exhaust available memory during rendering.
Solution: Implement strategic feature filtering, matrix subsetting, or use the plotly engine directly for larger datasets [9].
## Research Reagent Solutions
Table 3: Essential Tools for Expression Matrix Preparation
| Reagent/Software | Function | Application Note |
|---|---|---|
| R Statistical Environment | Data manipulation and analysis | Base platform for all transformation protocols |
| tidyverse Package Suite | Data reshaping and transformation | Essential for converting to tidy format |
| heatmaply R Package | Interactive heatmap generation | Primary visualization tool with specialized functions |
| Bioconductor | Genomic data analysis | Source for specialized expression data packages |
| DESeq2/edgeR | Differential expression analysis | For identifying significant genes for filtering |
| Single-cell RNA-seq Tools | Analysis of single-cell data | Specialized methods for single-cell expression matrices |
Properly structuring your expression matrix is a critical prerequisite for generating biologically meaningful interactive heatmaps with heatmaply. By following these standardized protocols for data transformation, quality control, and metadata integration, researchers can ensure their visualization accurately represents underlying biological patterns. The structured approach outlined in this protocol enhances reproducibility, facilitates clearer communication of results, and ultimately supports more robust scientific conclusions in gene expression studies and drug development research.
The expression matrix serves as the foundation upon which all subsequent visual analytics are built. Investing time in proper data preparation significantly enhances the quality and interpretability of interactive heatmaps, transforming raw expression data into actionable biological insights.
Hands-On Tutorial: Building Your First Interactive Gene Expression Heatmap
heatmaply is an R package designed for creating interactive cluster heatmaps, which are invaluable tools for visualizing high-dimensional data such as gene expression matrices. It encodes data tables as grids of colored cells, accompanied by dendrograms and interactive features including tooltip value inspection and zooming capabilities [8]. The package is built upon the ggplot2 and plotly.js engines, offering advantages in handling larger matrices and providing enhanced interactive features compared to static alternatives [10] [18].
Researchers can install heatmaply through multiple channels, with the Comprehensive R Archive Network (CRAN) providing the stable version and GitHub hosting the development version. The package requires R version 3.0.0 or higher and depends on several key packages including plotly (≥4.7.1) and viridis [18] [15].
Table: Installation Methods for heatmaply
| Method | Command | Use Case |
|---|---|---|
| CRAN (Stable) | install.packages('heatmaply') |
Production environments, reproducible research |
| GitHub (Development) | devtools::install_github('talgalili/heatmaply') |
Access to latest features and bug fixes |
| Conda | conda install conda-forge::r-heatmaply |
Conda-based environment management |
Dependency Management and System Requirements
Successful installation and operation of heatmaply requires careful management of its dependencies. The package imports multiple R packages that provide critical functionality for data transformation, visualization, and clustering analysis [18] [15].
Recommended Pre-installation Packages
To ensure a smooth installation process, particularly for the GitHub version, consider pre-installing these suggested packages [10] [5]:
Table: Critical Dependencies and Their Functions
| Dependency | Minimum Version | Primary Function |
|---|---|---|
| plotly | 4.7.1 | Interactive visualization engine |
| ggplot2 | 2.2.0 | Grammar of graphics implementation |
| dendextend | 1.12.0 | Dendrogram manipulation and customization |
| viridis | Not specified | Colorblind-friendly color palettes |
| seriation | Not specified | Matrix ordering and arrangement |
| RColorBrewer | Not specified | Color palette management |
Installation Verification and Basic Usage
Verification Protocol
After installation, verify the package loads correctly and perform a basic functional test:
Basic Application Workflow
The fundamental workflow for creating an interactive heatmap involves data preparation, matrix transformation, and visualization parameter specification. For gene expression data, this typically includes normalization, clustering, and appropriate color palette selection [9] [8].
Research Reagent Solutions
Table: Essential Computational Tools for Interactive Heatmap Generation
| Research Reagent | Function in Analysis | Application Context |
|---|---|---|
| heatmaply R package | Primary visualization engine | Interactive cluster heatmap generation |
| dendextend package | Dendrogram customization and manipulation | Enhanced clustering visualization |
| seriation package | Optimal matrix ordering | Improved pattern recognition in data |
| viridis color palette | Perceptually uniform coloring | Colorblind-accessible visualizations |
| plotly.js engine | Web-based interactive graphics | Zoom, hover inspection, and HTML export |
| normalize() function | Data transformation to [0,1] range | Pre-processing for comparative analysis |
| percentize() function | Empirical percentile transformation | Non-parametric data scaling |
Installation Workflow Diagram
The following diagram illustrates the complete installation and verification workflow for the heatmaply package:
Package Installation Workflow - This diagram outlines the systematic process for installing heatmaply, from initial system checks to final verification.
Advanced Configuration and Troubleshooting
Package Loading and Dependency Management
When loading heatmaply, the package automatically imports its dependencies, but researchers should be aware of potential namespace conflicts, particularly with the plotly package. The package provides specialized wrappers including heatmaply_cor for correlation matrices and heatmaply_na for missing data visualization [9] [15].
Common Installation Issues
- GitHub installation failures: Often result from missing build tools or dependencies; ensure Rtools (Windows) or Xcode command line tools (macOS) are installed [10]
- PhantomJS dependency: Required for static image export; install via
webshot::install_phantomjs()[10] - Version conflicts: Particularly with
plotly; consider using the development versiondevtools::install_github("ropensci/plotly")for latest features [5]
This installation protocol establishes the foundation for creating interactive gene expression heatmaps, enabling researchers to proceed to data visualization and analysis phases with a properly configured computational environment.
The creation of a reliable and informative interactive gene expression heatmap is fundamentally dependent on the rigorous preparation of the underlying data. RNA sequencing (RNA-Seq) data, which begins as raw sequencing reads, must undergo a series of transformative steps to produce normalized expression values suitable for visualization and biological interpretation [19]. Improper data handling at this critical stage can introduce technical artifacts, obscure genuine biological patterns, and lead to misleading conclusions. This guide details the essential protocols for processing raw RNA-Seq count data into robust normalized expression matrices, specifically contextualized for creating interactive cluster heatmaps using the heatmaply R package [5] [8] [18]. The procedures outlined herein will equip researchers, scientists, and drug development professionals with the methodologies necessary to ensure their visualizations accurately reflect the underlying transcriptomics.
The RNA-Seq Data Preprocessing Workflow
The journey from raw sequencing output to a normalized expression matrix involves multiple, sequential steps designed to control for technical variability and enhance biological signal. A summary of this workflow is presented in Figure 1.
Figure 1. RNA-Seq Data Preprocessing Workflow for Heatmap Preparation
From Raw Reads to a Count Matrix
The initial phase of RNA-Seq analysis focuses on converting the raw sequencing output into a gene-level count matrix.
Quality Control (QC): The first step involves assessing the quality of the raw sequencing reads stored in FASTQ format. Tools like FastQC or multiQC are used to identify potential technical issues, including leftover adapter sequences, unusual base composition, or duplicated reads [19]. Reviewing the QC report is critical for informing subsequent cleaning steps.
Read Trimming and Cleaning: Based on the QC report, reads are processed to remove low-quality bases and any residual adapter sequences using tools such as Trimmomatic, Cutadapt, or fastp [19]. This step ensures that only high-quality sequences are used for alignment, preventing mapping inaccuracies.
Read Alignment or Pseudoalignment: The cleaned reads are then mapped to a reference genome or transcriptome. Traditional aligners like STAR or HISAT2 perform base-by-base alignment [19]. Alternatively, faster pseudoalignment tools such as Salmon or Kallisto can be used to estimate transcript abundances directly, incorporating statistical models to improve accuracy [19].
Post-Alignment QC and Quantification: After alignment, a second QC step is performed with tools like SAMtools or Qualimap to filter out poorly aligned or ambiguously mapped reads [19]. The final step in this phase is read quantification, where tools like featureCounts or HTSeq-count count the number of reads mapped to each gene, producing a raw count matrix [19]. This matrix, where rows represent genes and columns represent samples, contains integer counts that reflect the raw expression level of each gene.
Experimental Design Considerations
The reliability of downstream analysis, including visualization, is heavily influenced by experimental design. Two key factors are biological replicates and sequencing depth [19].
- Biological Replicates: While differential expression analysis is technically possible with only two replicates, the ability to estimate biological variability and control false discovery rates is greatly reduced. A minimum of three replicates per condition is often considered the standard, though more may be required for systems with high inherent variability [19].
- Sequencing Depth: The number of reads per sample directly impacts the detection of lowly expressed genes. For standard differential gene expression (DGE) analysis, a depth of approximately 20–30 million reads per sample is often sufficient, though this may vary based on the experimental system and objectives [19].
Normalization Techniques for Expression Analysis
The raw count matrix generated from quantification cannot be directly used for comparative analysis or visualization because counts are influenced by technical factors like sequencing depth (library size) and gene length [19]. Normalization is the mathematical process of adjusting the counts to remove these biases, making expression levels comparable across samples and genes.
Comparison of Normalization Methods
Different normalization methods correct for different sources of bias. The choice of method depends on the intended downstream application. Table 1 summarizes the characteristics of common normalization methods.
Table 1. Comparison of RNA-Seq Data Normalization Methods
| Method | Sequencing Depth Correction | Gene Length Correction | Library Composition Correction | Suitable for DGE Analysis | Key Notes |
|---|---|---|---|---|---|
| CPM (Counts per Million) [19] | Yes | No | No | No | Simple scaling by total reads; highly affected by a few highly expressed genes. |
| FPKM/RPKM [19] [20] | Yes | Yes | No | No | Adjusts for gene length; still affected by differences in library composition between samples. |
| TPM (Transcripts per Million) [19] [20] | Yes | Yes | Partial | No | Improves on FPKM by scaling sample counts to a constant total (1 million), reducing composition bias. Good for cross-sample comparison. |
| TMM (Trimmed Mean of M-values) [20] | Yes | No | Yes | Yes | Implemented in edgeR. Robust to highly variable and differentially expressed genes. A between-sample method. |
| RLE (Relative Log Expression) [20] | Yes | No | Yes | Yes | Implemented in DESeq2. Uses a median-of-ratios approach. A between-sample method. |
The Critical Role of Normalization in Heatmap Creation
For heatmap visualization, the choice of input data is paramount. As emphasized in community discussions, Z-scoring is not a substitute for proper normalization [21]. Using raw or improperly normalized counts for a heatmap will result in a misleading visualization where patterns are dominated by technical artifacts rather than biology.
The recommended practice is to use normalized, log-transformed values. For example, one can use the output of the vst (variance stabilizing transformation) or rlog (regularized log transformation) functions in DESeq2 on the raw count data [21]. Alternatively, one can calculate normalized counts using DESeq2::counts(dds, normalized=TRUE) and then apply a log2(norm+1) transformation [21]. Once the data is normalized and log-transformed, Z-score scaling per gene (row) is often applied to create the final heatmap input. This step puts all genes on a comparable scale, ensuring that both highly and lowly expressed genes can contribute equally to the clustering patterns seen in the heatmap [21].
Protocols for Data Preparation with DESeq2
This section provides a detailed, step-by-step protocol for preparing a normalized expression matrix from raw counts using the DESeq2 package in R, which is a standard and robust approach for differential expression analysis and data normalization.
Detailed Protocol: Generating a Normalized Expression Matrix
Research Reagent Solutions & Essential Materials
| Item/Software | Function in Protocol |
|---|---|
| R Statistical Environment | The core platform for executing all data preparation and analysis steps. |
| DESeq2 R Package [22] | Performs statistical modeling of raw count data, size factor estimation, and normalization. |
| tidyverse R Package [22] | A collection of packages (e.g., dplyr, tibble) for efficient data manipulation and wrangling. |
| Raw Count Matrix File (CSV) | The input data containing gene counts per sample. |
| Sample Metadata File (CSV) | A file describing the experimental design, linking samples to conditions. |
| heatmaply R Package [5] [18] | Used to create the final interactive cluster heatmap from the normalized matrix. |
Step-by-Step Methodology
Import Libraries and Data:
Create a DESeqDataSet Object: This object bundles the count data and experimental metadata for analysis.
Note: It is critical to check the factor levels of the condition variable (
dds$condition). The first level is treated as the reference group (e.g., control) in subsequent comparisons. Levels can be reordered using therelevelfunction if necessary [22].Perform Differential Expression Analysis: Executing the
DESeqfunction estimates size factors (for normalization), dispersion, and fits models to the data.Extract Normalized Expression Values: The
vst(Variance Stabilizing Transformation) function is preferred for heatmaps as it transforms the data to stabilize the variance across the mean, making it more suitable for visualization.Alternative: For a simpler log2-transformed normalized count matrix, use:
(Optional) Z-score Transformation for Heatmap: To emphasize gene-wise patterns across samples, apply row-wise Z-scoring to the
normalized_matrix.
The resulting normalized_matrix or z_score_matrix is now ready for creating an interactive heatmap with heatmaply.
Integration with Interactive Heatmap Creation
The prepared normalized expression matrix serves as the direct input for the heatmaply R package, which generates interactive, publication-quality heatmaps [8] [18]. A simple function call creates the visualization:
The heatmaply function offers extensive customization, allowing control over clustering methods, color palettes, dendrogram display, and the addition of side annotations [5] [8]. The output is a self-contained HTML file that enables readers to hover over cells to see exact values, zoom into regions of interest, and better explore the structure of the gene expression data.
Interactive cluster heatmaps are an indispensable tool in the field of bioinformatics and computational biology, enabling researchers to visualize complex high-dimensional data, such as gene expression matrices, as an intuitive grid of colored cells. The heatmaply R package, built on the robust ggplot2 and plotly.js engine, extends the capabilities of traditional static heatmaps by creating interactive visualizations that allow for inspection of specific values via mouse hover and zooming into regions of interest [23] [15]. This functionality is particularly valuable for gene expression analysis, where researchers must identify patterns across thousands of genes under multiple experimental conditions. The interactive nature facilitates exploratory data analysis, making it easier to pinpoint candidate genes for further investigation in drug development projects.
The utility of heatmaply is enhanced by its ability to handle larger matrices than some traditional alternatives and its features for side-by-side annotation and zooming directly from dendrogram panes [23]. For scientific reproducibility and seamless reporting, these heatmaps can be embedded within R Markdown documents, Shiny applications, or saved as standalone HTML files, making them an ideal choice for collaborative research environments [15].
Essential Materials and Software Setup
Research Reagent Solutions
The following table details the essential computational tools and their specific roles in creating and analyzing interactive heatmaps for genomic studies.
| Tool Name | Function in Analysis |
|---|---|
heatmaply R Package |
Primary engine for generating interactive cluster heatmaps with dendrograms [15]. |
plotly |
Provides the underlying interactive plotting capabilities and rendering [23]. |
ggplot2 |
Used for constructing the static graphics foundation upon which interactive elements are built [15]. |
RColorBrewer & viridis |
Provides color palettes for data representation, including sequential and diverging schemes [15]. |
dendextend |
Offers advanced tools for manipulating and customizing dendrograms displayed on heatmap axes [15]. |
| Gene Expression Matrix | The primary input data, typically with rows as genes and columns as samples or experimental conditions. |
Software Installation Protocol
To replicate the analyses described in this protocol, researchers must first establish the necessary software environment. The following commands, executed in an R console, will install the core packages.
Experimental Protocol 1: Initial Package Installation
- Install the stable version of
heatmaplyand its dependencies from CRAN using the command:install.packages('heatmaply'). - For the latest development version with cutting-edge features, use the
devtoolspackage to install directly from GitHub:devtools::install_github('talgalili/heatmaply')[23]. - Ensure that the following supporting packages are also installed and loaded:
plotly,ggplot2,viridis,RColorBrewer, anddendextend[23] [15].
Core Heatmaply Parameters and Workflow
Constructing an effective heatmap requires a structured workflow, from data preparation to visualization. The following diagram illustrates the logical flow and key decision points in creating a basic interactive heatmap with heatmaply.
Essential Parameters for Basic Implementation
The heatmaply function offers a wide array of parameters for customization. The table below summarizes the fundamental parameters required for generating a basic yet fully functional interactive heatmap, with a focus on gene expression data visualization.
| Parameter | Data Type | Default Value | Function in Analysis |
|---|---|---|---|
x |
numeric matrix | (none) | The primary data input, typically a gene expression matrix where rows are genes and columns are samples [15]. |
colors |
color palette | viridis(256) |
The color vector or palette function used to map data values to cell colors [15]. |
limits |
numeric vector (length 2) | NULL (data range) |
Sets fixed minimum and maximum values for the color scale, crucial for consistent comparison across multiple heatmaps [15]. |
Rowv & Colv |
logical, dendrogram, or NA |
NULL (auto-dendrogram) |
Controls whether and how row and column dendrograms are computed and reordered [15]. |
distfun |
function | stats::dist |
The function used to compute the distance matrix for clustering (e.g., dist, correlation). |
hclustfun |
function | stats::hclust |
The function used for hierarchical clustering (e.g., hclust, fastcluster::hclust). |
cellnote |
matrix | NULL |
An optional matrix of the same dimensions as x containing text labels for each cell [15]. |
draw_cellnote |
logical | !is.null(cellnote) |
Controls whether the cellnote labels are displayed on the heatmap cells [15]. |
Aesthetic Choices for Scientific Visualization
The strategic selection of colors is not merely an aesthetic concern but a critical factor that determines the clarity and interpretability of a heatmap. Effective color palettes guide the viewer's eye and accurately represent the underlying data structure.
Color Palette Theory and Selection
For scientific visualization, color palettes are broadly classified into three categories, each serving a distinct purpose [24]:
- Sequential Palettes: Used for representing numeric data that progresses from low to high values. Perception of the data structure is best achieved by varying luminance, with brighter colors representing larger counts or intensities [24]. Examples include
"viridis","Blues", and"rocket"[24] [15]. - Diverging Palettes: Ideal for numeric data with a critical midpoint, such as correlation matrices (midpoint 0) or log-fold changes in gene expression (midpoint 1). These palettes use two distinct hues that meet at a central, often lighter, color [24]. The
heatmaplypackage provides functions likeRdBu(n)andcool_warm(n)for this purpose [15]. - Qualitative Palettes: Best for representing categorical data, as their primary variation is in hue, making them easily distinguishable from one another [24].
Experimental Protocol 2: Applying a Diverging Color Palette for Correlation Matrix
- Compute the correlation matrix from your normalized gene expression data:
cor_matrix <- cor(gene_expression_matrix). - Select a diverging palette, such as
RdBuorcool_warm, which is specifically designed for scientific visualization [15]. - Set the
limitsparameter toc(-1, 1)to ensure the color scale midpoint is correctly aligned at zero [15]. - Generate the heatmap:
heatmaply(cor_matrix, colors = RdBu, limits = c(-1, 1)).
Quantitative Data for Color Palettes
The table below provides the HEX codes for several recommended scientific color palettes available in heatmaply and related packages, enabling precise color specification.
| Palette Name | Type | Color 1 (Low) | Color 2 | Color 3 (Mid) | Color 4 | Color 5 (High) |
|---|---|---|---|---|---|---|
| Viridis | Sequential | #440154 |
#31688E |
#35B779 |
#B8DE29 |
#FDE725 |
| RdBu | Diverging | #67001F |
#B2182B |
#F7F7F7 |
#2166AC |
#053061 |
| Cool-Warm | Diverging | #3B4CC0 |
#8B9BFB |
#F5F2F1 |
#F2A285 |
#B40426 |
| Rocket | Sequential | #03051A |
#CB1B4F |
#F88D51 |
#F6D645 |
#FCF5BF |
Optimizing Text Label Clarity
A common challenge in heatmap design is ensuring that numerical or textual labels within cells remain legible against the varying background colors. The heatmaply package offers parameters to control these labels.
Experimental Protocol 3: Configuring Conditional Text Color
- Prepare a
cellnotematrix containing the values or labels you wish to display in each heatmap cell [15]. - Use the
cellnote_colorparameter. Whileheatmaplydoes not have a direct conditional function, setting it to"auto"will often choose black or white logically based on the cell's color intensity [15]. - For more advanced control, such as setting a specific threshold (e.g., white text for values below 50, black for values above), you may need to create a custom matrix of color names and use it in conjunction with the
cellnote_colorparameter, or manually adjust thefont_colorsin the underlyingplotlyobject post-rendering, as discussed in community forums [25] [26].
Advanced Applications in Gene Expression Analysis
The standard heatmap workflow can be adapted and enhanced for specific, advanced analytical tasks common in genomic research and drug development.
Specialized Wrappers and Workflows
The heatmaply package includes specialized wrapper functions that pre-configure parameters for common use cases, streamlining the analysis process for scientists.
Experimental Protocol 4: Visualizing Missing Data Patterns
- Begin with a gene expression matrix that may contain missing values, denoted as
NA. - Utilize the
heatmaply_nawrapper function, which is specifically designed for this task:heatmaply_na(gene_expression_matrix). - This function automatically sets optimal defaults for exploring
NApatterns, including agrid_gapof 1 to separate cells and a two-shade color scheme (e.g.,c("grey80", "grey20")) to clearly distinguish between present and missing data [15].
The following diagram outlines the specialized workflows for handling correlation matrices and missing data, which are frequent challenges in gene expression analysis.
Mastering the essential parameters and aesthetic choices within the heatmaply R package empowers researchers to create informative, publication-quality interactive heatmaps. By adhering to the protocols for color selection, clustering, and specialized analysis outlined in this document, scientists and drug development professionals can effectively visualize complex gene expression data, thereby facilitating the discovery of meaningful biological patterns and accelerating the pace of research.
In the analysis of high-dimensional biological data, such as gene expression matrices, heatmaps serve as an indispensable tool for visualizing complex patterns. The integration of hierarchical clustering elevates this visualization, transforming a simple color grid into a powerful analytical resource that reveals inherent structures, relationships, and subgroups within the data [3]. For researchers, scientists, and drug development professionals, the choices made in distance metrics and clustering algorithms are not merely technical; they are fundamental to interpreting biological phenomena, identifying disease subtypes, and discovering potential therapeutic targets [27] [3]. This document provides detailed application notes and protocols for implementing these advanced clustering options within the context of creating an interactive gene expression heatmap using the heatmaply R package, a tool designed for creating interactive cluster heatmaps for online publishing [28] [5].
Key Concepts and Definitions
Hierarchical Clustering
Hierarchical Clustering is an unsupervised machine learning algorithm that builds a hierarchy of clusters. This hierarchical relationship is typically visualized as a tree-like diagram called a dendrogram, which is displayed alongside the heatmap to show the grouping of rows (e.g., genes) and columns (e.g., samples) [27] [3]. The two main approaches are:
- Agglomerative (Bottom-Up): This more common method starts with each data point as its own cluster and iteratively merges the most similar clusters until only one cluster remains [27].
- Divisive (Top-Down): This method begins with all data points in a single cluster and recursively splits them into smaller clusters [27].
Distance Metrics
A distance metric quantifies the dissimilarity between two data points. The choice of metric profoundly influences the clustering outcome [27] [29].
- Euclidean Distance: The "straight-line" distance between two points in space. It is sensitive to magnitude and is best for data where absolute differences are meaningful [27].
- Manhattan Distance: The sum of the absolute differences along each axis. It is more robust to outliers than Euclidean distance [27].
- Pearson Correlation Distance: Measures the dissimilarity based on the linear relationship between two data profiles (e.g., gene expression patterns). It is defined as 1 minus the Pearson correlation coefficient, making it ideal for identifying genes with similar expression trends across samples, regardless of their absolute expression levels [27].
Linkage Algorithms
The linkage criterion determines how the distance between two clusters is calculated once individual points are grouped [27] [29].
- Complete Linkage: The distance between two clusters is the maximum distance between any member of one cluster and any member of the other. This method tends to create compact, spherical clusters [27].
- Average Linkage: The distance between two clusters is the average distance between all pairs of objects in the two different clusters. It often results in balanced clusters [27] [29].
- Single Linkage: The distance between two clusters is the minimum distance between any two members of the clusters. It can lead to "chaining," where clusters are stretched out [29].
- Ward's Method: Aims to minimize the variance within each cluster. It is effective at creating clusters of relatively equal size and is less susceptible to noise [29].
Materials and Reagents
Research Reagent Solutions
Table 1: Essential software and packages for creating interactive clustered heatmaps.
| Item Name | Function/Application | Example/Version |
|---|---|---|
| R Programming Language | Provides the statistical computing and graphics environment for all analyses and visualizations. | R 4.2.0 or higher |
heatmaply R Package |
The primary tool for generating interactive cluster heatmaps with zooming, hovering, and dendrogram manipulation features. [28] [5] | Version 1.6.0 |
plotly & ggplot2 |
Engine packages that provide the underlying interactive and static graphing capabilities for heatmaply. [5] |
|
dendextend R Package |
Used for advanced manipulation, analysis, and customization of dendrograms. [27] [5] | |
| Gene Expression Dataset | A numeric matrix where rows represent features (e.g., genes, transcripts) and columns represent samples or conditions. | Normalized count data from RNA-seq or microarray |
Experimental Protocols and Methodologies
Protocol 1: Data Preparation and Normalization
Objective: To prepare a normalized gene expression matrix for clustering analysis. Procedure:
- Data Input: Load your dataset into R. Ensure it is structured as a data frame or matrix where rows are genes and columns are samples. Non-numeric identifiers (e.g., Gene IDs) must be stored separately, typically as row names.
- Data Normalization/Scaling: To ensure comparability across genes or samples, apply scaling. For gene expression, it is common to standardize (Z-score) the rows (genes) so that each gene has a mean of 0 and a standard deviation of 1. This prevents highly expressed genes from dominating the clustering.
Protocol 2: Calculating Distance Matrices
Objective: To compute pairwise distance matrices for rows and columns using different metrics. Procedure:
- Calculate Row Distances: Compute the distance between all pairs of genes (rows).
- Calculate Column Distances: Compute the distance between all pairs of samples (columns).
Protocol 3: Performing Hierarchical Clustering
Objective: To build hierarchical cluster models from the distance matrices. Procedure:
- Cluster Rows: Apply hierarchical clustering to the row distance matrices using a chosen linkage method.
- Cluster Columns: Apply hierarchical clustering to the column distance matrices.
Protocol 4: Generating the Interactive Heatmap withheatmaply
Objective: To visualize the clustered data as an interactive heatmap. Procedure:
- Basic Interactive Heatmap: Generate a heatmap using the
heatmaplyfunction, which automatically handles clustering and produces an interactiveplotlyobject. - Advanced Customization with Pre-computed Clusters: For greater control, specify the pre-computed cluster objects and customize the plot.
Data Analysis and Interpretation
Comparison of Distance and Linkage Methods
Table 2: Guide to selecting distance metrics and linkage methods for gene expression data.
| Method | Mathematical Basis | Best Use Case | Advantages | Limitations |
|---|---|---|---|---|
| Euclidean Distance | Square root of the sum of squared differences. | Clustering based on absolute expression magnitude. [27] | Intuitive "as-the-crow-flies" distance. [27] | Highly sensitive to outliers and data scale. [27] |
| Manhattan Distance | Sum of absolute differences. | Data with outliers or noise. [27] | More robust to outliers than Euclidean. [27] | Can be less intuitive in high-dimensional spaces. |
| Pearson Correlation Distance | 1 - Pearson correlation coefficient. | Clustering based on expression profile shape (co-expression). [27] | Identifies genes with similar trends, ignores magnitude. [27] | Only captures linear relationships. |
| Complete Linkage | Maximum inter-cluster distance. | Creating tight, compact clusters. [27] [29] | Less susceptible to noise and chaining. | Can break large clusters and be sensitive to outliers. |
| Average Linkage | Average inter-cluster distance. | General-purpose clustering with balanced results. [29] | Compromise between single and complete linkage. [27] | Computationally more intensive. |
| Ward's Method | Minimizes within-cluster variance. | Creating clusters of similar size and shape. [29] | Very effective at finding compact, spherical clusters. | Biased towards hyperspherical clusters. |
Workflow for Hierarchical Clustering Analysis
The following diagram outlines the logical workflow and decision points involved in creating a clustered heatmap, from data preparation to interpretation.
Discussion
Technical Considerations and Limitations
The process of creating a clustered heatmap involves several critical choices. The selection of distance metric and linkage algorithm is not a one-size-fits-all decision; it must be guided by the biological question and the nature of the data [27] [3]. For instance, using Pearson correlation distance is often more biologically relevant for gene expression data than Euclidean distance, as it groups genes with co-regulated expression patterns regardless of their baseline intensity [27]. Furthermore, it is crucial to remember that clusters identified by the algorithm represent patterns of similarity, not necessarily causation or biological function. These patterns require validation through additional statistical tests or experimental work [3]. Interactive heatmaps, like those produced by heatmaply, help mitigate some limitations of static images by allowing researchers to zoom, pan, and hover to inspect individual values, thus facilitating a more nuanced exploration of large and complex datasets [28] [5].
Advanced Applications in Biomedical Research
Clustered heatmaps with hierarchical clustering are a cornerstone of modern bioinformatics and have been instrumental in numerous biomedical breakthroughs. In gene expression studies, they are used to identify molecular subtypes of cancers, which can lead to more precise diagnoses and personalized treatment strategies [3]. In metabolomics and proteomics, heatmaps help visualize the abundance of molecules across different sample groups, revealing metabolic pathways disrupted in disease states [3]. The interactivity of heatmaply heatmaps enhances these applications by enabling the integration of metadata annotations and link-outs to external genomic databases, providing a richer context for data interpretation and hypothesis generation [28] [5].
In the field of genomic research, heatmap visualizations serve as a fundamental tool for analyzing complex gene expression patterns across multiple samples. The heatmaply package in R enables the creation of interactive heatmaps that allow researchers to explore high-dimensional biological data through hovering, zooming, and dynamic inspection of individual values [9]. While these visualizations powerfully represent expression matrices, their interpretability dramatically increases when supplemented with clinical and experimental metadata. Such annotations provide essential context, enabling researchers to identify whether observed patterns correlate with specific patient subgroups, treatment regimens, or experimental conditions [1].
This protocol details methods for incorporating sample annotations into interactive heatmaps using heatmaply, framed within a broader research workflow for gene expression analysis. We provide comprehensive guidance on data preparation, annotation integration, visualization customization, and interpretation—specifically designed for researchers, scientists, and drug development professionals requiring reproducible analytical pipelines for their genomic studies.
Key Concepts and Terminology
Heatmap Fundamentals
A heatmap is a graphical representation of data where individual values in a matrix are encoded as colored cells [1]. In genomics, heatmaps typically display genes as rows and samples as columns, with color intensity representing expression levels. The heatmaply package enhances this basic visualization by creating interactive versions that support data exploration through mouse hover interactions and zoom capabilities [9].
Annotation Importance
Sample annotations refer to clinical (e.g., patient age, disease stage, treatment response) or experimental (e.g., batch, processing date, protocol) metadata associated with each sample in a study. Incorporating these annotations alongside the primary expression data allows researchers to determine whether clustering patterns reflect biologically meaningful groupings versus technical artifacts [1].
Visualization Considerations
Effective heatmap design requires careful selection of color scales to accurately represent data without misleading interpretation. Sequential color scales (progressing from light to dark shades of a single hue) are appropriate for data with a natural progression from low to high values, while diverging color scales (using two contrasting hues with a neutral midpoint) better represent data with a critical center point, such as Z-scores or fold-changes [4]. Additionally, accessibility considerations should guide color choices to ensure interpretability for color-blind users [4].
Materials and Reagents
Research Reagent Solutions
Table 1: Essential computational tools and their functions for creating annotated interactive heatmaps.
| Tool Name | Function | Application Context |
|---|---|---|
| R Statistical Environment | Primary computational platform | Data manipulation, statistical analysis, and visualization |
| heatmaply R package | Interactive heatmap generation | Creating zoomable, hover-responsive heatmaps with dendrograms |
| ggplot2 R package | Foundation for graphics | Underlying plotting system for heatmaply visualizations |
| dendextend R package | Dendrogram customization | Enhancing cluster visualizations with color-coded branches |
| seriation R package | Optimal ordering algorithms | Improving pattern recognition through matrix arrangement |
| RColorBrewer package | Color palette management | Providing color-blind friendly and perceptually uniform schemes |
| Normalized expression matrix | Primary quantitative data | Input values for heatmap visualization (e.g., TPM, FPKM, log2CPM) |
| Clinical metadata table | Sample annotations | Patient demographics, treatment groups, outcome measures |
Experimental Protocol
Data Preparation and Preprocessing
Expression Matrix Standardization
Begin with a normalized expression matrix (e.g., log2 counts per million, TPM, or FPKM) where rows represent features (genes, transcripts) and columns represent samples. To enable meaningful comparisons across genes with different expression ranges, apply appropriate data transformation:
Scaling prevents variables with larger values from dominating the pattern recognition and ensures that genes with lower expression levels can contribute meaningfully to cluster formation [1].
Annotation Data Structure
Prepare metadata as a data frame with samples as rows and annotation variables as columns. Ensure sample identifiers match exactly between the expression matrix and metadata:
Interactive Heatmap Generation with Annotations
Basic Heatmap Construction
Create an interactive heatmap using the core heatmaply() function with appropriate color mapping:
The heatmaply_cor() function is specifically optimized for correlation matrices with built-in diverging color scales appropriate for the -1 to 1 value range [15].
Incorporating Sidebar Annotations
Add clinical or experimental annotations as colored sidebars using the side_color arguments. This requires creating a color mapping function for each annotation variable:
Customizing Hover Information
Enhance interactivity by incorporating metadata displays in hover tooltips using the custom_hovertext parameter:
This approach, adapted from a Stack Overflow solution for incorporating custom hover text [30], provides contextual information when users hover over specific heatmap cells.
Enhanced Visualization Configuration
Optimizing Color Scales
Select color palettes based on data characteristics and accessibility requirements:
Avoid rainbow color scales which can create misperceptions of magnitude due to abrupt changes between hues and inconsistent brightness interpretation across the data range [4].
Cluster Configuration
Customize clustering behavior to highlight biologically relevant patterns:
The seriation parameter controls the arrangement of rows and columns to optimize the visualization by minimizing the Hamiltonian path length restricted by the dendrogram structure [9].
Workflow and Data Integration
The following diagram illustrates the complete workflow for creating annotated interactive heatmaps, from data preparation to final visualization:
Workflow for Annotated Heatmap Creation: This diagram outlines the sequential process for integrating sample annotations with gene expression data to create interactive heatmaps, highlighting the key computational steps from raw data to final visualization.
Results and Data Interpretation
Annotation Integration Outcomes
Successful implementation of the protocol yields an interactive heatmap with the following characteristics:
- Colored sidebars adjacent to row and/or column dendrograms representing sample metadata
- Hierarchical clustering of samples and genes based on expression similarity
- Hover tooltips displaying both expression values and associated metadata
- Zoom capability for exploring specific regions of interest
- Dynamic reordering through dendrogram manipulation
Quantitative Output Specifications
Table 2: Configuration parameters for optimized heatmap visualization.
| Parameter | Recommended Setting | Impact on Visualization |
|---|---|---|
| Color Scale Type | Sequential for expression, Diverging for Z-scores | Ensures appropriate data representation [4] |
| Contrast Ratio | ≥ 3:1 for adjacent colors | Meets accessibility standards [13] |
| Cluster Method | Ward.D2 or complete | Balances cluster cohesion and separation |
| Distance Metric | Euclidean for samples, correlation for genes | Appropriate for respective data types |
| Annotation Position | Row and/or column sidebars | Clear association with relevant dimension |
| Hover Information | Expression + key metadata | Contextualizes individual values [30] |
Interpretation Guidelines
When analyzing the annotated heatmap, focus on these key aspects:
Cluster-Annotation Correspondence: Check whether sample clusters align with clinical annotations (e.g., treatment groups, disease stage), which may indicate biologically meaningful patterns.
Batch Effects: Identify whether technical annotations (e.g., processing date, sequencing batch) explain clustering patterns, suggesting potential technical artifacts requiring statistical correction.
Expression Patterns: Note genes with similar expression profiles across sample groups, potentially indicating co-regulation or shared functional pathways.
Outlier Identification: Detect samples that don't cluster with their expected groups, which may represent mislabeling, unique biological characteristics, or quality issues.
Discussion
Advanced Applications
The integration of sample annotations with interactive heatmaps enables several advanced analytical scenarios:
Treatment Response Analysis: Visualize whether gene expression patterns correlate with clinical response to therapeutic interventions.
Biomarker Discovery: Identify genes whose expression consistently associates with specific patient subgroups or disease characteristics.
Quality Assessment: Detect batch effects or technical artifacts that might confound biological interpretation.
Hypothesis Generation: Generate new research questions based on observed relationships between clinical metadata and expression patterns.
Technical Considerations
Several factors require particular attention during implementation:
Color Selection: Choose color palettes that provide sufficient contrast (≥3:1 ratio) while remaining interpretable for color-blind users [4] [13]. Avoid red-green combinations, which are problematic for approximately 5% of the population [4].
Data Scaling: Select appropriate transformation methods based on data distribution and analytical goals. Z-score standardization emphasizes relative differences across genes, while normalization to 0-1 range preserves original distribution shapes [9].
Computational Efficiency: For large datasets (e.g., whole transcriptome with thousands of samples), consider preliminary dimensionality reduction (e.g., filtering low-variance genes) to improve rendering performance.
Troubleshooting Common Issues
Table 3: Solutions to frequent challenges in annotated heatmap generation.
| Problem | Potential Cause | Solution |
|---|---|---|
| Mismatched annotations | Non-aligned sample identifiers | Verify consistent ordering between expression matrix and metadata |
| Poor color differentiation | Inadequate contrast between adjacent colors | Implement color-blind friendly palettes with sufficient luminance difference |
| Uninformative clustering | Inappropriate distance metric or clustering method | Experiment with alternative algorithms (e.g., Euclidean, Manhattan, correlation) |
| Overwhelming hover information | Excessive metadata in tooltips | Prioritize most relevant annotations for display |
| Slow rendering performance | Large input matrices | Implement data subsetting or use plotly optimization options |
Incorporating clinical and experimental metadata into interactive heatmaps significantly enhances their utility for genomic research and drug development. The protocols outlined here provide a comprehensive framework for transforming raw expression data into biologically insightful visualizations that facilitate pattern recognition, hypothesis generation, and communication of findings. By implementing these methods using the heatmaply package in R, researchers can create accessible, interactive visualizations that effectively integrate multiple data dimensions, ultimately supporting more informed scientific decisions and accelerating discovery processes.
The flexibility of the described approach allows adaptation to diverse research contexts, from exploratory analysis of novel datasets to publication-quality figures for scientific communications. As genomic technologies continue to evolve, these methods for visual integration of complex data types will remain essential for extracting meaningful biological insights from high-dimensional datasets.
In the field of genomic research, effective visual communication of complex data is paramount. Heatmaps serve as fundamental tools for visualizing high-dimensional gene expression data, where colored grids represent expression levels across multiple samples and conditions. The choice of color palette in these visualizations directly impacts analytical accuracy and interpretive reliability. Within the R ecosystem, the heatmaply package enables creation of interactive cluster heatmaps using 'plotly' and 'ggplot2' engines, providing researchers with powerful capabilities for exploring patterns in gene expression data through intuitive color encodings [15]. This protocol focuses specifically on optimizing color scale selection and implementation for enhanced scientific communication in gene expression analysis.
The psychological and perceptual aspects of color influence how data patterns are recognized and interpreted. Appropriate color schemes can highlight biologically significant patterns in gene expression data, while poor color choices may obscure critical findings or introduce interpretive bias. Furthermore, accessibility considerations for color vision deficiencies ensure research findings are communicable to the entire scientific community. This document provides comprehensive application notes for selecting, implementing, and validating color palettes within interactive heatmaps generated via the heatmaply package for gene expression studies.
Theoretical Foundations: Color Palette Taxonomy and Applications
Palette Classification for Scientific Visualization
Color palettes for scientific visualization fall into three primary categories, each with distinct applications in gene expression analysis:
Sequential Palettes: Progress from low to high saturation of single or multiple hues, representing ordered data from low to high values. Examples include viridis, Blues, and Greens. These are ideal for displaying non-negative gene expression values where the magnitude represents significance [31] [15].
Diverging Palettes: Emphasize deviation from a critical midpoint, typically using contrasting hues on opposite ends of the spectrum with a neutral central color. Examples include RdBu, PiYG, and cool_warm. These are particularly valuable for displaying gene expression fold-changes relative to a control condition or for z-score normalized expression matrices [15].
Qualitative Palettes: Employ distinct colors without inherent ordering, best suited for categorical annotations in heatmaps such as sample groups, tissue types, or experimental conditions [31].
Accessibility Considerations in Scientific Communication
Accessibility guidelines from WCAG 2.1 mandate minimum contrast ratios for visual information: 3:1 for graphical objects and user interface components, and 4.5:1 for normal text [13] [32]. These requirements ensure scientific visualizations are interpretable by researchers with visual impairments or color vision deficiencies. For gene expression heatmaps, sufficient contrast between adjacent colors in the scale ensures that expression gradients remain distinguishable across the entire data range. Tools such as the WebAIM Contrast Checker provide quantitative assessment of color pair contrast ratios [33].
Color vision deficiency (CVD) affects approximately 8% of the male population and 0.5% of the female population, necessitating palette selections that remain discriminable under common CVD conditions. Simulations using tools like colorspace::deutan() can verify palette effectiveness for CVD audiences.
Experimental Protocols: Implementation in Heatmaply
Protocol 1: Basic Color Scale Customization
Objective: Implement a sequential color palette for non-negative gene expression values.
Materials and Reagents:
- R statistical environment (version ≥ 3.0.0)
- heatmaply package (version ≥ 1.6.0)
- Gene expression matrix (rows = genes, columns = samples)
Methodology:
- Install and load required packages:
Create a basic sequential heatmap:
Customize with colorRampPalette:
Validation: Verify that color progression intuitively represents expression magnitude, with higher expression values corresponding to darker/more saturated colors.
Protocol 2: Diverging Palettes for Fold-Change Visualization
Objective: Implement a diverging color palette for z-score normalized gene expression data.
Methodology:
- Normalize expression data:
Apply diverging palette with symmetrical limits:
Implement custom diverging palette:
Troubleshooting: If colors appear washed out, verify that limits parameter encompasses the full data range. As noted in Stack Overflow discussions, improper limit specification can result in suboptimal color distribution [34].
Protocol 3: Advanced Palette Configuration with Accessibility Validation
Objective: Implement an accessible diverging palette with contrast validation.
Methodology:
- Create optimized diverging palette:
Add value annotations for critical regions:
Validate color contrast:
Validation: Use WebAIM Contrast Checker to verify adjacent colors in the palette achieve at least 3:1 contrast ratio [33].
Data Presentation: Color Palette Specifications
Table 1: Sequential Color Palettes for Gene Expression Heatmaps
| Palette Name | Color Range | Code Implementation | Best Use Case | Accessibility Rating |
|---|---|---|---|---|
| Viridis | #F1F3F4 to #4285F4 | viridis(256) |
General expression data | Excellent |
| Blues | #FFFFFF to #4285F4 | Blues(256) |
Non-negative values | Good |
| Custom Blue-White | #FFFFFF to #4285F4 | colorRampPalette(c("#FFFFFF", "#4285F4"))(256) |
Publication figures | Good |
| Greys | #FFFFFF to #202124 | Greys(256) |
Black & white publication | Fair |
Table 2: Diverging Color Palettes for Fold-Change Visualization
| Palette Name | Color Range | Code Implementation | Midpoint Handling | CVD Accessibility |
|---|---|---|---|---|
| RdBu | #EA4335 to #4285F4 | RdBu(256) |
Neutral (white) | Good |
| Cool-Warm | #EA4335 to #4285F4 | cool_warm(256) |
Perceptually neutral | Excellent |
| Custom Magenta-Black-Yellow | #EA4335 to #FBBC05 | colorRampPalette(c("#EA4335", "#202124", "#FBBC05"))(256) |
Explicit midpoint | Good |
| PiYG | #34A853 to #EA4335 | PiYG(256) |
Neutral (white) | Fair |
Table 3: Research Reagent Solutions for Heatmap Visualization
| Reagent/Software | Function | Application Note |
|---|---|---|
| heatmaply R package | Interactive heatmap generation | Enables zooming, hovering, and dendrogram manipulation [15] |
| colorRamp2 (circlize) | Smooth color interpolation | Essential for creating continuous color mappings [35] |
| viridis palette | Perceptually uniform sequential colors | Addresses color vision deficiency limitations [15] |
| WCAG 2.1 Guidelines | Accessibility standards | Ensure 3:1 contrast ratio for graphical objects [13] |
| WebAIM Contrast Checker | Color contrast validation | Quantitative verification of palette accessibility [33] |
Visual Integration: Workflow and Logical Relationships
Color Scale Selection Workflow for Gene Expression Heatmaps
Advanced Applications: Annotation Integration and Cluster Visualization
Integrated Annotation with Custom Color Schemes
Objective: Enhance heatmap interpretability through coordinated annotation colors.
Methodology:
Optimizing for Differential Expression Visualization
Objective: Highlight statistically significant differential expression patterns.
Methodology:
Validation and Quality Control
Perceptual Uniformity Assessment
Protocol: Verify that perceptual distance between color steps corresponds to equal data intervals.
Methodology:
- Generate a linear gradient test image with known values
- Convert to grayscale and measure luminance profile
- Verify linear luminance progression using photometric measurement
- Adjust palette if nonlinearities exceed 10% deviation
Cross-Platform Color Consistency
Validation Steps:
- Export heatmap to multiple formats (PDF, PNG, SVG)
- Verify color fidelity across display technologies
- Test print reproduction using standard color profiles
- Validate interactive behavior in web contexts
Effective color scale implementation in gene expression heatmaps requires thoughtful consideration of data characteristics, analytical objectives, and audience needs. The protocols outlined herein provide researchers with comprehensive methodologies for selecting, implementing, and validating color palettes that enhance scientific communication while maintaining accessibility standards. The integration of these practices within the heatmaply framework ensures creation of publication-quality visualizations that faithfully represent biological patterns while remaining interpretable across diverse audience capabilities. As interactive visualization technologies evolve, these fundamental principles of color science and accessibility will continue to underpin effective scientific communication in genomics and drug development research.
Within gene expression research, the transition from data analysis to publication and presentation necessitates robust methods for saving visualizations. Creating an interactive heatmap with heatmaply is only the first step; effectively exporting it for different contexts—whether as an interactive HTML file for exploration or a high-resolution static image for a manuscript—is crucial. This document provides detailed Application Notes and Protocols for exporting visualizations, framed within the broader thesis of creating an interactive gene expression heatmap, ensuring your research is shareable and reproducible.
Experimental Protocols
Protocol 1: Exporting an Interactive HTML File
Principle: Save the heatmaply object as a self-contained HTML file that preserves interactive features like zooming, tooltips, and clicking. This is ideal for exploratory data analysis, sharing with collaborators, or embedding in web reports [36].
Procedure:
- Create Heatmap Object: Generate your interactive cluster heatmap using the
heatmaply()function, specifying your gene expression matrix and desired parameters (e.g., clustering method, colors). - Export with
htmlwidgets: Use thesaveWidget()function from thehtmlwidgetspackage to export the plot. Troubleshooting: If the file size is large, consider usingsaveWidget(..., selfcontained = FALSE), which saves dependencies in a separate directory.
Protocol 2: Exporting a High-Resolution Static Image
Principle: Render the heatmap as a PNG file for inclusion in scientific publications, presentations, or posters. This protocol ensures high quality and sufficient resolution [37].
Procedure:
- Open Graphics Device: Initiate a PNG graphics device using the
png()function. Critically specify thewidth,height, andres(resolution) parameters to control the output dimensions and quality. - Render and Save Plot: The act of creating the heatmap while the device is open will direct the output to the file.
- Close Graphics Device: Use
dev.off()to finalize the file save. Failing to do this will result in an incomplete or empty file [37]. Troubleshooting: If an empty file is generated, ensure you are usingdraw()fromComplexHeatmapand have correctly closed the device withdev.off()[37]. Adjustwidth,height, andresto prevent overcrowding of labels.
Protocol 3: Generating a Static Image from an InteractiveheatmaplyObject
Principle: Directly export a static image from a heatmaply plot object using the export() function from the plotly package, which leverages the underlying plotting engine [36].
Procedure:
- Create Plotly Object: Create your heatmap using
heatmaply, which returns aplotlyobject. - Export the Plot: Use the
export()function orsave_image()from theplotlypackage to save the plot as a PNG or SVG. Note: This method may require additional system dependencies (e.g., theorcacommand-line utility) for offline export. Thewebshotpackage can serve as an alternative.
Table 1: Comparison of Heatmap Export Methods in R
| Method | Output Format | Key Features | Ideal Use Case | Critical Parameters |
|---|---|---|---|---|
htmlwidgets::saveWidget() |
Interactive HTML | Preserves zoom, tooltips, click events [36] | Collaborator review, web dashboards, data exploration | selfcontained = TRUE/FALSE |
png() + dev.off() |
Static PNG | High-resolution, publication-ready [37] | Manuscript figures, poster presentations | width, height, res [37] |
plotly::export() |
Static PNG/SVG | Direct export from plotly/heatmaply object |
Quick static snapshot of interactive plot | (Requires orca or webshot) |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software Tools for Heatmap Visualization and Export
| Tool / Reagent | Function / Role in Experiment | Key Feature for Export |
|---|---|---|
heatmaply R Package |
Generates interactive cluster heatmaps using plotly [36] |
heatmaply() function returns a plotly object suitable for both HTML and static export [36]. |
ComplexHeatmap R Package |
Provides highly customizable static heatmaps [37] | draw() function is essential for rendering the heatmap to a graphics device for saving [37]. |
htmlwidgets R Package |
Framework for embedding interactive JavaScript visualizations in R | saveWidget() function is the standard method for saving interactive widgets as HTML files. |
plotly R Package |
Underlying engine for interactivity in heatmaply |
export() or save_image() functions enable direct static image export from interactive objects [36]. |
| R Graphics Device | (e.g., png, pdf, svg) |
Controls the format and dimensions (width, height, res) of saved static images [37]. |
Workflow Diagram
The following diagram outlines the logical decision process and workflow for saving your gene expression heatmap visualizations, incorporating both interactive and static export paths.
Solving Common Challenges: Optimization Strategies for Large-Scale Data
The creation of interactive gene expression heatmaps is a cornerstone of genomic analysis, enabling researchers to visualize complex patterns in high-dimensional data. The heatmaply R package is a powerful tool for this purpose, generating interactive cluster heatmaps using the plotly and ggplot2 engines [5] [15]. However, as the scale of genomic studies expands—encompassing thousands of genes across hundreds or thousands of samples—researchers face significant computational challenges related to memory management, processing speed, and visualization clarity. This application note provides detailed protocols and optimization strategies for managing these computational resources effectively when working with large gene sets and sample sizes within the heatmaply framework, ensuring analyses remain both feasible and interpretable.
Optimization Strategies for Large-Scale Data
Computational Enhancements and Data Handling
Table 1: Optimization Strategies for Large-Scale Heatmaps
| Strategy | Implementation | Benefit |
|---|---|---|
| Parallel Processing | Utilize built-in parallelization in GeneSetCluster 2.0 [38] | Significantly reduces execution time for clustering operations |
| Data Subsetting | Filter to top variable genes or most significant hits prior to visualization | Reduces matrix dimensions and memory footprint |
| Dendrogram Control | Set Rowv = FALSE or Colv = FALSE in heatmaply() to suppress dendrogram calculation [15] |
Eliminates computationally expensive hierarchical clustering |
| Data Aggregation | Average expression across sample replicates or group genes by functional clusters | Decreases the number of data points without losing biological signal |
| Interactive Inspection | Leverage plotly's hover and zoom features for large matrices [5] | Avoids creating multiple static plots for different regions of interest |
Addressing Data Redundancy
Large-scale gene-set analysis (GSA) often identifies thousands of overlapping processes, complicating both computation and interpretation [38]. GeneSetCluster 2.0 introduces a "Unique Gene-Sets" methodology to address duplicated gene-sets (e.g., identical Gene Ontology IDs) that appear across multiple GSA results. This method detects repeated gene-sets and merges them into a single entry containing the union of all associated genes, thereby eliminating clustering bias caused by duplications and simplifying the input data for heatmap visualization [38].
Step-by-Step Protocol for Large-Scale Heatmap Creation
Data Preprocessing and Dimensionality Reduction
- Filter Gene Sets: Begin with a curated list of non-redundant gene sets. Apply the "Unique Gene-Sets" method from GeneSetCluster 2.0 to merge duplicates, or filter gene lists based on statistical significance (e.g., FDR-adjusted p-value < 0.05) and effect size (e.g., fold-change > 2) [38] [39].
- Reduce Sample Dimensionality: If working with an extremely large number of samples, consider aggregating technical replicates or using dimensionality reduction techniques (e.g., PCA) to group similar samples before heatmap generation.
- Normalize Data: Ensure the data matrix (e.g., gene expression values) is properly normalized to facilitate accurate color encoding and comparison across the heatmap.
Creating an Optimized Heatmap withheatmaply
The following code block demonstrates the creation of an interactive heatmap with parameters optimized for larger datasets.
Protocol Notes:
- Dendrograms: Suppressing dendrograms (
Rowv = FALSE,Colv = FALSE) provides a major speed increase for very large matrices [15]. - Clustering: The
distfunandhclustfunarguments allow the use of alternative, faster distance and clustering functions if needed. - Saving Output: For easy sharing and viewing outside of R, save the heatmap as an HTML file using the
fileparameter. For static images (PNG/PDF), use thewebshotpackage as outlined in theheatmaplydocumentation [5].
Workflow for Iterative Refinement and Sub-Clustering
Interpreting large heatmaps can be challenging. The following workflow, also depicted in the diagram below, allows for iterative refinement.
Workflow for Iterative Heatmap Refinement
- Generate Initial Heatmap: Create a comprehensive heatmap of the preprocessed, filtered data.
- Identify Cluster of Interest: Use the interactive features to hover and zoom, identifying specific gene or sample clusters that display interesting patterns.
- Extract and Sub-cluster: Apply the
BreakUpClusterfunction from GeneSetCluster 2.0 to select a gene-set cluster and identify finer sub-clusters within it [38]. This targeted refinement allows for detailed exploration of specific relationships without recomputing the entire original analysis. - Refine Biological Insight: Create a new, focused heatmap of the sub-cluster to gain deeper biological interpretation. This process can be repeated iteratively.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Software and Packages for Interactive Heatmap Creation
| Item | Function | Application in Protocol |
|---|---|---|
| heatmaply R Package | Primary engine for creating interactive cluster heatmaps using 'plotly.js' [5] [15]. | Core visualization tool for generating the final interactive output. |
| plotly Library | Provides the underlying interactive graphics engine for heatmaply [5]. |
Enables hover-inspection, zooming, and panning within the heatmap. |
| GeneSetCluster 2.0 | An R package for summarizing and integrating gene-set analysis results, with optimized parallel processing [38]. | Used upstream for de-duplicating gene-sets and performing sub-cluster analysis. |
| seriation Package | Provides algorithms for reordering matrices and dendrograms to highlight patterns [15]. | Used internally by heatmaply to improve the visual structure of the heatmap. |
| dendextend Package | Tools for manipulating and visualizing dendrograms in R [15]. | Enhances the customization and appearance of dendrograms in heatmaply plots. |
| viridis Color Palette | Provides perceptually uniform and colorblind-friendly color maps [15]. | Default color scale in heatmaply for accurately representing data magnitude. |
| RColorBrewer Palettes | Provides sequential, diverging, and qualitative color schemes suitable for data visualization. | Can be passed to the colors argument in heatmaply for custom color scales [15]. |
Effectively managing computational resources is paramount for the successful visualization of large-scale genomic data. By integrating the interactive capabilities of the heatmaply package with strategic data management practices—including pre-filtering, redundancy handling with tools like GeneSetCluster 2.0, and selective suppression of computational overhead—researchers can navigate the challenges posed by massive gene sets and sample sizes. The protocols outlined herein provide a robust framework for creating performant and interpretable interactive heatmaps, thereby facilitating the extraction of meaningful biological insights from complex omics datasets.
Z-score normalization, or standardization, is a fundamental statistical preprocessing technique that transforms data to have a mean of zero and a standard deviation of one [40]. In the context of gene expression analysis, this method enables meaningful comparison of expression levels across different samples and experimental conditions by removing systematic biases and variations [41] [42]. This protocol details the theoretical foundations, practical implementation, and specific applications of Z-score normalization for creating interactive gene expression heatmaps using the heatmaply R package, providing researchers and drug development professionals with a standardized framework for reproducible analysis.
Gene expression data generated through high-throughput technologies like RNA sequencing (RNA-Seq) and microarrays typically contains measurements on different scales, making direct comparisons problematic [41] [42]. Z-score normalization addresses this challenge by transforming the data into a common scale, allowing researchers to identify patterns and relationships that would otherwise be obscured by technical variations.
The mathematical foundation of Z-score normalization is expressed by the formula:
Z = (X - μ) / σ
where X represents the original data point, μ signifies the mean of the dataset, σ denotes the standard deviation, and Z is the resulting normalized value [41] [40]. This transformation expresses each data point in terms of its distance from the mean in units of standard deviation, creating a dimensionless measure that facilitates comparison across different datasets and experimental conditions [43].
In biological applications, Z-scores for gene expression are typically calculated on a gene-by-gene basis across samples [44]. This row-wise normalization enables researchers to determine whether a gene is expressed higher (positive Z-score) or lower (negative Z-score) in specific samples compared to the average across all samples [43]. A Z-score of zero indicates expression identical to the mean expression level of that gene across all measured samples.
Table 1: Interpretation of Z-score Values in Gene Expression Analysis
| Z-score Range | Interpretation | Biological Significance |
|---|---|---|
| Z ≥ 2.0 | Significantly high expression | Potential up-regulation |
| 0.5 ≤ Z < 2.0 | Moderately high expression | Possible biological relevance |
| -0.5 < Z < 0.5 | Average expression | Baseline expression level |
| -2.0 < Z ≤ -0.5 | Moderately low expression | Possible biological relevance |
| Z ≤ -2.0 | Significantly low expression | Potential down-regulation |
When to Apply Z-score Normalization
Appropriate Use Cases
Z-score normalization is particularly beneficial in several scenarios common to gene expression analysis:
Clustering Analysis: Z-score normalization is crucial for clustering algorithms (K-means, hierarchical clustering) and principal component analysis (PCA) that rely on distance calculations [45] [1]. Without normalization, genes with naturally higher expression levels would dominate the distance metrics, potentially obscuring biologically relevant patterns.
Cross-Platform Comparisons: When integrating gene expression data from different platforms (RNA-Seq, microarrays) or laboratories, Z-score transformation standardizes data across experiments, making them comparable despite differences in original hybridization intensities or measurement techniques [41].
Heatmap Visualization: For interactive heatmap generation with heatmaply, Z-score normalization ensures color intensity accurately reflects relative expression patterns rather than absolute abundance [45] [44]. This is particularly important when visualizing genes with different baseline expression levels.
Outlier Detection: Z-scores beyond ±3 standard deviations from the mean may indicate potential outliers that warrant further investigation, whether due to technical artifacts or biologically significant extreme values [40].
Limitations and Alternatives
While powerful, Z-score normalization is not universally appropriate:
Normal Distribution Assumption: Z-score normalization performs optimally when data approximates a normal distribution [40]. For heavily skewed distributions, alternative transformations like log transformation may be preferable as an initial step.
Small Sample Considerations: With small sample sizes (n < 30), Z-score estimates may be unstable. In such cases, alternative methods like quantile normalization may be more robust [42].
Binary Variables: For binary variables (e.g., presence/absence indicators), Z-score normalization may not be appropriate, and percentile transformation often provides better performance [9] [45].
Table 2: Data Normalization Methods Comparison
| Method | Formula | Best Use Cases | Limitations |
|---|---|---|---|
| Z-score Normalization | Z = (X - μ)/σ | Distance-based algorithms, normally distributed data | Assumes normal distribution |
| Min-Max Normalization | (X - min)/(max - min) | Neural networks, image processing | Sensitive to outliers |
| Quantile Normalization | Rank-based redistribution | Microarray data, removing technical biases | Assumes same distribution shape |
| Percentile Transformation | ecdf(X) | Non-normal distributions, ordinal data | Not symmetric for binary variables |
| Log Transformation | log(X) | Skewed data, RNA-Seq counts | Cannot handle zero values without adjustment |
Experimental Protocols
Protocol 1: Z-score Normalization of RNA-Seq Data for Heatmap Visualization
This protocol describes the complete workflow for normalizing RNA-Seq count data and generating an interactive heatmap using the heatmaply package.
Materials and Reagents
Table 3: Essential Research Reagent Solutions
| Item | Function | Example/Specification |
|---|---|---|
| R Statistical Software | Data analysis environment | Version 4.0.0 or higher |
| heatmaply R Package | Interactive heatmap generation | Version 1.3.0 or higher [9] |
| Normalized Count Matrix | Input expression data | DESeq2 or edgeR normalized counts [44] |
| Annotation Data | Sample metadata | Clinical variables, treatment groups |
| plotly R Package | Interactive visualization backend | Required for heatmaply rendering [46] |
Procedure
Data Preprocessing
- Begin with normalized count data (e.g., DESeq2-normalized counts or log2-CPM values) [46] [44]. Normalization accounts for differences in library size and RNA composition.
- Filter genes to include those of biological interest (e.g., differentially expressed genes, pathway-specific genes).
- For RNA-Seq count data, a log2 transformation (log2(count + 0.5)) is recommended before Z-score normalization to address skewness [46].
Z-score Normalization Implementation
- Calculate Z-scores for each gene across all samples using the formula in Section 1.
In R, this can be accomplished with the
scale()function, which centers (subtracts mean) and scales (divides by standard deviation) the data:Alternatively, perform row-wise Z-score normalization manually:
Interactive Heatmap Generation with heatmaply
Create a basic interactive heatmap:
Enhance interpretation by adding sample annotations:
Interpretation and Validation
- In the resulting heatmap, red hues typically indicate positive Z-scores (higher expression), blue hues indicate negative Z-scores (lower expression), and white represents average expression [44].
- Validate normalization by examining whether replicate samples cluster together in the dendrogram.
- Identify potential batch effects that may require additional correction.
Protocol 2: Microarray Data Normalization and Cross-Experiment Comparison
This protocol adapts Z-score normalization for microarray data, enabling comparison across multiple experiments.
Procedure
Data Preprocessing
- Begin with background-corrected and log-transformed microarray intensity values [41].
- Apply quality control filters to remove problematic probes or arrays.
Experiment-Specific Z-score Normalization
For each experiment separately, apply Z-score normalization to all probe intensities:
This within-experiment normalization corrects for technical variations between hybridizations [41].
Cross-Experiment Comparison
- Combine Z-score normalized data from multiple experiments.
- Compare gene expression patterns using Z-ratios (differences between Z-scores) or statistical tests such as the two-sample Z-test [41].
Visualization of Combined Dataset
- Generate a heatmap that includes samples from all normalized experiments:
Implementation Workflow
The following diagram illustrates the complete Z-score normalization and heatmap generation workflow for gene expression data:
Technical Considerations and Troubleshooting
Data Quality Assessment
Before applying Z-score normalization, assess data quality through:
- Sample Correlation Analysis: Generate a correlation heatmap of samples using
heatmaply_cor()to identify outliers or mislabeled samples [9]:
- Missing Data Evaluation: Visualize missing data patterns with
heatmaply_na()to identify systematic missingness [9].
Advanced heatmaply Customization
Enhance biological interpretability through advanced heatmaply features:
- Dendrogram Customization: Adjust cluster analysis using seriation methods:
- Color Scheme Selection: Use colorblind-friendly palettes for publication:
Z-score normalization provides an essential preprocessing step for gene expression analysis, particularly when generating interactive heatmaps with heatmaply. By transforming data to a common scale, this method enables meaningful comparison of expression patterns across genes, samples, and experiments. The protocols outlined in this document establish a standardized approach for researchers and drug development professionals to implement Z-score normalization in their gene expression workflows, facilitating reproducible and biologically insightful visualization of high-dimensional data.
Dendrograms are tree-like diagrams that visually represent hierarchical clustering results, showing the relationships and similarity between data points. In gene expression analysis, they are indispensable tools for identifying patterns, subgroups, and anomalies within complex biological datasets. When combined with heatmaps, dendrograms provide a powerful visualization that reveals how both samples and genes cluster based on expression profiles, enabling researchers to identify co-expressed genes, sample subgroups, and potential outliers that may represent novel biological insights or data quality issues.
The interpretation of these dendrogram patterns requires understanding several computational approaches. Hierarchical clustering employs various distance metrics (such as Euclidean or correlation distance) to quantify similarity between expression profiles, and different clustering algorithms (including average, complete, or Ward's method) to build the tree structure. The resulting dendrogram branches represent the degree of similarity - shorter branches indicate higher similarity, while longer branches suggest greater divergence. This structural information helps researchers determine natural groupings in their data and identify potential anomalies that warrant further investigation.
Fundamental Concepts and Terminology
Dendrogram Structure and Components
A dendrogram consists of several key components that encode clustering information:
- Leaves: Terminal endpoints representing individual data points (samples or genes)
- Nodes: Branch points where clusters merge, representing the union of two or more leaves
- Branches: Lines connecting nodes to leaves or other nodes
- Branch Height: Represents the distance or dissimilarity between merging clusters
- Clusters: Groups formed by cutting the dendrogram at a specific height
The structure reveals both grouping patterns and relative similarities, with vertical position indicating similarity level (lower merges indicate higher similarity). The overall topology provides insights into the data's organization, including potential subgroups and outliers.
Distance Metrics and Clustering Methods
The appearance and interpretation of dendrograms depend heavily on the chosen distance metric and clustering method:
Table 1: Common Distance Metrics in Gene Expression Analysis
| Distance Metric | Calculation Approach | Best Use Cases |
|---|---|---|
| Euclidean Distance | Straight-line distance between points in n-dimensional space | When absolute expression differences matter |
| Correlation Distance | 1 - correlation coefficient between profiles | When expression pattern similarity is important |
| Manhattan Distance | Sum of absolute differences along each dimension | When dealing with high-dimensional data |
| Maximum Distance | Maximum difference between any corresponding dimensions | When conservative distance estimates are needed |
Table 2: Hierarchical Clustering Methods
| Clustering Method | Approach to Defining Cluster Similarity | Tendency in Cluster Formation |
|---|---|---|
| Average Linkage (UPGMA) | Mean distance between all pairs of elements | Balanced cluster sizes |
| Complete Linkage | Maximum distance between elements | Compact, similarly-sized clusters |
| Single Linkage | Minimum distance between elements | Elongated, "chained" clusters |
| Ward's Method | Minimizes within-cluster variance | Spherical, tight clusters |
The choice of distance metric significantly affects results. Euclidean distance works well when absolute expression values are important, while correlation distance (1 - correlation coefficient) better captures pattern similarity regardless of absolute expression levels [47]. For gene expression data, correlation distance often provides more biologically meaningful clusters as it groups genes with similar expression patterns across conditions.
Experimental Protocols for Dendrogram Analysis
Data Preprocessing and Scaling Protocol
Proper data preprocessing is essential for meaningful clustering results:
Data Import and Validation
- Load normalized expression data (e.g., log2 CPM, TPM, or FPKM values)
- Verify data integrity and absence of missing values
- Transform count data using appropriate methods (e.g., log2 for RNA-seq)
Data Scaling and Centering
Hierarchical Clustering and Dendrogram Generation
This protocol generates dendrograms using the pheatmap package with appropriate parameters:
For interactive exploration using heatmaply:
Workflow for Systematic Dendrogram Interpretation
The following diagram illustrates the comprehensive workflow for generating and interpreting dendrograms:
Interpreting Dendrogram Patterns and Identifying Anomalies
Normal Dendrogram Patterns and Biological Significance
Well-structured dendrograms reveal several characteristic patterns with specific biological interpretations:
Tight Sample Clustering: Samples with short branch lengths and early merging typically represent biological replicates or samples from the same experimental condition. In the airway dataset analysis, control samples and dexamethasone-treated samples formed distinct clusters, validating the experimental treatment effect [1].
Co-expressed Gene Modules: Genes that cluster together with short branch lengths often represent functionally related genes, such as those in the same pathway or regulated by the same transcription factors. The PAM50 gene set visualization showed characteristic clustering patterns corresponding to breast cancer molecular subtypes [48].
Stratified Sample Grouping: Clear separation of samples into major branches often corresponds to major experimental variables or biological subtypes. In TCGA BRCA data, samples frequently cluster by PAM50 subtype (Luminal A, Luminal B, HER2-enriched, Basal-like) when using appropriate distance metrics [48].
Graduated Branching Patterns: Progressive branching with increasing height suggests a continuum of expression states rather than discrete classes, which is common in developmental timecourses or gradual treatment responses.
Common Anomalies and Their Interpretation
Unexpected dendrogram patterns often reveal technical artifacts or novel biological insights:
Outlier Samples: Isolated samples with long branch lengths connecting to main clusters may indicate:
- Sample mislabeling or contamination
- Unique biological states or rare subtypes
- Technical artifacts during library preparation or sequencing
Inconsistent Replicate Clustering: Biological replicates that don't cluster together suggest:
- Batch effects requiring statistical correction
- Inconsistent experimental conditions
- High biological variability
Unexpected Cross-Group Clustering: Samples from different conditions clustering together may indicate:
- Misannotation of sample metadata
- Shared biological processes across presumed distinct conditions
- Inadequate experimental separation between conditions
Unbalanced Tree Topology: Markedly asymmetric dendrogram structure suggests:
- Presence of dominant subgroups with internal heterogeneity
- Inappropriate choice of distance metric or clustering method
- Need for data transformation or normalization
Table 3: Dendrogram Anomalies and Recommended Actions
| Anomaly Type | Potential Causes | Investigation Approaches |
|---|---|---|
| Isolated Outlier Samples | Sample quality issues, rare cell types | Check RNA quality metrics, explore as potential novel subtype |
| Replicates Not Co-clustering | Batch effects, technical variability | Perform PCA, implement batch correction, examine QC metrics |
| Weak Cluster Separation | Insufficient treatment effect, high noise | Increase sample size, check power, consider alternative normalization |
| Unexpected Sample Grouping | Sample mislabeling, shared biology | Verify sample metadata, explore marker genes for unexpected groups |
Diagnostic Procedures for Anomaly Investigation
When anomalies are detected, systematic investigation is essential:
Technical Quality Control Review
- Examine RNA integrity numbers (RIN) and library quality metrics
- Verify alignment rates and sequencing depth uniformity
- Check for spatial or temporal batch effects
Biological Plausibility Assessment
- Verify cluster patterns against known biological expectations
- Examine expression of housekeeping and marker genes
- Correlate with clinical or phenotypic metadata
Methodological Robustness Testing
- Test multiple distance metrics and clustering algorithms
- Assess stability through bootstrap or subsampling approaches
- Compare with alternative visualization methods (PCA, t-SNE)
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Tools for Dendrogram Analysis in Gene Expression Studies
| Tool/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Heatmap Generation Packages | pheatmap, ComplexHeatmap, heatmap.2 | Static heatmap and dendrogram generation | Publication-quality figures, routine analysis |
| Interactive Visualization | heatmaply, plotly, shiny | Interactive data exploration | Data quality assessment, hypothesis generation, collaboration |
| Distance Metrics | Euclidean, correlation, Manhattan | Quantifying sample/gene similarity | Varies by data structure and biological question |
| Clustering Algorithms | Average, complete, Ward linkage | Defining cluster relationships | Dependent on expected cluster characteristics |
| Color Palettes | Spectral, RdBu, BrBG, custom | Visual encoding of expression values | Optimized for color vision deficiency, publication requirements |
| Data Scaling Methods | Z-score, median centering, normalization | Making expression profiles comparable | Essential for cross-sample/gene comparison |
Advanced Analysis: Correlation Heatmaps and Sample Relationships
Beyond standard expression heatmaps, correlation heatmaps with dendrograms provide valuable diagnostic information about sample relationships:
Correlation-based dendrograms are particularly valuable for identifying:
- Batch effects through temporal or processing-based clustering
- Sample mix-ups or mislabeling
- Outlier samples with poor correlation to expected groups
- Biological replicate consistency within experimental conditions
In the airway dataset analysis, correlation heatmaps confirmed that biological replicates showed higher correlation with each other than with samples from different treatment conditions, validating experimental integrity [1].
Validation and Biological Interpretation Framework
Robust interpretation requires validating dendrogram patterns through multiple approaches:
Statistical Support Assessment
- Apply bootstrap resampling to estimate cluster stability
- Calculate approximately unbiased (AU) p-values using pvclust
- Assess cluster robustness to parameter changes
Biological Context Integration
- Annotate clusters with gene ontology enrichment analysis
- Correlate with clinical outcomes or phenotypic measurements
- Compare with established molecular classifications
Multi-Method Verification
- Compare with k-means or PAM clustering results
- Verify patterns using dimensionality reduction (PCA, t-SNE, UMAP)
- Assess consistency across independent datasets
The PAM50 breast cancer analysis demonstrated this approach, where dendrogram patterns were interpreted in the context of established subtype classifications and clinical outcomes [48]. This multi-faceted validation ensures that observed patterns represent biologically meaningful relationships rather than technical artifacts or random noise.
Successful interpretation of dendrogram patterns and anomalies enables researchers to generate robust biological hypotheses, identify potential data quality issues, and make informed decisions about subsequent analytical approaches and experimental validation.
Overplotting presents a significant challenge in the visual analysis of high-dimensional biological data, such as gene expression studies. It occurs when a large number of data points overlap in a visualization, obscuring patterns and relationships essential for scientific discovery. This issue is particularly prevalent in traditional scatter plots representing hundreds or thousands of genes across multiple samples. Heatmaps effectively address this limitation by transforming numerical data into a grid of colored cells, where color intensity represents values, enabling researchers to discern patterns that would otherwise remain hidden in overplotted displays [31]. Within the R ecosystem, the heatmaply package provides an implementation for creating interactive cluster heatmaps, which are particularly valuable for genomic research applications [5] [9].
The fundamental strength of the heatmap for addressing overplotting lies in its data aggregation approach. Rather than plotting individual points that inevitably overlap, a heatmap divides the data space into discrete bins and represents the density or summary statistic of each bin through color [31]. This transformation from point-based to area-based visualization preserves the overall distribution and correlation patterns while eliminating visual congestion. When implemented interactively, as with heatmaply, this approach allows researchers to maintain a comprehensive overview of their dataset while retaining the ability to inspect individual values through hover effects and zooming capabilities [9].
Practical Strategies for Overplotting Mitigation
Data Transformation Techniques
Effective management of overplotted data requires appropriate transformation techniques to enhance pattern recognition. The heatmaply package provides multiple transformation functions to prepare data for optimal visualization. The normalize function rescales variables to a 0-1 range by subtracting the minimum and dividing by the maximum, preserving distribution shapes while enabling direct comparison across measurements [9]. The percentile function converts values to their empirical percentile, providing an intuitive interpretation where each value represents the percentage of observations at or below that level [9]. For normally distributed data, the scale parameter performs column-wise or row-wise Z-score standardization, centering data around zero with standard deviation units [9].
Different transformation methods reveal distinct aspects of the data. The normalize function is particularly valuable for preserving the shape of non-normal distributions, while percentile handling of tie values may affect clustering outcomes. The choice of transformation should align with both the data characteristics and the biological question, as each method emphasizes different patterns and relationships within the dataset [9].
Dimensionality Organization with Seriation
The arrangement of rows and columns significantly impacts a heatmap's interpretability. The heatmaply package implements several seriation algorithms through the seriation package to optimize element ordering [9]. The "OLO" (Optimal Leaf Ordering) method computes the optimal Hamiltonian path length restricted by the dendrogram structure, effectively arranging elements to minimize the sum of distances between adjacent leaves [9]. The "GW" (Gruvaeus and Wainer) method provides a faster heuristic approach to similar optimization, while the "mean" method replicates the default ordering found in traditional heatmap functions like gplots::heatmap.2 [9].
For genomic applications, seriation works in concert with clustering distance metrics to group genes with similar expression patterns and samples with similar expression profiles. The combination of appropriate distance metrics (Euclidean, Manhattan, correlation-based) with optimized ordering enables researchers to identify co-expressed gene modules and sample subgroups that might remain hidden in overplotted alternative visualizations [1].
Table 1: Data Transformation Functions in heatmaply
| Function | Method | Best Use Cases | Considerations |
|---|---|---|---|
normalize |
Scales to [0,1] by (x-min(x))/max(x) | Non-normal distributions, preserving original shape | Maintains distribution shape but not always ideal for clustering |
percentile |
Converts to empirical percentile (ECDF) | Intuitive interpretation, non-parametric approach | Handling of ties may affect clustering results |
scale |
Z-score standardization (center and scale) | Normally distributed data, parametric tests | Sensitive to outliers, assumes approximate normality |
Implementation Protocol: Interactive Gene Expression Heatmap
Data Preparation and Loading
Begin by installing and loading required packages in R. The core functionality resides in heatmaply, with complementary packages enhancing its utility for genomic applications [6]:
For gene expression data, import normalized expression values with genes as rows and samples as columns. The data should be structured as a numeric matrix or data frame, with appropriate row and column names [1]:
Interactive Heatmap Generation
Create a basic interactive heatmap with default parameters to initially explore the data structure and identify potential clustering patterns [9]:
For specialized applications, employ wrapper functions optimized for specific analytical contexts. The heatmaply_cor function simplifies correlation matrix visualization, while heatmaply_na effectively highlights missing data patterns [9]:
Table 2: heatmaply Parameters for Genomic Data Visualization
| Parameter | Function | Recommended Setting for Gene Expression |
|---|---|---|
scale |
Data standardization | "row" for gene expression (genes as rows) |
seriate |
Row/column ordering | "OLO" (optimal leaf ordering) |
k_col/k_row |
Predefined clusters | Determined by experimental design |
colors |
Color palette | viridis, RdBu, or custom diverging palette |
dendrogram |
Tree display | "both" for full hierarchical clustering |
showticklabels |
Label visibility | c(TRUE, FALSE) for large matrices |
Advanced Customization for Publication
Customize the heatmap for publication-quality figures or interactive supplements. Incorporate sample annotations, adjust color schemes for color vision deficiency, and optimize layout parameters [9]:
Visual Workflow and Research Toolkit
Experimental Workflow Diagram
Research Reagent Solutions
Table 3: Essential Computational Tools for Interactive Heatmap Generation
| Tool/Resource | Function | Application Context |
|---|---|---|
heatmaply R package |
Interactive heatmap generation | Primary visualization engine with ggplot2/plotly backend |
dendextend R package |
Dendrogram customization | Enhanced tree visualization and manipulation |
seriation R package |
Matrix ordering algorithms | Optimized arrangement of rows and columns |
RColorBrewer |
Color palette management | Colorblind-friendly and perceptual color schemes |
| Normalized expression matrix | Input data structure | Gene-by-sample tabular format with normalized values |
| Sample annotation data frame | Experimental metadata | Treatment groups, batches, phenotypic information |
Technical Specifications and Accessibility
Color Contrast and Visualization Accessibility
Effective heatmaps require careful attention to color contrast to ensure accessibility and interpretability. The WCAG 2.2 guidelines recommend a minimum contrast ratio of 3:1 for graphical components, with higher ratios for text elements [49]. The color palette specified for these visualizations has been selected to provide sufficient contrast between adjacent colors while maintaining color vision deficiency compatibility [50] [51].
When creating heatmaps for publication or web deployment, several contrast considerations apply. For text elements within the visualization (axis labels, legends), maintain a contrast ratio of at least 4.5:1 against the background [52] [53]. For large-scale text (approximately 18pt or 14pt bold), a slightly lower ratio of 3:1 may be acceptable, though higher ratios improve readability across viewing conditions [49]. The heatmaply default viridis palette provides excellent perceptual characteristics and color vision deficiency compatibility, though custom palettes should be verified for contrast adequacy [9].
Optimization for High-Density Genomic Data
Visualizing genome-scale datasets (thousands of genes across hundreds of samples) requires computational optimization to maintain interactivity. The heatmaply package leverages the plotly.js engine, which efficiently handles larger matrices compared to alternative implementations [5]. For extremely large datasets, strategic subsetting approaches may be necessary, such as filtering by variance or focusing on differentially expressed gene subsets [1].
Performance optimization strategies include adjusting the plot_method parameter to "plotly" for reduced memory footprint, limiting the number of simultaneously displayed labels through the showticklabels parameter, and utilizing the file parameter to export static versions for archival purposes [9]. When computational resources are constrained, the pheatmap package provides a high-quality static alternative with similar clustering capabilities [1].
In gene expression analysis, heatmaps serve as a fundamental tool for visualizing complex patterns of differential expression across multiple experimental conditions. The choice of color scale is not merely an aesthetic decision but a critical factor influencing the interpretation of biological data. Unlike general-purpose graphics, scientific heatmaps must convey quantitative information accurately, ensuring that visual perception aligns with underlying statistical values. This application note examines color scale optimization within the heatmaply R package, an environment for creating interactive heatmaps, with emphasis on maintaining data integrity across various output devices and for diverse audiences, including those with color vision deficiencies.
The fundamental challenge in heatmap visualization lies in creating a mapping between expression values and color that is both intuitively understandable and scientifically precise. Research indicates that inconsistent color application can lead to misinterpretation of data patterns, particularly when analyses are reviewed across research teams using different display technologies or when published in various formats. Within genomics and drug development, where heatmaps frequently represent log-fold changes in gene expression, appropriate color scaling directly impacts biological interpretation and subsequent research decisions.
Established Color Conventions in Bioinformatics
Current Practices and Historical Context
Despite widespread use of color-coded heatmaps in bioinformatics, no formal universal standard governs color assignment in differential gene expression visualization. Community practice has led to several conventions, though inconsistencies persist. Surveys of bioinformatics practitioners reveal approximately equal division between those who associate red with upregulated genes and those who intuitively expect the opposite mapping, demonstrating the absence of consensus [54].
Traditionally, microarray and early RNA-seq analyses frequently employed a red-black-green scheme, with red indicating upregulation, black representing neutral values, and green signifying downregulation [54]. This convention emerged during the microarray era and persists as default in some software packages. However, this scheme presents significant accessibility concerns and has been largely superseded by more perceptually uniform alternatives.
More recently, the field has shifted toward red-white-blue or red-yellow-blue schemes, which offer improved accessibility for colorblind users and better visual distinction on white backgrounds commonly used in publications [54]. These divergent colormaps place neutral values at an intermediate color (white or yellow), with progression to red for positive values and blue for negative values, creating intuitive "hot" and "cold" associations.
Accessibility Considerations
The prevalent red-green color scheme presents significant challenges for individuals with color vision deficiencies (affecting approximately 8% of the male population). Consequently, major bioinformatics resources and publications increasingly recommend avoiding red-green combinations in favor of colorblind-friendly alternatives [54]. The heatmaply package facilitates this transition through built-in perceptually uniform colormaps and customizable scale options.
Color Scale Implementation in Heatmaply
Core Color Functions and Parameters
The heatmaply package provides multiple interfaces for color control, ranging from simple preset colormaps to fully customized gradient definitions. Implementation occurs primarily through the col and scale_fill_gradient_fun parameters, which enable different levels of specification complexity.
Preset Colormaps: heatmaply includes several built-in colormaps optimized for scientific visualization:
viridis- Perceptually uniform, colorblind-friendly (default)cool_warm- Divergent scheme with cyan to magenta transitionPurpleAndYellow- Custom divergent schemeRdBu- Red to blue divergent scheme
Implementation requires simple parameter assignment:
Custom Gradient Specification: For precise control, researchers can define custom color gradients using scale_fill_gradient_fun with explicit low, mid, and high color definitions:
This approach provides explicit control over the critical midpoint value and range limits, ensuring consistent mapping between expression values and colors across multiple visualizations.
Critical Parameter Configuration
Table 1: Essential Color Parameters in Heatmaply
| Parameter | Function | Recommended Setting |
|---|---|---|
limits |
Defines value range for color mapping | Should encompass data range: limits = c(min_val, max_val) |
midpoint |
Sets neutral value position | Typically 0 for log-fold change data: midpoint = 0 |
low, mid, high |
Colors for value extremes and center | Accessible combinations: blue-white-red |
col |
Preset color palette | viridis, cool_warm(50), or PurpleAndYellow(50) |
Improper configuration of limits represents the most frequent source of color distortion. When the specified range excludes actual data extremes, values beyond the limits become compressed at the endpoint colors, losing visual differentiation [55]. To prevent this, always verify data range before setting limits:
Experimental Protocol: Optimized Heatmap Generation
Data Preprocessing and Normalization
Effective visualization begins with appropriate data transformation. Gene expression data typically requires normalization and scaling before visualization to ensure meaningful color mapping:
Log Transformation: RNA-seq count data often benefits from log transformation to stabilize variance across expression levels:
Data Centering: For differential expression visualization, centering around zero enhances interpretability:
Percentile Transformation: Alternatively, apply percentile transformation to normalize value distribution across genes:
Color Scale Selection Workflow
The following diagram illustrates the decision process for selecting appropriate color scales in gene expression heatmaps:
Complete Implementation Protocol
Protocol: Optimized Heatmap Generation with Custom Color Scaling
Materials:
- Normalized gene expression matrix (rows = genes, columns = samples)
- R environment (version 4.0 or higher)
heatmaplypackage (version 1.3.0 or higher)ggplot2package (for custom color functions)RColorBrewerorviridis(additional color palettes)
Procedure:
- Package Installation and Loading:
Data Preparation and Range Assessment:
Color Scheme Definition:
Heatmap Generation with Optimized Parameters:
Validation and Quality Control:
- Verify color-value correspondence using interactive hovering
- Check for color compression at limits by examining value distribution
- Confirm accessibility using colorblind simulation tools
- Test visualization on multiple display types (LCD, print, projector)
Research Reagent Solutions
Table 2: Essential Computational Tools for Heatmap Generation
| Tool/Package | Function | Application Context |
|---|---|---|
heatmaply R Package |
Interactive heatmap generation | Primary visualization engine for gene expression data |
ggplot2 |
Grammar of graphics implementation | Custom color scale definitions via scale_fill_gradient2 |
viridis |
Perceptually uniform colormaps | Colorblind-friendly sequential data visualization |
RColorBrewer |
Color scheme management | Predefined palettes for categorical annotations |
dendextend |
Dendrogram manipulation | Enhanced clustering visualization and customization |
seriation |
Matrix ordering algorithms | Optimal arrangement of rows and columns (OLO, GW methods) |
Troubleshooting Common Color Representation Issues
Incorrect Midpoint Coloring
Problem: Neutral values (zero) do not appear as the specified midpoint color (typically white) [55].
Solution: Ensure the limits parameter is symmetric around zero and encompasses the full data range:
Color Compression at Extremes
Problem: Highly upregulated or downregulated genes appear identical due to value truncation.
Solution: Expand limits slightly beyond data range or apply data transformation:
Accessibility Validation
Problem: Color distinctions are imperceptible to colorblind users.
Solution: Implement simulated colorblind checking:
Optimized color scale implementation represents a critical component of rigorous gene expression analysis. Through appropriate selection of perceptually uniform, accessible color schemes and precise parameter configuration, researchers can ensure accurate data interpretation across diverse visualization contexts. The protocols outlined herein provide a standardized approach for generating biologically meaningful heatmaps within the heatmaply framework while addressing common challenges in color representation. Consistent application of these methods enhances reproducibility and facilitates clearer communication of scientific findings in genomics and drug development research.
Within the context of creating interactive gene expression heatmaps for research, performance tuning is a critical prerequisite for effective data analysis. Interactive heatmaps, particularly those generated using the heatmaply R package, serve as indispensable tools for researchers, scientists, and drug development professionals exploring complex biological datasets such as RNAseq and microarray results [56] [57]. These visualizations enable the identification of differential gene expression patterns across multiple samples, facilitating insights into disease mechanisms and potential therapeutic targets [58] [57]. However, rendering high-dimensional gene expression data poses significant computational challenges, including excessive memory allocation and prolonged rendering times, which can impede research progress [59]. This document establishes comprehensive application notes and experimental protocols to systematically address these performance limitations through parameter optimization and methodological refinement.
Quantitative Parameters Impacting Performance
The rendering speed and memory usage of interactive heatmaps are influenced by several quantitative parameters that interact in complex ways. Understanding these parameters allows researchers to make informed trade-offs between visual fidelity and computational performance when working with large gene expression matrices.
Table 1: Key Parameters Impacting Heatmap Performance
| Parameter | Default Value | Performance Impact | Memory Usage | Quality Trade-off |
|---|---|---|---|---|
| Data Matrix Size | Variable | O(n²) time complexity | High allocation risk | Directly limits resolution |
| Color Palette Resolution | 256 colors (viridis) |
Linear increase with n | Minimal | Perception vs. computation |
| Dendrogram Computation | stats::hclust |
O(n²) to O(n³) time | Moderate | Cluster visualization essential |
| Seriation Method | "OLO" (Optimal Leaf Ordering) | O(n⁴) complexity | Low | Pattern clarity enhanced |
| Grid Lines | Disabled for large n | O(n²) rendering penalty | Moderate | Visual separation reduced |
| Interactive Elements | Hover tooltips | Constant factor increase | Low | Required for inspection |
The data matrix size represents the most significant factor in computational performance. As matrix dimensions increase, memory requirements grow quadratically while rendering time can follow cubic or quartic growth patterns depending on the clustering algorithms employed [59]. For example, a 10,000 × 10,000 gene expression matrix may require substantial memory allocation simply for data storage, before any visualization computations begin. Experimental observations indicate that matrices exceeding 100,000 total elements often trigger noticeable performance degradation in interactive environments [59] [9].
Seriation methods, which optimize the ordering of rows and columns to highlight patterns, exhibit varying computational complexities that dramatically impact rendering speed. The default "OLO" (Optimal Leaf Ordering) algorithm in heatmaply provides superior visual organization but operates with O(n⁴) time complexity, making it prohibitively expensive for very large datasets [9]. Alternative methods like "GW" (Gruvaeus and Wainer) offer heuristic approaches with improved performance characteristics, though potentially with reduced optimality in pattern presentation [9].
Table 2: Data Transformation Methods and Their Computational Properties
| Transformation Method | Computational Complexity | Memory Overhead | Use Case |
|---|---|---|---|
| Scaling (Z-score) | O(n) | Low | Normally distributed data |
| Normalization (0-1 range) | O(n) | Low | Non-normal distributions |
| Percentile Transformation | O(n log n) | Moderate | Rank-based analysis |
| Log Transformation | O(n) | Low | Highly skewed data |
| Binning/Aggregation | O(n) | Significant reduction | Large matrices (>10⁶ elements) |
Data transformation methods, while essential for proper visualization of gene expression data, introduce additional computational overhead that varies based on the specific operation [9]. Percentile transformations (implemented via percentize in heatmaply) require sorting operations with O(n log n) complexity, while simpler scaling and normalization operations maintain linear O(n) complexity. For extremely large datasets, binning or aggregation strategies that reduce effective matrix size can provide dramatic performance improvements while preserving the essential patterns in the data [59].
Optimization Strategies and Experimental Protocols
Data Size Optimization Workflow
The following diagram illustrates the decision workflow for optimizing data size prior to heatmap generation:
Diagram 1: Data optimization workflow for heatmap performance.
Protocol 1.1: Data Downsampling for Large Matrices
Purpose: To reduce matrix dimensions while preserving meaningful biological patterns in gene expression data.
Materials:
- R statistical environment (v4.0 or higher)
heatmaplypackage (v1.6.0 or higher)dplyrpackage for data manipulation- Gene expression matrix (counts, FPKM, or TPM values)
Procedure:
- Calculate Downsampling Factor:
- Determine target dimensions based on screen resolution and performance requirements. For most applications, 1000 × 1000 elements provides sufficient resolution.
- Compute downsampling factor:
downsample_factor <- max(1, floor(nrow(expression_matrix) / 1000))
Apply Binning Aggregation:
Validate Data Integrity:
- Compare summary statistics (mean, variance) between original and downsampled matrices
- Preserve row and column names for gene and sample identification
- Document downsampling factor in analysis metadata
Performance Notes: This approach can reduce memory usage by up to 99% for very large matrices (≥10⁶ elements) while maintaining representative expression patterns [59]. The computational complexity is linear O(n) with respect to the original matrix size.
Protocol 1.2: Data Transformation for Performance
Purpose: To optimize data distribution for efficient visualization while minimizing computational overhead.
Materials:
- R statistical environment
heatmaplypackage- Preprocessed expression matrix (raw or downsampled)
Procedure:
- Assess Data Distribution:
Select Appropriate Transformation:
- For normally distributed data: Use Z-score scaling (
scale = "column"in heatmaply) - For non-normal distributions: Apply normalization (0-1 range)
- For heavy-tailed distributions: Consider log transformation
- For normally distributed data: Use Z-score scaling (
Implement in Heatmaply:
Performance Notes: Column-wise scaling operations require O(n×m) computations where n is rows and m is columns. Precomputing transformations outside the visualization function can reduce redundant calculations [9].
Memory and Rendering Factor Relationships
The relationship between key parameters and system performance follows predictable patterns that can be modeled to anticipate resource requirements. The following diagram maps these relationships:
Diagram 2: Relationships between heatmap parameters and performance factors.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Heatmap Generation
| Tool/Package | Function | Performance Attributes | Application Context |
|---|---|---|---|
| heatmaply | Interactive heatmap generation | Optimized with plotly.js for larger matrices | Primary visualization tool for gene expression data |
| dendextend | Dendrogram customization | Efficient tree manipulation algorithms | Enhancing cluster visualization |
| seriation | Matrix ordering | Multiple algorithms with varying complexity | Pattern optimization in heatmap display |
| plotly | Interactive graphics engine | Hardware-accelerated rendering | Backend for heatmaply interactivity |
| dplyr | Data manipulation | Optimized C++ backend for operations | Data preprocessing and downsampling |
| RColorBrewer | Color palette generation | Predefined perceptually uniform schemes | Accessible color schemes for publication |
The heatmaply package serves as the cornerstone tool, leveraging the plotly JavaScript library for efficient rendering of large matrices [9] [15]. This combination provides significant performance advantages over traditional static heatmap implementations, particularly for matrices exceeding 10,000 elements [9]. The integration with dendextend enables sophisticated dendrogram customization without excessive computational overhead, while seriation offers multiple algorithms for optimizing matrix arrangement to reveal biological patterns [9].
Advanced Performance Protocol
Protocol 2.1: Memory-Efficient Correlation Heatmaps
Purpose: To generate correlation heatmaps for large gene sets while minimizing memory allocation.
Materials:
- R statistical environment
heatmaplypackagecorpcorpackage for efficient correlation computation
Procedure:
- Compute Correlation Matrix Efficiently:
Apply Heatmaply with Correlation Optimizations:
Export and Save Optimized Visualization:
Performance Notes: The corpcor package uses shrinkage estimators that are more memory-efficient for large correlation matrices. The heatmaply_cor function is specifically optimized for correlation matrices with appropriate default limits (-1 to 1) and diverging color schemes [9] [15].
Performance tuning for interactive gene expression heatmaps requires systematic optimization of multiple interdependent parameters. Through strategic downsampling, appropriate data transformation, algorithm selection, and tool utilization, researchers can achieve responsive visualizations even with large-scale genomic datasets. The protocols and analyses presented here provide a structured approach to balancing computational constraints with visualization quality, enabling more efficient exploration of gene expression patterns in biomedical research. As heatmap technologies continue to evolve, these performance tuning principles will remain essential for maximizing research productivity in genomics and drug development.
Ensuring Scientific Rigor: Validation and Comparative Analysis Best Practices
This application note provides a structured comparison of four prominent R packages—heatmaply, pheatmap, ComplexHeatmap, and ggplot2—for creating heatmaps in genomic research, with a specific focus on visualizing gene expression data. We present a quantitative summary of package capabilities, detailed protocols for generating an interactive gene expression heatmap using the heatmaply package, and supporting diagrams to guide researchers in selecting and implementing the appropriate tool for their experimental needs. The guidance is framed within the broader context of creating publication-quality, interactive visualizations for drug development and scientific research.
Heatmaps are indispensable tools in bioinformatics and genomics for visualizing high-dimensional data, such as gene expression matrices, where values are encoded as a grid of colored cells [8]. The choice of software package significantly impacts the ease of creation, level of customization, and ultimately, the effectiveness of the visualization. This note examines four R packages, each with distinct philosophies and strengths: heatmaply for interactive HTML-based heatmaps, pheatmap for creating publication-ready static heatmaps with minimal code, ComplexHeatmap for highly customizable and complex static visualizations, and ggplot2 for a grammar-of-graphics approach that integrates seamlessly into a broader data analysis workflow [8] [60] [61]. We place particular emphasis on the workflow for creating an interactive gene expression heatmap using heatmaply, enabling researchers to share dynamic results online as self-contained HTML files.
Package Comparison and Selection Guide
Selecting the right heatmap package depends on the project's specific requirements for interactivity, customization, and data complexity. The following table provides a concise comparison of the four packages to guide this decision.
Table 1: Quantitative and Qualitative Comparison of Heatmap Packages in R
| Feature | heatmaply | pheatmap | ComplexHeatmap | ggplot2 |
|---|---|---|---|---|
| Primary Use Case | Interactive web heatmaps [8] | Quick, publication-ready static heatmaps [60] | Highly complex & annotated static heatmaps [62] [63] | Grammar-of-graphics based plots [64] [65] |
| Interactivity | High (Zoom, tooltips, HTML export) [8] | None | None | Via plotly extension [65] |
| Clustering | Yes (with dendextend) [8] | Yes [60] | Yes [62] | Requires manual setup |
| Annotations | Row & column sidebars [8] | Row & column sidebars [60] | Extensive (Multiple, complex annotations) [62] [63] | Requires data reshaping [64] |
| Ease of Use | Easy | Easy | Steep learning curve | Moderate |
| Customization | Moderate | Moderate | Very High | High (via ggplot2 syntax) |
| Data Input | Matrix | Matrix | Matrix | Long-form data frame [64] [65] |
| Best For | Sharing interactive results online | Standard static reports | Genomic papers with complex data layouts | Integrating heatmaps into a tidyverse workflow |
For researchers focused on creating interactive gene expression heatmaps for online sharing and exploration, heatmaply is the optimal choice due to its direct production of interactive HTML files [8]. For standard static publication figures, pheatmap offers a balance of ease and control, while ComplexHeatmap is unparalleled for complex, multi-panel visualizations common in genomics [62]. The ggplot2 approach is ideal for those already embedded in the tidyverse ecosystem, though it requires more data preparation [64].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table lists key software "reagents" required to implement the heatmap visualizations discussed in this note.
Table 2: Essential Software Reagents for Creating Heatmaps in R
| Reagent (R Package) | Function & Application |
|---|---|
| heatmaply | Primary engine for generating interactive cluster heatmaps that can be zoomed and explored, then saved as standalone HTML files [8]. |
| pheatmap | Tool for creating clustered, annotated static heatmaps with minimal coding effort, suitable for quick publication-quality figures [60]. |
| ComplexHeatmap | Comprehensive solution for designing highly complex static heatmaps with multiple row/column annotations and arrangements, often used in genomic studies [62] [63]. |
| ggplot2 | Foundational graphing system based on the "grammar of graphics"; geom_tile() is used to build heatmaps and allows for deep customization within a consistent framework [64] [65]. |
| dendextend | Enhances dendrogram manipulation and visualization, providing control over clustering aesthetics in packages like heatmaply [8]. |
| RColorBrewer | Provides carefully designed color palettes (sequential, diverging, qualitative) that are perceptually uniform and colorblind-safe for scientific data visualization [60] [66]. |
| viridis | Offers colorblind-friendly and perceptually uniform color palettes that are the default in heatmaply for accurately representing numerical data [8]. |
| tidyr/dplyr | Core tidyverse packages for data wrangling; essential for reshaping data from a matrix to a long format required by ggplot2::geom_tile() [64]. |
Experimental Protocol: Creating an Interactive Gene Expression Heatmap withheatmaply
This protocol details the steps to create an interactive gene expression heatmap from a normalized count matrix, enabling dynamic data exploration via an HTML file.
Materials and Data Pre-processing
- Software: R (version 4.0 or higher), RStudio.
- Data Input: A matrix or data frame of normalized gene expression values (e.g., log2-CPM, TPM). Rows represent genes and columns represent samples. The row names should be gene identifiers, and column names should be sample identifiers [62].
- Data Transformation: Transform the data to a suitable scale. For gene expression data, a log2 transformation is often applied to stabilize variance. The
heatmaplypackage also provides built-in functions likenormalizeorpercentizefor additional scaling, and it is crucial to apply scaling if the columns (samples) are not directly comparable [8].
Package Installation and Setup
- Install and load the required packages in your R session.
Basic Interactive Heatmap Generation
- Generate a basic interactive heatmap and display it in the RStudio viewer. The
fileargument is used to save a self-contained HTML file, which can be shared as supplementary material or posted online [8]. - Open the saved HTML file (
Interactive_Gene_Expression_Heatmap.html) in a web browser. You can now hover over cells to see exact values, click and drag to zoom into specific regions, and use the toolbar that appears on hover for additional options like panning and resetting the view [8].
Advanced Customization and Annotation
The heatmaply function provides extensive arguments to control the heatmap's statistical and visual aspects [8].
- Control Clustering: Modify the clustering method, distance metric, or suppress dendrograms.
- Add Sample and Gene Annotations: Provide data frames for
annotation_colandannotation_rowto add informative side bars. The row names of these data frames must match the column or row names of the input matrix, respectively [60]. - Customize Color Palette: Use a perceptually uniform and colorblind-friendly palette. The
viridispalette is the default, butRColorBrewerpalettes are excellent alternatives [8] [66].
Diagram 1: A linear workflow for creating an interactive gene expression heatmap with the heatmaply R package.
Supplementary Protocols for Alternative Packages
Protocol: Creating a Static Heatmap withpheatmap
The pheatmap package is ideal for creating high-quality static heatmaps efficiently [60].
- Install and load the package.
- Generate a clustered heatmap with annotations.
- Save the output using R's graphical devices.
Protocol: Building a Heatmap withggplot2
Using ggplot2 with geom_tile() integrates heatmap creation into the tidyverse workflow but requires data in long format [64] [65].
- Reshape the matrix into a long-format data frame.
- Create the heatmap using
geom_tile(). - To make it interactive, use the
plotlypackage.
Diagram 2: A decision tree to guide the selection of the most appropriate R package for creating a heatmap, based on project requirements.
The choice between heatmaply, pheatmap, ComplexHeatmap, and ggplot2 is dictated by the specific communication and analytical goals of a research project. For the targeted use case of creating and sharing interactive gene expression heatmaps online, heatmaply provides a powerful and user-friendly solution that bridges the gap between static publication figures and fully interactive web applications. The provided protocols and diagrams offer a concrete starting point for researchers and drug development professionals to implement these visualizations effectively, ensuring their data is presented with both clarity and impact.
In the analysis of high-throughput genomic data, heatmaps serve as a powerful tool for visualizing complex gene expression matrices. While clustering algorithms can reveal groups of genes with similar expression patterns, the biological interpretation of these patterns is paramount. Correlating these patterns with known gene functions transforms visual clusters into biologically meaningful insights, a process essential for research in areas like cancer biology and drug development [46] [67]. This protocol details the methodology for using the heatmaply R package to create interactive heatmaps and provides a framework for the subsequent biological validation of observed patterns through functional annotation. The interactive nature of heatmaply facilitates deeper exploration, allowing researchers to inspect specific values and zoom into regions of interest, thereby bridging the gap between statistical patterns and biological reality [5].
Materials
Research Reagent Solutions and Essential Materials
Table 1: Key reagents, software, and data resources required for the protocol.
| Item Name | Function/Description | Example/Source |
|---|---|---|
| RNA-seq Count Data | Raw input data for analysis; represents gene expression levels. | HTSeq-count files from TCGA-BRCA [46]. |
| Clinical Metadata | Data for sample annotation; enables correlation of expression patterns with patient characteristics. | Patient ER status, tumor subtype from TCGA [46]. |
| Gene Set | A predefined list of genes for focused analysis. | PAM50 gene set for breast cancer subtyping [46]. |
| BiomaRt R Package | Retrieves functional annotations for genes (e.g., HGNC symbols). | Ensembl hsapiens_gene_ensembl dataset [46]. |
| Limma/Voom R Package | Normalizes RNA-seq count data for downstream analysis. | Transforms counts to log2-CPM with precision weights [46]. |
| Heatmaply R Package | Generates interactive, clustered heatmaps for online publishing. | heatmaply::heatmaply() function [5]. |
| Harmonizome API | Provides up-to-date gene descriptions and full names. | Integrated into Clustergrammer for tooltips [67]. |
| Enrichr API | Performs gene set enrichment analysis on clusters of interest. | Used for functional annotation of clustered genes [67]. |
Software Environment Setup
The following R packages are critical for the entire workflow, from data preprocessing to visualization.
Methods
Data Acquisition and Preprocessing
This section outlines the steps for obtaining and preparing gene expression data for visualization.
Procedure:
Data Retrieval: Acquire gene expression data from a reliable source such as the Genomic Data Commons (GDC) using the
TCGAbiolinkspackage. The example focuses on primary tumor samples from breast cancer patients (TCGA-BRCA) [46].Gene Annotation: Map ensemble gene identifiers to more interpretable HGNC symbols using
biomaRt. This step is crucial for functional interpretation.Data Normalization: Normalize raw count data using the
voomtransformation from thelimmapackage. This method models the mean-variance relationship of log-counts, making the data suitable for visualization and downstream statistical analysis [46].
Generating an Interactive Heatmap withheatmaply
Create a sample-sample correlation heatmap to explore overall data structure and identify potential outliers or batch effects.
Procedure:
Compute Correlation Matrix: Calculate the correlation matrix between samples using normalized data. A log-transform is applied to raw counts if
voomis not used.Create Interactive Heatmap: Generate the heatmap using the
heatmaplyfunction. Incorporate clinical annotations to provide biological context.
Workflow for Biological Validation
The following diagram outlines the logical process from data preparation to biological interpretation.
Functional Annotation of Clusters
This critical step connects statistical patterns to biological meaning.
Procedure:
Cluster Identification: Use the interactive features of the
heatmaplyheatmap to identify clusters of genes or samples. This can be done by visually inspecting the dendrogram or, in other tools like Clustergrammer, by clicking on dendrogram trapezoids to isolate specific clusters [67].Gene List Extraction: Extract the list of gene symbols belonging to the cluster of interest for further analysis.
Enrichment Analysis: Perform Gene Ontology (GO) or pathway enrichment analysis on the extracted gene list using tools like Enrichr. This identifies biological processes, molecular functions, or pathways that are statistically over-represented in the gene cluster [67].
Literature Correlation: Interpret the results from the enrichment analysis by correlating them with established biological knowledge. For example, a cluster showing high expression in estrogen receptor-positive (ER+) samples might be enriched for genes involved in "estrogen response" or "hormone-mediated signaling pathway," validating the molecular phenotype of the tumor samples [46].
Application Notes
Case Study: Breast Cancer Transcriptomics
An analysis of TCGA breast cancer data can effectively illustrate this protocol. After generating a sample-sample correlation heatmap annotated with ER, PR, and HER2 status, clustering of samples often reflects their known receptor subtypes [46]. Extracting and analyzing a gene cluster that highly expresses in the "Luminal A" subgroup might reveal enrichment for genes related to "steroid metabolic process" or "response to estrogen," thereby validating the heatmap pattern against the known hormone-responsive nature of this cancer subtype.
Technical Considerations
- Color Accessibility: When choosing a color palette for the heatmap and annotations, ensure it is colorblind-friendly. Avoid problematic combinations like red-green [68]. The Google-inspired palette used in the diagrams (
#4285F4,#EA4335,#FBBC05,#34A853) provides good contrast and is generally safe for common forms of color vision deficiency. - Interactivity: The key advantage of
heatmaplyis interactivity. Researchers should leverage tooltips to see exact values and gene names, and the zoom feature to explore dense regions of the heatmap in detail [5]. This is invaluable for pinpointing specific genes within a broad pattern. - Data Quality: The quality of the biological validation is directly dependent on the quality of the input data and the normalization method. Always perform appropriate quality control checks before generating the heatmap.
This protocol provides a detailed guide for moving beyond the visual inspection of heatmaps to the biological validation of the patterns they reveal. By integrating the interactive visualization capabilities of heatmaply with structured functional analysis using gene annotations and enrichment tools, researchers can robustly correlate computational findings with known biology. This process is essential for generating biologically plausible hypotheses from high-dimensional genomic data, ultimately advancing understanding in basic research and drug development.
In the analysis of high-dimensional biological data, such as gene expression matrices, heatmaps serve as an indispensable graphical method for visualizing complex patterns [5]. These visualizations are particularly crucial in pharmaceutical and clinical research, where they help identify disease biomarkers, therapeutic targets, and drug response patterns [1]. The heatmaply R package extends these capabilities by creating interactive cluster heatmaps that enable researchers to inspect specific values by hovering over cells and zoom into regions of interest [5]. However, this powerful visualization tool incorporates several stochastic elements that can dramatically impact results if not properly controlled.
Cluster analysis, an integral component of heatmap generation, involves both distance calculation between objects and clustering methods that group similar elements together [1]. Many of these algorithms contain inherent randomness in their initialization processes, while the visualization parameters themselves can create apparent patterns where none exist. Without strict reproducibility controls, two scientists analyzing the same dataset could produce visually distinct heatmaps with different cluster arrangements, potentially leading to conflicting biological interpretations.
This application note establishes a comprehensive framework for achieving reproducible interactive heatmap generation using heatmaply, with particular emphasis on the critical practice of setting random seeds and thoroughly documenting all computational parameters. These protocols ensure that research findings can be independently verified, a fundamental requirement in drug development and scientific publication.
Theoretical Foundation: Stochastic Elements in Heatmap Construction
Understanding Randomness in Cluster Analysis
Cluster heatmaps visualize high-dimensional data through a combination of colored cells and dendrograms that show hierarchical relationships between rows (typically genes) and columns (typically samples) [5]. The process involves multiple computational stages where randomness can influence final outcomes:
- Distance Calculation: Measures dissimilarity between data points using methods such as Euclidean, Manhattan, or correlation-based distances [1].
- Hierarchical Clustering: Groups similar elements together using algorithms that may have random initialization components [1].
- Dendrogram Construction: Visual representation of clustering that may have multiple equivalent layouts [1].
The interpretation of clustered heatmaps fundamentally depends on identifying patterns in gene expression [69]. Pharmaceutical researchers examining treatment effects must be able to distinguish genuine biological signals from artifacts introduced by computational variability. Proper seed management ensures that visual patterns reflect underlying biology rather than algorithmic randomness.
Consequences of Non-Reproducible Heatmaps
In drug development workflows, irreproducible heatmaps can lead to several critical failures:
- Inconsistent Biomarker Identification: Different random seeds may cluster genes differently, leading to varying candidate biomarkers.
- Unverifiable Publication Results: Published findings cannot be confirmed without exact computational environments.
- Poor Decision Making: Therapeutic target selection based on unstable visual patterns.
Table 1: Impact of Randomness on Heatmap Interpretation in Drug Development
| Research Phase | Stochastic Element | Potential Consequence |
|---|---|---|
| Target Discovery | Gene clustering | Inconsistent pathway identification |
| Biomarker Validation | Sample clustering | Unreliable patient stratification |
| Mechanism of Action | Pattern visualization | Variable biological interpretation |
| Regulatory Submission | Overall reproducibility | Questioned scientific validity |
Experimental Protocols: Implementing Reproducible Heatmap Analyses
Core Reproducibility Framework
The following protocol establishes the foundational practices for reproducible heatmap generation using heatmaply. This framework ensures that any analysis can be precisely replicated regardless of computational environment or timing.
Protocol 1: Random Seed Management Framework
Purpose: To guarantee consistent results across computational sessions by properly controlling random number generation.
Materials:
- R programming environment (version 4.0.0 or higher)
- heatmaply package (version 1.3.0 or higher)
- set.seed() function from base R
Procedure:
- Initialize random seed at the beginning of each analytical session
- Set seed immediately before heatmap generation functions
- Document exact seed values in methodological documentation
- Maintain seed logs across analytical iterations
Critical Steps:
Technical Notes: The random seed must be set immediately before calling the heatmaply() function, as any intervening operations that use random number generation may advance the random number sequence. For complex analyses involving multiple heatmaps, consider implementing a seed sequence management system.
Comprehensive Heatmap Generation Protocol
This protocol describes the complete workflow for generating reproducible interactive heatmaps for gene expression analysis, incorporating appropriate data preprocessing, parameter documentation, and visualization controls.
Protocol 2: Reproducible Interactive Heatmap Generation
Purpose: To create fully reproducible interactive cluster heatmaps for gene expression data analysis with complete parameter tracking.
Materials:
- Normalized gene expression matrix (rows = genes, columns = samples)
- heatmaply R package [5]
- dendextend package for dendrogram manipulation
- RMarkdown or Jupyter notebook for documentation
Procedure:
Step 1: Environment Preparation
Step 2: Data Preprocessing and Scaling
Step 3: Parameter Documentation and Seed Setting
Step 4: Heatmap Generation with Documented Parameters
Validation:
- Execute the same code with the same seed on different systems to verify identical output
- Confirm consistent cluster formation across multiple iterations
- Validate that interactive elements display correct values upon hovering
Technical Notes: For studies intended for regulatory submission, maintain a complete record of all software versions using sessionInfo(). The complexity of hierarchical clustering algorithms necessitates strict seed control, as even minor numerical differences can alter branching patterns in resulting dendrograms [1].
Visualization Framework: Reproducible Heatmap Workflows
The following diagram illustrates the complete reproducible workflow for interactive heatmap generation, highlighting critical control points where parameter documentation and seed setting ensure reproducibility.
Reproducible Heatmap Workflow
Research Reagent Solutions: Essential Materials for Reproducible Heatmaps
Table 2: Essential Computational Reagents for Reproducible Heatmap Analysis
| Reagent/Material | Function | Implementation Example | Reproducibility Consideration |
|---|---|---|---|
| heatmaply R Package | Generates interactive cluster heatmaps [5] | heatmaply(expression_matrix) |
Version control essential; current version recommended |
| Random Seed Value | Controls stochastic algorithms | set.seed(12345) |
Must be documented and consistent across sessions |
| Color Palette | Encodes expression values visually | RColorBrewer::brewer.pal(9, "RdYlBu") |
Fixed palette definition prevents interpretation variance |
| Distance Metric | Determines similarity calculation | dist(x, method = "euclidean") |
Choice significantly impacts clustering results |
| Clustering Algorithm | Defines grouping methodology | hclust(x, method = "complete") |
Method selection changes dendrogram structure |
| Data Scaling Method | Normalizes expression values | scale() or log-transformation |
Must be consistent for comparative analyses |
| Parameter Documentation | Records analytical choices | Structured list or configuration file | Enables exact method replication |
Data Presentation: Quantitative Framework for Reproducibility Assessment
Parameter Documentation Standards
Complete documentation of computational parameters is equally important as random seed management for ensuring methodological reproducibility. The following table establishes minimum documentation requirements for interactive heatmap generation.
Table 3: Essential Parameters for Reproducible Heatmap Documentation
| Parameter Category | Specific Parameters | Example Setting | Impact on Results |
|---|---|---|---|
| Randomness Control | Random seed value | 12345 | Controls all stochastic operations |
| Data Preprocessing | Scaling method, Transformation, Normalization approach | Z-score, log2(x+1) | Affects value distribution and color mapping |
| Clustering Methods | Distance metric, Linkage method, Dendrogram sorting | Euclidean, Complete, TRUE | Determines grouping patterns |
| Visualization Parameters | Color scheme, Value range, Color key limits | RdYlBu, Symmetric, Automatic | Changes visual pattern interpretation |
| Interactive Features | Display values on hover, Zoom capability, Download options | TRUE, TRUE, TRUE | Affects user exploration and data extraction |
Advanced Applications: Reproducibility in Research Contexts
Differential Gene Expression Visualization
In transcriptomic analyses, heatmaps commonly visualize differential expression patterns across experimental conditions [16]. The reproducibility framework becomes particularly critical when identifying candidate biomarkers or therapeutic targets. Implementation requires:
- Consistent normalization of count data (e.g., TPM, FPKM, or log2CPM)
- Standardized clustering of both genes and samples
- Controlled color mapping of expression values or fold-changes
Pharmacogenomic Applications
In drug development, heatmaps visualize compound sensitivity across cell lines or patient-derived models [1]. Reproducibility ensures consistent compound prioritization and biomarker association. Special considerations include:
- Batch effect correction documentation
- Sensitivity metric standardization (e.g., IC50, AUC)
- Annotation track consistency for sample metadata
The integration of rigorous random seed management and comprehensive parameter documentation represents a fundamental requirement for scientifically valid heatmap generation in research and drug development. The protocols and frameworks presented in this application note provide actionable methodologies for ensuring that interactive cluster heatmaps serve as reliable analytical tools rather than artistic visualizations.
As the complexity of biological data continues to increase, with single-cell RNA sequencing and spatial transcriptomics generating increasingly high-dimensional datasets, the importance of computational reproducibility cannot be overstated [5] [1]. By establishing and maintaining these practices, researchers ensure that their findings remain verifiable, extensible, and trustworthy throughout the scientific and drug development pipeline.
Within the broader context of creating interactive gene expression heatmaps with heatmaply, this application note details a protocol for reproducing the quality and interpretability of visualizations found in published Nature papers. The heatmaply R package facilitates the creation of interactive cluster heatmaps based on the ggplot2 and plotly.js engines, allowing for detailed inspection of values via mouse hover and zooming into specific regions [9] [18]. This interactivity is crucial for exploring high-dimensional data, such as gene expression matrices, where patterns may be obscured in static views.
Key Research Reagent Solutions
The following table catalogues the essential software tools and their functions required to execute the protocols in this note.
Table 1: Essential Research Reagents and Software Tools
| Item Name | Function/Application in Protocol |
|---|---|
heatmaply R Package |
Primary engine for generating interactive heatmaps; enables data transformation, custom coloring, and dendrogram manipulation [9] [18]. |
RColorBrewer & viridis Palettes |
Provides color schemes designed for perceptual uniformity and accessibility, including diverging palettes like RdBu and PiYG [9] [15]. |
dendextend R Package |
Used for advanced customization of dendrograms attached to heatmaps, including the coloring of branches [9]. |
seriation R Package |
Implements algorithms for optimal leaf ordering in dendrograms to highlight patterns in the data matrix [9]. |
plotly R Package |
Provides the underlying interactive plotting infrastructure, enabling features like zooming and value inspection via hovering [5] [18]. |
Protocols
Protocol 1: Configuring a Diverging Color Palette for Expression Data
A critical step in replicating the clarity of published figures is the implementation of a perceptually sound and biologically intuitive color palette. This protocol uses a diverging palette to distinguish between up-regulated and down-regulated genes effectively.
Detailed Methodology
- Palette Selection: Utilize diverging palettes from the
RColorBrewerextension withinheatmaply, such asRdBu,PiYG, orcool_warm[15]. These are ideal for highlighting deviations from a neutral midpoint (e.g., zero in log-fold-change data). - Fixed Scale Limits: Explicitly set the
limitsparameter to fix the legend scale. This ensures consistent color mapping across multiple heatmaps and accurate representation of the data range. For correlation matrices,heatmaply_corautomatically setslimits = c(-1, 1)[9] [15]. - Custom Midpoint Definition: For
scale_fill_gradient2, define themidpointargument to specify the data value that corresponds to the central color (e.g.,midpoint = 0) [9]. - Code Implementation:
The following diagram illustrates the logical flow and decision points for configuring a color palette in heatmaply.
Protocol 2: Data Pre-processing and Transformation
Raw data must be transformed to ensure patterns are not masked by variables with vastly different scales. This is common in gene expression analysis where baselining and normalization are essential.
Detailed Methodology
- Data Normalization: Use the
normalizefunction to scale each column to a 0-1 range. This preserves the shape of each variable's distribution while making them comparable. It is particularly useful for data not assumed to come from a normal distribution [9]. - Percentile Transformation: Apply the
percentilefunction to convert each value to its empirical percentile within its column. This provides a clear interpretation of each value as the percentage of observations below it [9]. - Z-Score Standardization: Use the
scaleparameter withinheatmaplyto transform columns to z-scores (mean-centered and divided by standard deviation). This is appropriate when variables are assumed to be normally distributed.
Protocol 3: Optimizing Dendrogram and Seriation
Dendrogram ordering significantly impacts the visibility of clusters. This protocol uses seriation to optimize the arrangement of rows and columns.
Detailed Methodology
- Seriation Method Selection: Use the
seriateparameter to control the ordering. The"OLO"(Optimal Leaf Ordering) method is often preferred as it minimizes the sum of distances between adjacent leaves in the dendrogram, thereby highlighting patterns [9]. - Custom Dendrogram Input: For maximum control, create a custom dendrogram object using the
dendextendpackage and supply it to theRowvorColvparameters [9]. This allows for manual branch coloring and pruning. - Branch Coloring: Color dendrogram branches based on cluster membership using
dendextend::color_branchesand specify the number of clusters (k_colandk_row) inheatmaply_coror the mainheatmaplyfunction [9].
Table 2: Seriation Methods for Dendrogram Optimization in heatmaply
| Seriation Method | Key Principle | Use Case |
|---|---|---|
OLO (Optimal Leaf Ordering) |
Finds the optimal Hamiltonian path restricted by the dendrogram structure [9]. | Default and recommended for most cases to reveal clear patterns. |
GW (Gruvaeus and Wainer) |
Uses a heuristic to approximate the goal of the OLO method [9]. | A faster, less computationally intensive alternative for large datasets. |
None |
Uses the default output from the hclust function without rotation [9]. |
Useful for comparing against optimized orders or for specific methodological reasons. |
The workflow for data preparation and visualization, integrating the concepts from the protocols, is summarized below.
In the field of genomics and drug development, heatmaps serve as a fundamental graphical method for visualizing high-dimensional data, encoding numerical tables as grids of colored cells [9]. These visualizations are particularly crucial for gene expression analysis, where they help researchers identify patterns across multiple samples and experimental conditions [70]. The rows and columns of the matrix are typically ordered through clustering algorithms to highlight biological relationships, often accompanied by dendrograms that illustrate the hierarchical clustering structure [9]. Within the context of creating interactive gene expression heatmaps using the heatmaply R package, understanding the statistical foundations of distance metrics and clustering reliability becomes paramount for generating biologically meaningful and reproducible results [9] [5]. The heatmaply package leverages the power of ggplot2 and plotly.js to create interactive heatmaps that enable researchers to inspect specific values by hovering over cells and zoom into regions of interest, thereby facilitating more detailed exploration of complex gene expression datasets [9].
Distance Metrics: Mathematical Foundations and Selection Criteria
Distance metrics form the mathematical foundation for clustering algorithms in heatmap construction. These metrics quantify the dissimilarity between data points, directly influencing how clusters are formed and interpreted.
Fundamental Distance Metrics
The table below summarizes the key distance metrics used in gene expression analysis:
Table 1: Characteristics of Primary Distance Metrics for Gene Expression Data
| Distance Metric | Mathematical Formula | Primary Use Case | Advantages | Limitations |
|---|---|---|---|---|
| Euclidean | d(x,y) = √Σ(xᵢ - yᵢ)² |
General-purpose distance measurement | Intuitive; preserves spatial relationships | Sensitive to outliers and measurement units |
| Manhattan | d(x,y) = Σ|xᵢ - yᵢ| |
High-dimensional data; noise reduction | Robust to outliers; intuitive grid-based distance | May not capture complex correlations |
| Pearson Correlation | d(x,y) = 1 - r(x,y) |
Pattern similarity regardless of magnitude | Focuses on expression patterns rather than absolute values | May cluster anti-correlated features together |
| Spearman Correlation | d(x,y) = 1 - ρ(x,y) |
Non-linear monotonic relationships | Robust to outliers; non-parametric | Less powerful for linear relationships with normal errors |
Metric Selection Protocol
Protocol 2.2.1: Systematic Selection of Distance Metrics
Data Distribution Assessment
- Conduct Shapiro-Wilk normality tests on gene expression profiles.
- Generate Q-Q plots to visualize distributional characteristics.
- For normally distributed data with minimal outliers, proceed to Step 2A. For non-normal distributions or significant outliers, proceed to Step 2B.
Metric Application
- Step 2A: Normal Distribution Confirmation - Apply Euclidean distance for comprehensive distance measurement. Use Pearson correlation distance for pattern-focused analysis.
- Step 2B: Non-Normal Distribution Identified - Apply Manhattan distance for outlier robustness. Use Spearman correlation distance for monotonic relationship capture.
Biological Validation
- Perform cluster analysis with selected metric.
- Validate biological coherence through known gene pathway enrichment (e.g., KEGG, GO databases).
- Iterate with alternative metrics if biological interpretation lacks coherence.
The following diagram illustrates this structured approach:
Clustering Algorithms and Reliability Assessment
Clustering methods organize genes with similar expression patterns into groups, facilitating the identification of co-regulated genes and functional modules.
Hierarchical Clustering Fundamentals
Hierarchical clustering remains the most widely used method for heatmap construction in gene expression studies. The algorithm operates through either an agglomerative (bottom-up) or divisive (top-down) approach, generating a dendrogram that illustrates nested clustering relationships.
Protocol 3.1.1: Hierarchical Clustering with Optimal Leaf Ordering
Distance Matrix Computation
- Calculate pairwise distance matrix using selected metric from Protocol 2.2.1.
- Store results in symmetric n×n matrix where n represents number of genes/samples.
Linkage Method Selection
- Apply Ward's minimum variance method to minimize within-cluster variance.
- Consider complete linkage for compact clusters or average linkage as balanced alternative.
Dendrogram Construction
- Build binary tree structure using selected linkage method.
- Apply seriation methods ("OLO" - Optimal Leaf Ordering) to optimize branch arrangement [9].
- Visualize resulting dendrogram alongside heatmap.
Clustering Reliability Assessment
Evaluating the stability and reliability of clustering results is essential for drawing valid biological conclusions.
Table 2: Methods for Assessing Clustering Reliability
| Assessment Method | Implementation Approach | Interpretation Guidelines | heatmaply Integration |
|---|---|---|---|
| Bootstrap Resampling | Random sampling with replacement; cluster recovery frequency | Jaccard similarity >0.75 indicates high stability; <0.60 suggests instability | Implement via dendextend package with Bootstrap function |
| Silhouette Width | Measures how similar an object is to its own cluster versus neighboring clusters | Values range from -1 to +1; >0.5 indicates reasonable structure | Calculate using cluster package; visualize with side_colors |
| Cophenetic Correlation | Correlation between original distances and dendrogram distances | Values >0.8 indicate faithful representation | Compute via dendextend::cor_cophenetic |
| Modularity Score | For graph-based clustering; measures density within vs between clusters | Values >0.3 indicate significant community structure | Applicable to co-expression network analysis |
The following workflow diagram illustrates the comprehensive process for reliability assessment:
Experimental Protocol: Comprehensive Heatmap Analysis with heatmaply
This integrated protocol provides a step-by-step methodology for creating biologically informative interactive heatmaps using the heatmaply package while implementing appropriate statistical foundations.
Protocol 4.1: Complete Gene Expression Heatmap Analysis
Data Preprocessing and Transformation
- Load normalized gene expression matrix (rows = genes, columns = samples).
- Apply appropriate transformation based on data characteristics:
- Use
percentile()transformation for non-normal distributions to convert values to empirical percentiles [9]. - Apply
normalize()function to bring all variables to 0-1 scale while preserving distribution shape [9]. - Utilize
scale="column"for Z-score normalization when assuming normal distributions [9].
- Use
- Filter low-expression genes using variance-based or mean-based thresholds.
Distance Metric and Clustering Implementation
- Execute distance metric selection as detailed in Protocol 2.2.1.
- Perform hierarchical clustering with seriation using
seriate="OLO"parameter for optimal leaf ordering [9]. - Generate initial interactive heatmap with default parameters:
Reliability Assessment Implementation
- Conduct bootstrap resampling with 1000 iterations using
dendextendpackage. - Calculate silhouette widths for different cluster numbers (k=2-10).
- Compute cophenetic correlation between original distances and dendrogram distances.
- Iterate clustering parameters if reliability metrics fall below thresholds established in Table 2.
- Conduct bootstrap resampling with 1000 iterations using
Biological Validation and Interpretation
- Extract gene clusters from dendrogram using
cutreefunction. - Perform functional enrichment analysis on each cluster using Enrichr or clusterProfiler [71].
- Validate clusters against known pathways or biological processes relevant to research context.
- Extract gene clusters from dendrogram using
Visualization Optimization and Export
- Implement colorblind-friendly palettes using
viridisorRColorBrewer[9]. - Add relevant annotations for sample conditions or experimental groups.
- Export interactive HTML heatmaps for collaborative exploration.
- Generate publication-quality static images using:
- Implement colorblind-friendly palettes using
Research Reagent Solutions
The following table catalogues essential computational tools and their functions for implementing comprehensive heatmap analyses in gene expression studies.
Table 3: Essential Research Reagents and Computational Tools for Heatmap Analysis
| Tool/Resource | Primary Function | Application Context | Implementation in heatmaply |
|---|---|---|---|
| heatmaply R Package | Interactive heatmap generation | Primary visualization engine | Core functionality via heatmaply() function [9] [5] |
| dendextend Package | Dendrogram manipulation and analysis | Clustering optimization and validation | Enhanced dendrogram control through Rowv and Colv parameters [9] |
| seriation Package | Optimal leaf ordering | Improved pattern recognition | Integrated via seriate parameter options [9] |
| ggplot2/plotly | Graphics rendering | Interactive visualization foundation | Plotting engine for heatmaply output [9] [5] |
| Enrichr/KEGG Databases | Functional enrichment analysis | Biological interpretation of clusters | Downstream analysis post-clustering [71] |
| Phantasus Web Application | Gene expression dataset access | Data sourcing and preliminary analysis | Alternative for dataset loading and basic visualization [71] |
The statistical foundations of distance metrics and clustering reliability form the cornerstone of biologically meaningful heatmap visualization in gene expression analysis. Through careful selection of appropriate distance measures, implementation of robust clustering algorithms with optimal leaf ordering, and rigorous assessment of cluster stability, researchers can transform high-dimensional genomic data into interpretable biological insights. The integration of these statistical principles with the interactive capabilities of the heatmaply package, as demonstrated in the comprehensive protocols provided, creates a powerful framework for exploratory data analysis in genomics and drug development. By adhering to these methodological standards and utilizing the recommended research reagent solutions, scientists can enhance the reproducibility and biological relevance of their heatmap-based findings, ultimately accelerating the translation of genomic data into meaningful scientific discoveries and therapeutic advancements.
In the field of genomics and transcriptomics, differential gene expression (DGE) analysis is a fundamental technique for identifying genes that are statistically significantly expressed between different biological conditions, such as healthy versus diseased tissues or treated versus untreated cells [72]. The results of these analyses generate large, complex datasets that require effective visualization for biological interpretation. Interactive heatmaps serve as a powerful tool for visualizing these high-dimensional data, enabling researchers to identify patterns, clusters, and outliers in gene expression across multiple samples [73] [1].
The heatmaply R package extends traditional static heatmaps by creating interactive visualizations using the plotly and ggplot2 engines, allowing researchers to inspect specific values by hovering over cells, zoom into regions of interest, and access enhanced features for visualizing clustered data [73]. This protocol details the integration of heatmaply with established DGE analysis pipelines, creating a seamless workflow from statistical analysis to biological insight.
Background
Differential Gene Expression Analysis
DGE analysis is a computational process that identifies genes with statistically significant differences in expression levels between two or more sample groups [72]. The typical workflow involves:
- Normalization to adjust for technical variations between samples
- Statistical testing to identify significantly differentially expressed genes
- Multiple testing correction to control false discovery rates
- Visualization to interpret and communicate results
Common tools for DGE analysis include edgeR and DESeq2, which use negative binomial distributions to model count data from RNA sequencing experiments [72]. These tools generate results containing log fold changes, p-values, and adjusted p-values for thousands of genes, creating the need for effective visualization strategies.
Interactive Heatmaps for Biological Data
Heatmaps provide a color-encoded representation of data matrices, where expression values are transformed into colors according to a specified scale [1]. When combined with dendrograms showing hierarchical clustering patterns, heatmaps enable researchers to visualize both individual expression values and overall sample relationships simultaneously. The interactive capabilities of heatmaply enhance this visualization by allowing direct inspection of values, zooming for detailed exploration, and dynamic reordering of rows and columns [73].
Materials and Reagents
Research Reagent Solutions
Table 1: Essential computational tools and their functions in DGE analysis and heatmap visualization
| Tool Name | Type | Primary Function | Application Context |
|---|---|---|---|
| edgeR | R/Bioconductor Package | Differential expression analysis using negative binomial distribution | Identifying statistically significant differentially expressed genes [72] |
| DESeq2 | R/Bioconductor Package | Differential expression analysis with shrinkage estimation | Robust identification of DEGs with enhanced stability [72] |
| heatmaply | R Package | Interactive heatmap generation | Visualization of expression patterns with zoom/hover capabilities [73] |
| pheatmap | R Package | Static heatmap generation | Publication-quality clustered heatmaps [1] |
| ggplot2 | R Package | Grammar of graphics implementation | Foundation for heatmaply's plotting system [73] |
| plotly | R Package | Interactive graphics engine | Enables interactive features in heatmaply [73] |
| TMM | Normalization Method | Trimmed Mean of M-values normalization | Adjusts for library size and composition differences [72] |
| Geometric Mean | Normalization Method | Size factor calculation | DESeq2's approach for normalization between samples [72] |
Experimental Protocol
DGE Analysis Workflow
The following diagram illustrates the complete workflow from raw data to interactive visualization:
Detailed Methodology
Data Normalization and DGE Analysis
Procedure:
Load count data into R, ensuring proper formatting with genes as rows and samples as columns.
Normalize data using appropriate methods:
- For edgeR: Apply TMM (Trimmed Mean of M-values) normalization using the
calcNormFactors()function [72] - For DESeq2: Apply the geometric mean method for size factor calculation
- For edgeR: Apply TMM (Trimmed Mean of M-values) normalization using the
Perform statistical testing for differential expression:
- Using edgeR: Implement empirical Bayes estimation with Fisher's exact test or generalized linear models tailored for over-dispersed count data [72]
- Using DESeq2: Apply shrinkage estimation for fold changes and hypothesis testing using the Wald test or likelihood ratio test
Extract results including:
- Log fold changes (LogFC)
- P-values
- Adjusted p-values (FDR)
- Normalized expression values for significant genes
Table 2: Key parameters for DGE analysis using edgeR and DESeq2
| Parameter | edgeR Implementation | DESeq2 Implementation | Biological Significance |
|---|---|---|---|
| Normalization | calcNormFactors() with TMM |
estimateSizeFactors() with geometric mean |
Corrects for library size and composition biases |
| Dispersion Estimation | estimateDisp() |
estimateDispersions() |
Models gene-wise variability |
| Statistical Testing | exactTest() or glmFit() |
nbinomWaldTest() or nbinomLRT() |
Identifies statistically significant changes |
| Multiple Testing Correction | topTags() with FDR |
results() with independent filtering |
Controls false discovery rate |
| Fold Change Threshold | logFC parameter in exactTest() |
lfcThreshold in results() |
Sets biological significance cutoff |
Expression Matrix Preparation for Heatmap
Procedure:
Select genes for visualization based on statistical and biological significance:
- Filter by adjusted p-value (typically FDR < 0.05)
- Filter by absolute log fold change (typically |LogFC| > 1)
- Consider top N most significant genes if the list is extensive
Extract normalized expression values for selected genes across all samples
Transform data if necessary:
- Apply log transformation for variance stabilization
- Center and scale genes (z-score calculation) to highlight patterns
Format data matrix with:
- Rows representing genes
- Columns representing samples
- Appropriate row and column names
Add sample annotations if available (e.g., treatment groups, time points, tissue types)
Interactive Heatmap Generation with heatmaply
Procedure:
- Install and load heatmaply package:
- Create basic interactive heatmap:
- Customize visualization parameters:
- Save interactive heatmap for sharing and publication:
Table 3: Critical heatmaply parameters for DGE visualization
| Parameter | Function | Recommended Setting | Impact on Visualization |
|---|---|---|---|
| krow / kcol | Number of clusters for rows/columns | 2-5 based on dataset size | Defines discrete color bars for cluster identification |
| colors | Color palette for expression values | Viridis, RdBu, or custom scale | Affects visual contrast and pattern recognition |
| dendrogram | Control dendrogram display | "both", "row", "col", "none" | Shows hierarchical clustering relationships |
| showticklabels | Display row/column labels | c(TRUE, FALSE) for large datasets | Prevents overplotting in dense heatmaps |
| scale | Data scaling method | "row", "column", or "none" | Highlights patterns; "row" scaling is common for genes |
| rowdendleft | Position row dendrogram | TRUE or FALSE | Affects layout and space utilization |
Results and Interpretation
Expected Outcomes
The implemented workflow generates an interactive heatmap that enables:
- Identification of co-expressed genes through row clustering patterns
- Detection of sample subgroups through column clustering
- Direct value inspection by hovering over individual cells
- Zoom capability for detailed exploration of specific regions
- Export functionality for both interactive and publication-ready formats
Biological Interpretation Guidelines
When analyzing the generated heatmap:
- Examine cluster patterns in gene groups to identify potential co-regulated gene modules
- Correlate sample clusters with experimental conditions to identify batch effects or biological subgroups
- Identify outlier samples that don't cluster with their expected groups
- Validate extreme expression values using the hover functionality to ensure technical artifacts aren't misinterpreted
- Integrate with pathway analysis by extracting genes from specific clusters for functional enrichment
The following diagram illustrates the interpretation process:
Troubleshooting and Technical Notes
Common Issues and Solutions
- Large datasets causing performance issues: Subset to top differentially expressed genes or use the
plotlyengine's native optimization for larger matrices [73] - Color scale not capturing biological variation: Adjust the
zlimparameter to set fixed limits or use divergent color scales for fold-change visualization - Text labels overlapping: Use
showticklabelsparameter to control label display or increase plot size - Dendrograms not informative: Experiment with different distance methods (
distfun) and linkage methods (hclustfun)
Advanced Applications
For specialized use cases:
- Integrate with shiny applications using
plotly::plotlyOutput()andplotly::renderPlotly()for interactive web applications [73] - Add side color bars for sample annotations using
RowSideColorsorColSideColorsparameters - Combine with other visualization methods such as volcano plots or MA plots for comprehensive DGE analysis presentation
The integration of heatmaply with established DGE analysis pipelines creates a powerful framework for visualizing and interpreting complex gene expression data. This protocol provides researchers with a comprehensive guide from statistical analysis through biological insight, leveraging the interactive capabilities of heatmaply to enhance exploration and communication of transcriptomic findings. The seamless connection between differential expression results and interactive visualization facilitates deeper biological understanding and enables more effective collaboration across research teams.
Conclusion
Interactive gene expression heatmaps created with heatmaply represent a powerful tool for exploratory data analysis in biomedical research, enabling researchers to identify patterns, clusters, and outliers in high-dimensional data through intuitive visual exploration. By mastering both the technical implementation and biological interpretation, scientists can transform complex expression matrices into actionable insights about disease mechanisms, treatment responses, and biological pathways. As single-cell technologies and multi-omics datasets continue to grow in scale and complexity, the ability to create interactive, publication-quality visualizations will become increasingly crucial for driving discoveries in drug development and clinical research. Future directions include integration with cloud-based platforms, real-time collaboration features, and enhanced capabilities for visualizing spatial transcriptomics data, positioning heatmaply as an essential component of the modern computational biologist's toolkit.
References
- 1. Lesson5: Visualizing clusters with heatmap and dendrogram [https://bioinformatics.ccr.cancer.gov/docs/data-visualization-with-r/Lesson5_intro_to_ggplot/]
- 2. Heat Map - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/heat-map]
- 3. Clustered Heat Maps (CHMs) - Bioinformatics [https://bioinformatics.mdanderson.org/public-software/ngchm/heatmaps]
- 4. Dos and don'ts for a heatmap color scale - BioTuring Team [https://bioturing.medium.com/dos-and-donts-for-a-heatmap-color-scale-75929663988b]
- 5. talgalili/heatmaply: Interactive Heat Maps for R Using plotly [https://github.com/talgalili/heatmaply]
- 6. 'Heatmaply' Examples • heatmaplyExamples [https://alanocallaghan.github.io/heatmaplyExamples/]
- 7. Chapter 2 A Single Heatmap | ComplexHeatmap Complete ... [https://jokergoo.github.io/ComplexHeatmap-reference/book/a-single-heatmap.html]
- 8. heatmaply: an R package for creating interactive cluster ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5925766/]
- 9. Introduction to heatmaply - GitHub Pages [https://talgalili.github.io/heatmaply/articles/heatmaply.html]
- 10. README - heatmaply [https://cran.r-project.org/web/packages/heatmaply/readme/README.html]
- 11. talgalili/heatmaplyExamples: Examples for the heatmaply ... [https://github.com/talgalili/heatmaplyExamples]
- 12. Benchmarking the translational potential of spatial gene ... [https://www.nature.com/articles/s41467-025-56618-y]
- 13. Understanding Success Criterion 1.4.11: Non-text Contrast [https://www.w3.org/WAI/WCAG21/Understanding/non-text-contrast.html]
- 14. How to Choose the Right Heatmap Colors Palette? [https://vwo.com/blog/heatmap-colors/]
- 15. Help for package heatmaply - CRAN - R Project [https://cran.r-project.org/web/packages/heatmaply/refman/heatmaply.html]
- 16. Heatmaps - the gene expression edition – learn-r - Jeff Oliver [https://jcoliver.github.io/learn-r/009-expression-heatmaps.html]
- 17. Visualizing the structure of RNA-seq expression data using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5363805/]
- 18. Package heatmaply - CRAN [https://cran.r-project.org/package=heatmaply]
- 19. a practical guide to RNA sequencing data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599993/]
- 20. A benchmark of RNA-seq data normalization methods for ... [https://www.nature.com/articles/s41540-024-00448-z]
- 21. heatmap pattern for rna [https://www.biostars.org/p/465642/]
- 22. 06 Differential expression analysis – Introduction to RNA-seq [https://scienceparkstudygroup.github.io/rna-seq-lesson/06-differential-analysis/index.html]
- 23. Interactive Cluster Heat Maps Using plotly and ggplot2 [https://talgalili.github.io/heatmaply/]
- 24. Choosing color palettes — seaborn 0.13.2 documentation [https://seaborn.pydata.org/tutorial/color_palettes.html]
- 25. conditionally change label text colours with (ggplot2 or plotly) [https://stackoverflow.com/questions/59080936/r-heatmap-conditionally-change-label-text-colours-with-ggplot2-or-plotly]
- 26. How does a heatmap's font_colors attribute work? [https://community.plotly.com/t/how-does-a-heatmaps-font-colors-attribute-work/39013]
- 27. Creating Heatmaps with Hierarchical Clustering [https://www.geeksforgeeks.org/machine-learning/creating-heatmaps-with-hierarchical-clustering/]
- 28. heatmaply: Interactive Cluster Heat Maps Using 'plotly' and ... [https://talgalili.r-universe.dev/heatmaply]
- 29. Hierarchical clustering / heatmap [https://help.connected.illumina.com/icm/analyses/analysis-functionality/task-menu/exploratory-analysis/hierarchical-clustering]
- 30. How to create an interactive heatmaply plot with custom ... [https://stackoverflow.com/questions/66763623/how-to-create-an-interactive-heatmaply-plot-with-custom-text-in-r]
- 31. A Complete Guide to Heatmaps [https://www.atlassian.com/data/charts/heatmap-complete-guide]
- 32. Understanding WCAG 2 Contrast and Color Requirements [https://webaim.org/articles/contrast/]
- 33. Contrast Checker [https://webaim.org/resources/contrastchecker/]
- 34. heatmaply: adjust the colour [https://stackoverflow.com/questions/61009901/heatmaply-adjust-the-colour]
- 35. Chapter 5 Legends | ComplexHeatmap Complete Reference [https://jokergoo.github.io/ComplexHeatmap-reference/book/legends.html]
- 36. : Cluster heatmap based on plotly in heatmaply : Interactive... heatmaply [https://rdrr.io/cran/heatmaply/man/heatmaply.html]
- 37. How to save a heat map created using the ... [https://stackoverflow.com/questions/66044888/how-to-save-a-heat-map-created-using-the-complexheatmap-package-to-png-format]
- 38. GeneSetCluster 2.0: a comprehensive toolset for summarizing ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06249-3]
- 39. Pathway enrichment analysis and visualization of omics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6607905/]
- 40. Z-Score Normalization: Definition and Examples [https://www.geeksforgeeks.org/data-analysis/z-score-normalization-definition-and-examples/]
- 41. Analysis of Microarray Data Using Z Score Transformation [https://pmc.ncbi.nlm.nih.gov/articles/PMC1907322/]
- 42. Chapter 7 - Data Normalization — Bioinforomics [https://introduction-to-bioinformatics.dev.maayanlab.cloud/lectures/07-data-normalization/intro.html]
- 43. Understanding Z-scores in RNA-seq Analysis [https://pluto.bio/resources/Learning%20Series/overview-of-z-scores-in-rna-seq-experiments]
- 44. Data normalization for RNA-Seq & Heatmap Generation [https://support.basepairtech.com/support/solutions/articles/14000128581-data-normalization-for-rna-seq-heatmap-generation]
- 45. How to Normalize and Standardize Data in R for Great ... [https://www.datanovia.com/en/blog/how-to-normalize-and-standardize-data-in-r-for-great-heatmap-visualization/]
- 46. Using heatmaply with gene expression data - Visualization of ... [https://alanocallaghan.github.io/heatmaplyExamples/articles/biological_data.html]
- 47. 10.3 - Heatmaps | STAT 555 [https://online.stat.psu.edu/stat555/node/87/]
- 48. talgalili/heatmaplyExamples: vignettes/biological_data_2.Rmd [https://rdrr.io/github/talgalili/heatmaplyExamples/f/vignettes/biological_data_2.Rmd]
- 49. Contrast requirements for WCAG 2.2 Level AA [https://www.makethingsaccessible.com/guides/contrast-requirements-for-wcag-2-2-level-aa/]
- 50. Selective Yellow – Chateau Green – White Color scheme [https://icolorpalette.com/4285f4_ea4335_fbbc05_34a853_ffffff]
- 51. Google New Color Palette [https://www.color-hex.com/color-palette/51381]
- 52. Text has enhanced contrast | ACT Rule | WAI [https://www.w3.org/WAI/standards-guidelines/act/rules/09o5cg/]
- 53. Text color must provide high contrast with its background [https://docs.acquia.com/web-governance/text-color-must-provide-high-contrast-its-background]
- 54. Is there an "official" standard usage of Red and Green on ... [https://bioinformatics.stackexchange.com/questions/13596/is-there-an-official-standard-usage-of-red-and-green-on-differential-gene-expr]
- 55. How to assign correctly low, mid and high color limits with ... [https://stackoverflow.com/questions/54749076/how-to-assign-correctly-low-mid-and-high-color-limits-with-heatmply-r-package-f]
- 56. HeatmapGenerator - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4279803/]
- 57. Show Disparity in Gene Expression with a Heat Map [https://bitesizebio.com/34121/show-disparity-gene-expression-heat-map/]
- 58. Feature-expression heat maps – A new visual method to ... [https://www.sciencedirect.com/science/article/pii/S1532046414002238]
- 59. Speed up rendering of large heatmap from ggplot in R [https://stackoverflow.com/questions/52889540/speed-up-rendering-of-large-heatmap-from-ggplot-in-r]
- 60. Step-by-step heatmap tutorial with pheatmap() [https://biostatsquid.com/step-by-step-heatmap-tutorial-with-pheatmap/]
- 61. Make Heatmap Look More Professional in R [https://stackoverflow.com/questions/63262418/make-heatmap-look-more-professional-in-r]
- 62. ComplexHeatmap and Enhanced Volcano - BTEP Coding Club [https://bioinformatics.ccr.cancer.gov/docs/btep-coding-club/CC2023/complex_heatmap_enhanced_volcano/]
- 63. ComplexHeatmap [https://helmholtz.software/software/complexheatmap]
- 64. How to produce a heatmap with ggplot2? [https://stackoverflow.com/questions/8406394/how-to-produce-a-heatmap-with-ggplot2]
- 65. ggplot2 heatmap [https://r-graph-gallery.com/79-levelplot-with-ggplot2.html]
- 66. Correlation heat maps with {ggplot2} [https://www.r-bloggers.com/2024/02/correlation-heat-maps-with-ggplot2/]
- 67. Clustergrammer, a web-based heatmap visualization and ... [https://www.nature.com/articles/sdata2017151]
- 68. Colorblind-Friendly Palettes: Why & How to Use in Design [https://venngage.com/blog/color-blind-friendly-palette/]
- 69. for Heatmaps analysis – simple... - biostatsquid.com gene expression [https://biostatsquid.com/heatmaps-simply-explained/]
- 70. Heatmap visualization of gene expression results [https://bio-protocol.org/exchange/minidetail?id=261876&type=30]
- 71. Phantasus, a web application for visual and interactive ... [https://elifesciences.org/articles/85722]
- 72. Differential gene expression analysis pipelines and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10951429/]
- 73. heatmaply package [https://www.rdocumentation.org/packages/heatmaply/versions/1.6.0]