GATK Somatic Calling: Complete Guide to Input BAM Requirements and Best Practices
This comprehensive guide details the critical input BAM file requirements for successful somatic variant discovery with GATK's Mutect2.
GATK Somatic Calling: Complete Guide to Input BAM Requirements and Best Practices
Abstract
This comprehensive guide details the critical input BAM file requirements for successful somatic variant discovery with GATK's Mutect2. Covering foundational principles, methodological workflows for tumor-normal, tumor-only, and mitochondrial analyses, common troubleshooting scenarios, and validation strategies using benchmark datasets, it provides researchers and clinicians with the essential knowledge to optimize their sequencing data for accurate detection of cancer mutations and somatic mosaicism.
Understanding the Role of BAM Files in GATK Somatic Variant Discovery
GATK Mutect2 is a sophisticated computational tool designed for the discovery of somatic short variants, including single nucleotide variants (SNVs) and insertions or deletions (indels), from high-throughput sequencing data [1] [2]. Developed by the Broad Institute, it combines the DREAM challenge-winning somatic genotyping engine of the original MuTect with the assembly-based machinery of HaplotypeCaller, providing a powerful solution for detecting somatic mutations in cancer and other contexts [2].
Mutect2 operates using a Bayesian somatic genotyping model that differs from the original MuTect approach, performing local assembly of haplotypes to identify variants with high sensitivity and specificity [1]. This tool is featured prominently in the Somatic Short Mutation Calling Best Practice Workflow and has evolved significantly since its inception, with versions from v4.1.0.0 onwards enabling joint analysis of multiple samples and accommodating extreme high depths up to 20,000X [1].
Beyond its primary application in cancer genomics, Mutect2 has proven adaptable to other research contexts, including mitochondrial variant calling and detection of somatic mosaicism [1]. Its powerful processing engine and high-performance computing features make it capable of handling projects of any scale, from targeted panels to whole genomes.
Key Operational Modes
Tumor with Matched Normal
The tumor-normal mode represents the most robust configuration for somatic variant calling, where a tumor sample is analyzed alongside a matched normal sample from the same individual. In this mode, Mutect2 employs specific logic to avoid emitting variants that are clearly present in the germline based on the evidence from the matched normal, conserving computational resources by pre-filtering obvious germline events at an early stage [1]. When a variant's germline status is borderline, Mutect2 will emit the variant to the callset for subsequent filtering by FilterMutectCalls and manual review [1].
The basic command structure for this mode is:
Starting with v4.1, Mutect2 supports joint calling of multiple tumor and normal samples from the same individual, requiring only that additional samples be specified with -I and -normal parameters [1]. This multi-sample approach genotypes all tumor samples jointly, allowing them to share statistical power, which is particularly useful when analyzing several samples with low coverage or low variant allele fractions [3].
Tumor-Only Mode
When a matched normal sample is unavailable, Mutect2 can operate in tumor-only mode using a single sample's alignment data. This approach is inherently more challenging because the evidence from normal reads is missing, and the powerful normal artifact filter is not available [3]. The tool must rely more heavily on population germline resources and panels of normals to distinguish somatic variants from germline polymorphisms.
The basic command for tumor-only calling is:
For creating a panel of normals (PoN), researchers first call variants on each normal sample individually in tumor-only mode, then use CreateSomaticPanelOfNormals to generate the composite PoN [1] [2]. After FilterMutectCalls filtering, additional filtering by functional significance with Funcotator is recommended for tumor-only callsets [1].
Mitochondrial Mode
Mutect2 offers specialized processing for mitochondrial DNA through its mitochondrial mode, activated with the --mitochondria flag. This mode automatically optimizes parameters for calling variants on mitochondria, setting --initial-tumor-lod to 0, --tumor-lod-to-emit to 0, --af-of-alleles-not-in-resource to 4e-3, and the advanced parameter --pruning-lod-threshold to -4 [1]. This configuration enhances sensitivity for detecting variants in high-depth mitochondrial sequencing data.
The command structure for mitochondrial calling is:
This mode accepts only a single sample, though it can be provided in multiple files [1].
Force-Calling Mode
For scenarios where specific variants require investigation regardless of whether they would normally be detected, Mutect2 provides a force-calling mode that genotypes all alleles specified in a VCF file in addition to any other variants discovered during normal operation [1]. This is particularly useful for clinical applications where known variants of interest must be comprehensively assessed.
The command includes the -alleles parameter:
For samples suspected to exhibit orientation bias artifacts (such as FFPE tumor samples), researchers can add an --f1r2-tar-gz argument to collect F1R2 metrics for subsequent analysis with LearnReadOrientationModel [1].
Performance Characteristics and Benchmarking
Performance Across Sequencing Depths and Mutation Frequencies
Extensive benchmarking studies have systematically evaluated Mutect2's performance across different sequencing depths and variant allele frequencies (VAF). The relationship between these parameters and detection performance follows predictable patterns that should inform experimental design decisions.
Table 1: Performance Metrics of Mutect2 Across Different Sequencing Depths
| Sequencing Depth | Recall Rate Range | Precision Rate Range | F-score Range | Recommended Use Cases |
|---|---|---|---|---|
| 100X | 23-96% | >95% (in most samples) | 0.374-0.96 | Limited applications due to variable sensitivity |
| 200X | 48-97% | >95% | 0.63-0.96 | Standard for mutation frequency ≥20% |
| 300X | 50-97% | >95% | 0.65-0.96 | Moderate improvement over 200X |
| 500X | 60-97% | 93-95% | 0.70-0.96 | Beneficial for low frequency mutations (5-10%) |
| 800X | 70-97% | 93-95% | 0.75-0.96 | Optimal performance across all frequencies |
Higher sequencing depths generally improve recall rates, with increases of 0.6-44% observed when depth increases from 100X to 200-800X [4]. However, this comes with a marginal decrease in precision (less than 0.7%) when sequencing depth exceeds 200X [4]. For most applications targeting higher mutation frequencies (≥20%), sequencing depths of 200X provide sufficient sensitivity to call 95% of mutations [4].
Table 2: Mutect2 Performance Across Different Mutation Frequencies
| Mutation Frequency | Recall Rate | Precision | F-score | Recommended Depth |
|---|---|---|---|---|
| 1% (Very Low) | 2.7-34.5% | 68.9-100% | 0.05-0.51 | 500-800X |
| 5-10% (Low) | 48-96% | 95.5-95.9% | 0.65-0.95 | ≥500X |
| 20-40% (High) | 92-97% | >95% | 0.94-0.96 | ≥200X |
Mutation frequency substantially influences the performance benefits gained from increasing sequencing depth [4]. For low mutation frequencies (1%), detection performance remains poor even at high depths, with recall rates between 2.7-34.5% across all depths and tools [4]. At moderate frequencies (5-10%), recall improves substantially to 48-96%, while high mutation frequencies (20-40%) enable excellent recall rates of 92-97% [4].
Comparative Performance with Strelka2
Benchmarking studies have compared Mutect2 against other somatic callers, particularly Strelka2, revealing context-dependent performance differences:
Table 3: Performance Comparison Between Mutect2 and Strelka2
| Performance Metric | Mutation Frequency ≥20% | Mutation Frequency 5-10% | Mutation Frequency 1% |
|---|---|---|---|
| Mutect2 Precision | >95% | 95.5-95.9% | Variable |
| Strelka2 Precision | >95% | 96.2-96.5% | Variable |
| Mutect2 Recall | >90% | 50-96% | 2.7-34.5% |
| Strelka2 Recall | >90% | 48-93% | 2.7-34.5% |
| Relative Performance | Comparable (difference <1%) | Mutect2 has slightly better F-score | Mutect2 superior at 500X+ |
| Computational Speed | Reference | 17-22 times faster | 17-22 times faster |
For higher mutation frequencies (≥20%), both tools perform excellently with minimal differences (less than 1%) in precision, recall, and F-score [4]. At moderate mutation frequencies (5-10%), Strelka2 demonstrates slightly higher precision (96.2-96.5% vs. 95.5-95.9%) while Mutect2 achieves higher recall (50-96% vs. 48-93%), resulting in better overall F-scores for Mutect2 [4]. At the lowest mutation frequency (1%), Mutect2 surpasses Strelka2 when sequencing depth increases to 500X or higher [4]. Notably, Strelka2 is significantly faster, processing data 17-22 times quicker on average [4].
Input BAM Requirements and Preprocessing
BAM File Preparation Workflow
Proper preparation of input BAM files is crucial for optimal Mutect2 performance. The preprocessing workflow ensures that sequencing artifacts are minimized and data quality is maximized before variant calling.
Critical BAM Quality Metrics
For reliable somatic variant calling, input BAM files must meet specific quality standards. Several key metrics determine whether sequencing data is appropriate for Mutect2 analysis.
Sequence Alignment: The initial alignment of raw sequencing reads to a reference genome is a critical foundation for accurate variant calling [5]. Recommended aligners include BWA-Mem, which efficiently handles the characteristics of NGS data [5]. The resulting alignments are stored in BAM format, which has become the standard for storing and sharing NGS data due to compressed file size, indexed-access capabilities, and standardized format [5].
Duplicate Marking: PCR duplicates, representing 5-15% of sequencing reads in a typical exome, should be identified and marked using tools such as Picard or Sambamba [5]. These redundant reads originate from the same DNA sequence molecule and can be identified based on alignment position and read pairing information. Marking (rather than removing) duplicates preserves information for downstream QC metrics while preventing artificial inflation of coverage estimates.
Base Quality Score Recalibration (BQSR): This optional but recommended step adjusts the base quality scores of sequencing reads using an empirical error model [5]. While evaluations of variant calling accuracy before and after BQSR suggest improvements are marginal, it may help correct systematic errors in base quality scores [5].
Quality Control: Comprehensive QC of analysis-ready BAMs should be performed prior to variant calling to evaluate key sequencing metrics, verify sufficient sequencing coverage was achieved, and check samples for evidence of contamination [5]. For family studies and paired samples (e.g., tumor-normal), expected sample relationships should be confirmed with tools for relationship inference such as the KING algorithm [5].
Resource and Panel Requirements
Mutect2 does not strictly require a germline resource or panel of normals (PoN) to run, but both are strongly recommended for optimal performance [1]. The tool uses these resources to prefilter sites, saving computational resources and improving specificity.
Germline Resources: Population VCFs such as gnomAD provide allele frequencies that serve as prior probabilities for germline variation [1] [3]. When a variant is absent from the germline resource, Mutect2 uses an imputed allele frequency based on the --af-of-alleles-not-in-resource parameter [1]. The default values for this parameter are dynamically adjusted: tumor-only calling sets it to 5e-8, tumor-normal calling to 1e-6, and mitochondrial mode to 4e-3 [1].
Panel of Normals (PoN): The PoN captures technical artifacts and common germline variants present in a population of normal samples [3]. Mutect2 marks variants found in the PoN with the "PON" info field, which FilterMutectCalls then uses for filtering [3]. Most PON sites are silently pre-filtered without ending up in the output, though some may be emitted if they appear in active regions containing non-PON sites [3].
Advanced Filtering and Artifact Detection
Comprehensive Filtering Workflow
Raw variant calls from Mutect2 require sophisticated filtering to separate true somatic variants from technical artifacts and residual germline polymorphisms. The complete filtering workflow involves multiple specialized tools and considerations.
Orientation Bias Artifact Filtering
Sequence context-dependent artifacts represent a significant challenge in somatic variant calling, particularly for certain sample types. Mutect2 provides specialized tools to address these artifacts.
Common Artifact Types:
- OxoG Artifacts: Stem from oxidation of guanine to 8-oxoguanine, resulting in G to T transversions [6]. These are particularly common in certain sample preparation protocols.
- FFPE Artifacts: Result from formaldehyde deamination of cytosines in formalin-fixed paraffin-embedded samples, causing C to T transitions [6]. These are prevalent in archival clinical specimens.
The FilterByOrientationBias tool provides specialized filtering for these artifacts and should be run after FilterMutectCalls [6]. This tool requires pre-adapter detailed metrics calculated by Picard's CollectSequencingArtifactMetrics and specification of the base substitutions to consider for orientation bias [6].
The workflow for orientation bias filtering involves:
- Running CollectSequencingArtifactMetrics on the sample BAM
- Processing Mutect2 calls through FilterMutectCalls
- Applying FilterByOrientationBias with the appropriate artifact modes
Example command for OxoG artifact filtering:
For FFPE samples, researchers would specify --artifact-modes 'C/T' to address formalin-induced deamination artifacts [6].
Key Filtering Parameters
FilterMutectCalls employs a sophisticated statistical framework to distinguish true somatic variants from various false positive scenarios:
Normal Artifact LOD: While the normal log odds (NLOD) is calculated assuming a diploid genotype, the normal-artifact-lod uses the same variable ploidy assumption approach as tumor-lod [2]. If the normal artifact log odds becomes large, FilterMutectCalls applies the "artifact-in-normal" filter [2]. For matched normal samples with tumor contamination, consider increasing the normal-artifact-lod threshold [2].
Tumor LOD: The tumor log odds is calculated independently of any matched normal and determines whether to filter a tumor variant [2]. Variants with tumor LODs exceeding the threshold pass filtering [2].
Sensitivity Adjustment: The -f-score-beta parameter in FilterMutectCalls controls the balance between sensitivity and precision [3]. Increasing this parameter from its default value of 1 enhances sensitivity at the expense of precision, which may be appropriate for screening applications where false negatives are less desirable than false positives [3].
Research Reagent Solutions
Table 4: Essential Research Reagents and Resources for Mutect2 Analysis
| Resource Type | Specific Examples | Purpose and Function | Criticality |
|---|---|---|---|
| Germline Resource | af-only-gnomad.hg38.vcf | Provides population allele frequencies as prior probabilities for germline variation | Highly Recommended |
| Panel of Normals | 1000g_pon.hg38.vcf, Mutect2-WGS-panel-b37.vcf | Identifies technical artifacts and common germline variants present in normal populations | Highly Recommended |
| Reference Genome | GRCh38, HG19 | Standardized reference sequence for read alignment and variant calling | Required |
| Benchmarking Resources | GIAB Gold Standard Datasets (HG001-7) | Enables performance validation and optimization through comparison against truth sets | Recommended for QC |
| Alignment Tools | BWA-MEM | Efficient mapping of sequencing reads to reference genome | Required |
| Duplicate Marking | Picard MarkDuplicates | Identifies PCR duplicates to prevent coverage inflation | Required |
| Variant Filtering | FilterMutectCalls, FilterByOrientationBias | Removes false positives and technical artifacts from raw variant calls | Required |
These research reagents form the foundation of reproducible somatic variant detection. The germline resource, typically derived from gnomAD, provides population allele frequencies that serve as prior probabilities in Mutect2's statistical model [1] [3]. When a variant is absent from this resource, Mutect2 uses a small imputed allele frequency based on the --af-of-alleles-not-in-resource parameter [1]. The Panel of Normals captures systematic artifacts specific to sequencing protocols and analysis methods [3]. Publicly available PoNs include the 1000 Genomes Project panel for hg38 and Broad Institute-generated panels for both whole-genome and exome sequencing [3].
Benchmarking resources such as the Genome in a Bottle (GIAB) consortium's gold standard datasets enable objective performance assessment and pipeline optimization [5]. These resources provide high-confidence variant calls based on the consensus of multiple technologies and variant calling methods, establishing "ground truth" for evaluation [5]. For somatic calling, synthetic datasets or carefully characterized cell line mixtures can provide similar benchmarking capabilities.
Critical Importance of Properly Processed Input BAM Files
In genomic research, particularly in somatic variant discovery for cancer and drug development, the quality of the input data fundamentally determines the validity of all subsequent findings. Properly processed Binary Alignment Map (BAM) files represent the essential foundation upon which accurate variant identification rests. The Genome Analysis Toolkit (GATK), the industry standard for genomic analysis, imposes stringent requirements on input BAM files to ensure analytical validity [7]. Somatic variant calling with tools like Mutect2 requires BAM files that have undergone extensive pre-processing to correct for technical artifacts and biases inherent in sequencing technologies [8] [9]. When BAM files fail to meet these specifications, researchers risk introducing false positives, overlooking genuine somatic mutations, and ultimately generating unreliable results that can misdirect critical research pathways and therapeutic development programs.
The complex journey from raw sequencing reads to analysis-ready BAM files involves multiple computationally intensive steps, each designed to address specific technical challenges [9]. This whitepaper provides researchers and drug development professionals with a comprehensive technical framework for understanding, creating, and validating BAM files that meet the exacting standards required for robust somatic variant discovery using GATK's best practices workflow.
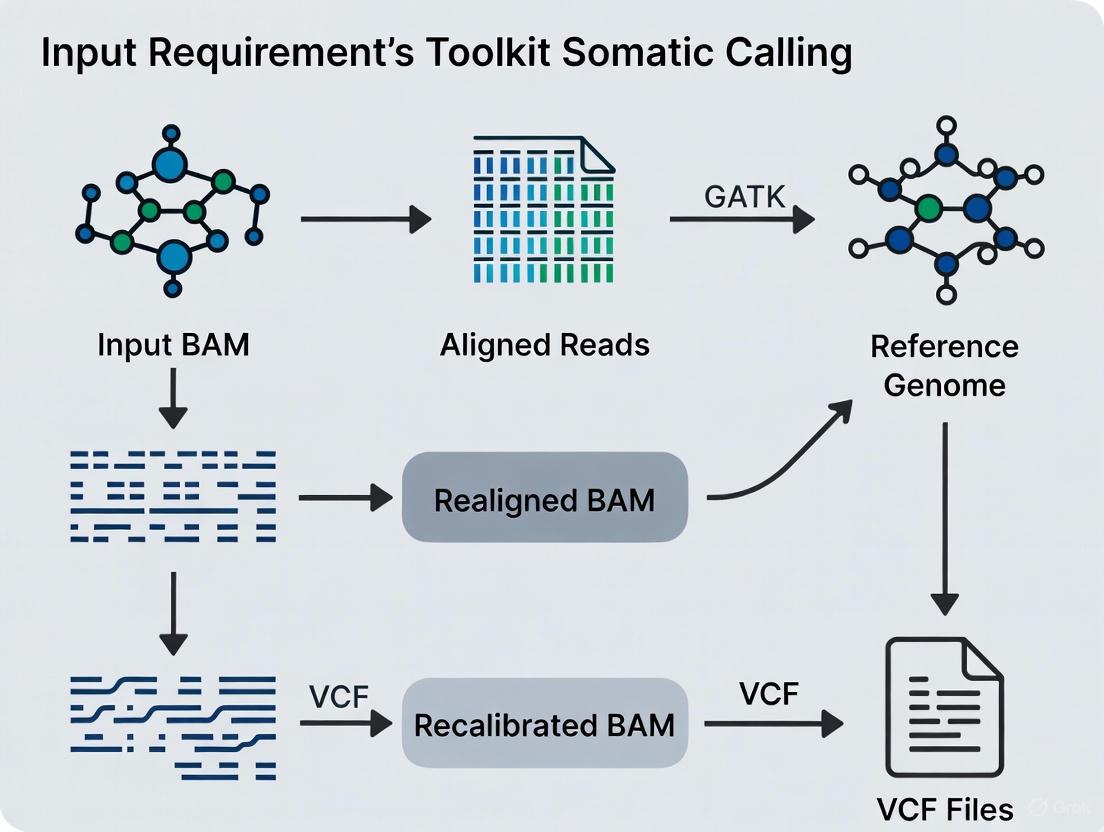
BAM File Specifications and GATK Requirements
Structural and Compositional Mandates
GATK maintains precise specifications for input BAM files to ensure compatibility and analytical accuracy. These requirements span file structure, content organization, and metadata completeness. According to GATK documentation, all SAM, BAM, or CRAM files must be accompanied by an index, which can be generated by Picard's BuildBamIndex tool [7]. The header must contain read groups with sample names, and every read in a BAM must belong to a read group listed in the header [7]. This read group information is critical for tracking the provenance of sequencing data and for downstream analysis steps.
A particularly critical requirement is that GATK tools require that reads be sorted in coordinate order—not by queryname and not "unsorted" [7]. This coordinate sorting enables efficient processing of genomic regions and is essential for proper function of GATK's variant discovery tools. The sort order must be explicitly declared in the header using the SO:coordinate flag [7]. Contig ordering must also be consistent across all files in an analysis, including FASTA and VCF files, with everything matching the sequence dictionary of the reference genome FASTA [7].
Table: Essential BAM File Requirements for GATK Somatic Variant Calling
| Requirement Category | Specific Specification | Purpose/Rationale |
|---|---|---|
| File Format | BAM (preferred) or CRAM | CRAM observed to have performance issues; BAM recommended for analysis [7] |
| Sort Order | Coordinate-sorted (SO:coordinate in header) |
Enables efficient regional processing; required by GATK tools [7] |
| Indexing | Must have accompanying index (.bai file) | Permits random access to genomic regions; required by GATK [7] |
| Read Groups | Required with sample names (SM tags) | Essential for sample tracking and downstream analysis [7] |
| Validation | Must pass ValidateSamFile (Picard) | Ensures file integrity and compliance with specifications [7] |
| Contig Order | Consistent with reference dictionary | Prevents analytical errors from coordinate mismatches [7] |
Technical Composition of BAM Files
Understanding the internal structure of BAM files is essential for troubleshooting and quality assurance. BAM files are the binary, compressed version of SAM (Sequence Alignment Map) files, which are tab-delimited text files containing alignment information [10]. The basic structure consists of a header section and alignment section. The header section begins with '@' characters and includes critical metadata such as @SQ lines for reference sequences and @RG lines for read groups [10].
The alignment section contains one line per read, with 11 mandatory fields that provide comprehensive mapping information [10]. Key fields include the SAM flag (which encodes multiple properties of the alignment in a single numerical value), mapping quality (MAPQ), and CIGAR string (which precisely describes the alignment including matches, mismatches, insertions, and deletions) [10]. The CIGAR string is particularly important for variant discovery as it enables the identification of insertion and deletion events.
The BAM Pre-processing Pipeline for Somatic Variant Discovery
Systematic Workflow from Raw Data to Analysis-Ready BAM
The transformation of raw sequencing data into analysis-ready BAM files requires a multi-stage pre-processing pipeline designed to correct systematic errors and prepare data for variant discovery. The GATK best practices workflow for data pre-processing involves three principal phases: mapping to a reference genome, duplicate marking, and base quality score recalibration [9]. Each stage addresses distinct technical challenges and introduces specific corrections to enhance data quality.
Mapping to Reference represents the initial processing step, performed per-read group, where individual read pairs are aligned to a reference genome [9]. This provides the fundamental coordinate system for all subsequent genomic analyses. Because the mapping algorithm processes each read pair in isolation, this step can be massively parallelized to increase throughput. The resulting file is in SAM/BAM format sorted by coordinate [9].
Marking Duplicates constitutes the second processing step, performed per-sample, where read pairs likely originating from duplicates of the same original DNA fragments are identified [9]. These represent non-independent observations that can bias variant calling, so the program tags all but a single read pair within each set of duplicates, causing the marked pairs to be ignored during variant discovery. GATK recommends using MarkDuplicatesSpark for this step, which utilizes Apache Spark to parallelize what has historically been a performance bottleneck [9].
Base Quality Score Recalibration (BQSR) forms the third and most computationally sophisticated pre-processing step, performed per-sample [9]. This procedure applies machine learning to detect and correct patterns of systematic errors in the base quality scores, which are confidence scores emitted by the sequencer for each base call. Since variant calling algorithms rely heavily on these quality scores, correcting systematic biases is essential for accurate variant discovery [9]. Biases can originate from various sources, including biochemical processes during library preparation, manufacturing defects in sequencing chips, or instrumentation defects in the sequencer.
Specialized Processing for Specific Applications
While the core pre-processing workflow remains consistent across applications, certain data types require specialized processing steps. For RNA sequencing data, an additional SplitNCigarReads step is necessary to split reads that contain Ns in their CIGAR string, which typically represent reads spanning splicing events [11]. This specialized processing ensures that variant calling algorithms can properly interpret splicing-aware alignments.
For tumor-normal paired analyses, the pre-processing pipeline must be applied consistently to both sample types, with particular attention to potential artifacts in formalin-fixed paraffin-embedded (FFPE) samples, which often exhibit specific sequencing artifacts that require specialized handling in the Mutect2 workflow [8] [12].
Quality Control and Validation of Processed BAM Files
Comprehensive Quality Assessment Metrics
Rigorous quality control of processed BAM files is essential before proceeding to variant discovery. Multiple quality assessment tools provide complementary perspectives on BAM file quality, with GATK's CollectMultipleMetrics and external tools like Qualimap offering comprehensive evaluation frameworks [11] [13]. These tools generate a suite of metrics that collectively characterize the technical quality of the sequencing data and its suitability for somatic variant calling.
Alignment metrics provide fundamental information about mapping success, including the total number of reads, mapping rate, and read distribution across genomic regions [13]. For targeted sequencing approaches, the percentage of reads on-target represents a critical quality indicator. Coverage metrics characterize the depth and uniformity of sequencing across the genome or target regions, with mean coverage, coverage uniformity, and the percentage of bases achieving minimum coverage thresholds (typically 20-30X for somatic variants) representing key quality benchmarks [13].
Library complexity metrics help identify potential issues with amplification bias, with duplication rates serving as a primary indicator [13]. Exceptionally high duplication rates may suggest insufficient starting material or amplification artifacts that could compromise variant detection. Insert size metrics provide information about library preparation quality, with deviations from expected insert size distributions potentially indicating fragmentation issues or other library preparation artifacts [13].
Table: Essential Quality Control Metrics for Processed BAM Files
| Metric Category | Specific Metrics | Acceptance Criteria |
|---|---|---|
| Alignment Quality | Mapping rate (>90%), Properly paired rate, Mean mapping quality (>Q30) | Indicates successful alignment to reference |
| Coverage Statistics | Mean coverage (≥30X for somatic), Uniformity, % bases at ≥20X (>85%) | Ensures sufficient depth for variant detection |
| Library Complexity | Duplication rate (varies by protocol), Estimated library size | Identifies potential amplification biases |
| Base Quality | Pre- and post-recalibration quality scores, Error rates | Confirms effective error correction |
| Sequence Content | GC distribution, Nucleotide composition | Detects sequence-specific biases |
| Insert Size | Mean insert size, Standard deviation | Verifies library preparation quality |
Specialized QC for Somatic Variant Discovery
Beyond standard quality metrics, BAM files intended for somatic variant discovery require additional, specialized quality assessments. Cross-sample contamination represents a particular concern in somatic analyses, as even low levels of contamination can generate false positive variant calls [8]. GATK provides specialized tools including GetPileupSummaries and CalculateContamination specifically designed to estimate contamination fractions, with CalculateContamination engineered to perform well even in samples with significant copy number variation [8].
For analyses involving FFPE-derived samples or those sequenced on platforms prone to specific artifacts, read orientation bias assessment becomes critical [12]. The Mutect2 workflow includes specialized tools (LearnReadOrientationModel) to detect and correct for substitution errors that occur predominantly on a single strand before sequencing [12]. This specialized quality control step is essential for minimizing false positive calls in challenging sample types.
Integration with GATK Mutect2 Somatic Variant Calling
Direct Pipeline Connectivity
Properly processed BAM files serve as the direct input to the GATK Mutect2 somatic variant calling workflow, which identifies somatic short variants (SNVs and Indels) via local assembly of haplotypes [8] [1]. The Mutect2 caller uses a Bayesian somatic genotyping model that differs from the original MuTect algorithm and employs assembly-based machinery similar to HaplotypeCaller [1]. When Mutect2 encounters regions showing evidence of somatic variation, it completely reassembles reads in that region to generate candidate variant haplotypes, then aligns each read to these haplotypes to obtain likelihood matrices [8].
The critical dependency between BAM quality and Mutect2 performance manifests in multiple aspects of the algorithm. The base quality scores directly influence the Bayesian genotyping model, with inaccurately calibrated quality scores potentially skewing variant likelihood calculations [9]. The presence of duplicate reads can artificially inflate evidence for variant alleles if not properly flagged [9]. Improperly sorted BAM files or those with incorrect read groups can disrupt the regional processing approach used by Mutect2, leading to analysis failures or incomplete variant calling [7].
Advanced Filtering Dependencies
The downstream filtering steps in the Mutect2 workflow exhibit direct dependencies on properly processed BAM files. FilterMutectCalls, which applies sophisticated filters to distinguish true somatic variants from artifacts, relies on multiple BAM-derived metrics [8] [12]. The contamination estimation derived from GetPileupSummaries and CalculateContamination directly influences filtering stringency [8]. The read orientation model learned from the BAM file data enables detection of strand-specific artifacts common in FFPE samples [12].
Recent advances in Mutect2 have introduced a somatic clustering model that leverages allele fraction patterns across variants to distinguish true somatic events from artifacts [12]. This model uses a Dirichlet process binomial mixture model to identify subclonal allele fraction clusters, which depends on accurate read alignment and quality calibration in the input BAM files to properly characterize allele fractions [12].
Successful somatic variant discovery requires not only proper BAM files but also a suite of reference resources and analytical tools that collectively ensure analytical validity. These reagents provide the necessary context for distinguishing true somatic variants from technical artifacts and population polymorphisms.
Table: Essential Research Reagents for Somatic Variant Discovery
| Resource Type | Purpose/Function | Examples/Sources |
|---|---|---|
| Reference Genome | Coordinate system for alignment and variant calling | GRCh37/hg19, GRCh38/hg38 (with consistent decorators) |
| Germline Resource | Provides population allele frequencies to distinguish rare germline variants from somatics | gnomAD, 1000 Genomes, population-specific databases |
| Panel of Normals (PON) | Identifies recurrent technical artifacts across normal samples | Created from 40+ normal samples using CreateSomaticPanelOfNormals [12] |
| Known Sites Resources | Enables base quality recalibration and contamination assessment | dbSNP, Mills indel gold standard, known indels sets [9] |
| Functional Annotation Databases | Provides biological context for identified variants | COSMIC, dbSNP, GENCODE, ClinVar [8] |
The germline resource, typically derived from large population databases like gnomAD, provides allele frequencies that enable Mutect2 to distinguish potential germline polymorphisms from true somatic events [1]. The Panel of Normals (PON) is particularly critical for tumor-only analyses, as it captures technical artifacts that recur across normal samples, allowing these to be filtered from the final variant call set [12]. Creation of a high-quality PON requires Mutect2 to be run on each normal sample in tumor-only mode, followed by GenomicsDBImport and CreateSomaticPanelOfNormals to aggregate artifacts [12].
Properly processed BAM files represent the non-negotiable foundation for robust somatic variant discovery in cancer research and therapeutic development. The intricate dependencies between BAM file quality, pre-processing methodologies, and downstream variant calling algorithms necessitate rigorous attention to each stage of the data preparation pipeline. From initial read alignment through duplicate marking, base quality recalibration, and comprehensive quality assessment, each step introduces specific corrections that collectively enable accurate distinction of true somatic mutations from technical artifacts.
The GATK Mutect2 workflow incorporates sophisticated statistical models and filtering approaches that presume properly processed, analysis-ready BAM files as input. Deviations from best practices in BAM processing propagate through the analytical pipeline, potentially compromising the sensitivity and specificity of variant detection. For researchers in drug development and clinical applications, where variant calls may inform therapeutic decisions and clinical trial design, adherence to these stringent BAM processing standards is not merely methodological refinement—it is an essential requirement for generating clinically actionable insights.
As somatic variant detection extends into increasingly challenging contexts, including low allele fractions, complex structural variants, and heterogeneous sample types, the importance of optimized BAM processing will only intensify. Future methodological advances will likely introduce additional preprocessing refinements, but the fundamental principle will endure: reliable somatic variant discovery depends irrevocably on properly processed input BAM files.
Within the framework of somatic variant discovery for cancer research and therapeutic development, data pre-processing constitutes the foundational phase that transforms raw sequencing data into analysis-ready aligned files. This process is particularly critical for drug development pipelines where accurate detection of somatic mutations directly informs target identification, patient stratification, and treatment response monitoring. The Genome Analysis Toolkit (GATK) Best Practices workflow establishes a standardized methodology for pre-processing sequence data prior to variant detection, ensuring optimal input quality for somatic callers like Mutect2 [9] [8]. For research teams in pharmaceutical and biotechnology settings, rigorous implementation of these steps reduces false positive rates in variant calling, thereby increasing confidence in identified biomarkers and therapeutic targets.
The pre-processing phase specifically addresses technical artifacts and systematic biases introduced during sequencing library preparation and instrumentation [9]. Through three coordinated procedures—read alignment, duplicate marking, and base quality score recalibration—the workflow produces BAM files that accurately represent the true biological sequence of the tumor and normal samples. This technical foundation is indispensable for subsequent somatic variant calling, where distinguishing true somatic mutations from sequencing artifacts requires carefully calibrated quality metrics [14]. The computational reproducibility afforded by standardized pre-processing directly supports regulatory requirements in clinical trial sequencing and diagnostic development.
Core Pre-processing Steps
Sequence Alignment to Reference
The initial pre-processing step involves aligning raw sequencing reads from FASTQ or unmapped BAM (uBAM) files to a reference genome, establishing genomic coordinates for all subsequent analyses [9]. This process utilizes optimized aligners such as BWA-MEM, which handles the mapping of individual read pairs to the reference genome in a highly parallelizable manner [15]. For somatic variant discovery in cancer research, consistent alignment across tumor-normal pairs is essential to ensure that observed differences reflect true biological variation rather than technical inconsistencies in mapping.
The alignment step is performed per read group, which corresponds to the intersection of library preparation and sequencing lane [16]. Each read group must be assigned appropriate identifiers (RGID) and sample names (SM) that distinguish biological samples from technical replicates. Following alignment, the MergeBamAlignments tool integrates mapping information with original read data to produce coordinate-sorted BAM files [9]. For multi-library or multiplexed sequencing designs, the Broad Institute's production pipeline processes each read group individually before merging, ensuring optimal handling of lane-specific effects while maintaining sample integrity [16].
Table 1: Key Alignment Tools and Their Applications in Somatic Research
| Tool | Primary Function | Role in Somatic Analysis |
|---|---|---|
| BWA-MEM | Read alignment to reference | Maps tumor and normal reads to common coordinate system |
| MergeBamAlignments | Integrates mapping with original read data | Preserves original read information while adding mapping coordinates |
| Samtools | BAM file manipulation and indexing | Provides utilities for file processing and quality checks [14] |
Duplicate Marking
Duplicate marking identifies non-independent observations arising from the same original DNA fragments, most commonly through PCR amplification during library preparation [9]. These artifacts artificially inflate coverage metrics and can mislead variant allele frequency calculations—a critical parameter in somatic variant calling where subclonal mutations may exist at low frequencies. The GATK workflow employs MarkDuplicatesSpark (or MarkDuplicates followed by SortSam) to identify and tag duplicate read pairs based on their alignment coordinates and pairing information [9].
For cancer genome analysis, duplicate marking is particularly important in tumor samples that often originate from limited biological material requiring significant amplification. The process distinguishes between PCR duplicates (occurring during library preparation) and optical duplicates (from sequencing cluster imaging), with the former being more prevalent in amplified samples [9]. Modern implementations perform duplicate marking after merging all read groups for a sample, enabling detection of duplicates that occur across different sequencing lanes [16]. This approach recognizes that the same DNA fragment might be sequenced in multiple lanes in multiplexed designs, and only cross-lane duplicate marking can identify these artifacts.
Diagram 1: Duplicate marking process flow. The algorithm compares alignment coordinates to identify duplicate reads before marking all but one representative read.
Base Quality Score Recalibration
Base quality score recalibration (BQSR) corrects systematic biases in the quality scores assigned by sequencing instruments [9]. These quality scores represent confidence values for each base call and profoundly influence variant calling algorithms, which use them as weights when evaluating evidence for or against potential variants [9]. BQSR employs machine learning to detect patterns of systematic errors based on covariates such as sequencing cycle, base composition, and machine characteristics, then adjusts quality scores to better reflect empirical error rates.
The BQSR process occurs in two distinct phases: BaseRecalibrator builds a recalibration model by analyzing all base calls in the dataset and identifying systematic errors, while ApplyBQSR applies the model to adjust quality scores in the BAM file [9]. For somatic variant discovery, this step is crucial because tumors often exhibit mutation patterns that can be confused with sequencing artifacts without proper quality adjustment. The procedure is performed per read group, recognizing that different lanes and libraries may exhibit distinct error profiles [16]. This granular approach ensures that lane-specific artifacts from sequencing chips or instrumentation defects are properly corrected.
Table 2: Base Quality Score Recalibration Components
| Component | Function | Technical Implementation |
|---|---|---|
| BaseRecalibrator | Builds error model from covariate data | Collects statistics by sequencing cycle, read group, and sequence context [9] |
| Recalibration Model | Mathematical representation of systematic errors | Applies machine learning to detect patterns across covariates [9] |
| ApplyBQSR | Adjusts base quality scores in BAM | Modifies QUAL fields based on model predictions [15] |
| AnalyzeCovariates (Optional) | Visualizes recalibration effects | Generates plots comparing pre- and post-recalibration quality metrics [9] |
Diagram 2: Base quality score recalibration workflow. The process involves analyzing systematic errors, building a correction model, and applying quality score adjustments.
Experimental Design Considerations
Multiplexed Sequencing and Multi-Library Designs
Complex experimental designs involving multiplexed sequencing or multiple libraries per sample require specific adaptations to the standard pre-processing workflow. For drug development studies analyzing large cohorts of tumor samples, multiplexing enables cost-effective sequencing but necessitates careful data handling [16]. The recommended approach processes each read group (representing a unique library-lane combination) individually through alignment and sorting before merging read groups belonging to the same sample during duplicate marking [16].
This strategy efficiently handles technical variability while ensuring biological consistency within samples. When multiple libraries are prepared from the same biological sample, they must maintain distinct read group identifiers to preserve their technical origin throughout analysis [16]. Base quality recalibration subsequently processes the merged BAM files while still distinguishing read groups internally, applying lane-specific corrections where necessary [16]. This balanced approach acknowledges that some artifacts are lane-specific while biological phenomena should be consistent across technical replicates.
Resource Planning for Large-Scale Studies
For pharmaceutical companies conducting large-scale cancer genomics studies, computational resource planning is essential for timely processing of hundreds or thousands of tumor-normal pairs. The GATK Best Practices reference implementations provide scalable solutions optimized for cloud execution [9]. The MarkDuplicatesSpark tool specifically addresses performance bottlenecks in duplicate marking by leveraging Apache Spark for parallel processing, significantly accelerating this computationally intensive step [9].
Base quality recalibration can be scattered across genomic regions (typically by chromosome) during the initial statistics collection phase, then gathered into a genome-wide model [9]. This parallelization strategy enables efficient processing of whole-genome sequencing data from large clinical trials. For organizations with established high-performance computing infrastructure, the GATK workflows can be distributed across clusters to meet the computational demands of drug discovery timelines.
Integration with Somatic Variant Discovery
Input Requirements for Mutect2
The Mutect2 somatic variant caller, which forms the core of the GATK somatic discovery workflow, requires properly pre-processed BAM files as input [8]. The pre-processing steps directly impact Mutect2's performance by ensuring that alignment artifacts do not masquerade as somatic variants and that base quality scores accurately reflect error probabilities [8] [1]. Specifically, duplicate marking prevents over-represented fragments from inflating variant evidence, while base quality recalibration enables the Bayesian caller to properly weigh evidence for and against potential mutations [9].
For tumor-normal analyses, both samples must undergo identical pre-processing procedures to ensure comparability [8]. The GATK somatic short variant discovery workflow explicitly states that "input BAMs should be pre-processed as described in the GATK Best Practices for data pre-processing" before variant calling with Mutect2 [8]. This consistency is particularly important for detecting subclonal variants with low allele frequencies, where subtle technical differences between tumor and normal processing could obscure true biological signals.
Quality Control Metrics
Rigorous quality control following pre-processing is essential before proceeding to variant discovery. The GATK Best Practices recommend verifying key sequencing metrics, assessing coverage uniformity, checking for sample contamination, and confirming expected sample relationships in family studies or tumor-normal pairs [14]. Tools such as Picard and BEDTools provide standardized metrics for evaluating pre-processing outcomes [14].
For cancer studies, additional checks should include comparing the overall quality profiles of tumor and normal samples, verifying expected contamination levels in tumor samples, and ensuring adequate coverage in genomic regions of therapeutic interest [14]. These QC measures provide confidence that pre-processed BAM files meet the quality standards required for reliable somatic variant detection, particularly in clinical contexts where results may inform treatment decisions.
Research Reagent Solutions
Table 3: Essential Computational Tools for Pre-processing in Somatic Variant Studies
| Tool/Resource | Function | Application Context |
|---|---|---|
| BWA-MEM | Read alignment to reference | Standard aligner for Illumina sequencing data [14] [15] |
| GATK MarkDuplicatesSpark | Identify and tag duplicate reads | Handles duplicate marking efficiently across multiple cores [9] |
| GATK BaseRecalibrator | Build base quality recalibration model | Detects patterns of systematic errors in sequencing data [9] [15] |
| GATK ApplyBQSR | Apply base quality adjustments | Implements model to correct systematic quality score biases [15] |
| Samtools | BAM file manipulation and indexing | Provides utilities for file processing, sorting, and indexing [14] |
| Picard Tools | Sequence data processing metrics | Generates quality control metrics for aligned reads [14] |
The pre-processing workflow encompassing alignment, duplicate marking, and base quality recalibration establishes the essential foundation for accurate somatic variant discovery in cancer research. By systematically addressing technical artifacts and biases inherent to sequencing technologies, these steps transform raw data into analysis-ready BAM files suitable for variant calling with Mutect2. For pharmaceutical and biotechnology researchers, rigorous implementation of these standardized procedures ensures the reliability of variant calls used to identify therapeutic targets, develop biomarkers, and stratify patient populations. The integration of these pre-processing steps within the broader context of somatic variant discovery represents a critical methodology supporting the advancing field of precision oncology.
In somatic variant analysis with the Genome Analysis Toolkit (GATK), the fidelity of the final variant callset is inextricably linked to the quality and correctness of the input alignment (BAM) files. Adherence to stringent input requirements is not merely a preliminary step but a critical determinant of success, directly impacting the sensitivity and specificity of mutation detection. This technical guide details the core input specifications—encompassing sample type, read group formatting, and alignment quality—within the framework of the GATK Best Practices workflow for somatic short variant discovery. It provides researchers and drug development professionals with the definitive protocols and quality control measures necessary to generate reliable, reproducible results in cancer genomics and therapeutic development.
Sample Type and Experimental Design
The foundational step in somatic variant analysis is the appropriate selection and pairing of samples. The GATK somatic short variant discovery workflow is designed to identify somatically acquired mutations by comparing sequence data from a tumor sample against a matched normal sample from the same individual [8]. The matched normal sample is crucial for identifying and filtering out germline variants present in the patient's constitutive DNA, thereby isolating the true somatic changes that have occurred in the tumor tissue.
The workflow offers flexibility to accommodate different research scenarios [1]:
- Tumor-Normal Matched Pairs: This is the standard and most recommended approach. It involves processing both the tumor BAM and the matched normal BAM simultaneously. In this mode, Mutect2, the primary variant calling engine in GATK, uses the normal sample to aggressively filter out germline events at an early stage, conserving computational resources and improving the purity of the initial somatic callset [1].
- Tumor-Only Analysis: It is possible to run Mutect2 on a single tumor sample without a matched normal. However, this approach requires the use of a Panel of Normals (PoN) and a germline resource (e.g., gnomAD) to help identify and remove common technical artifacts and population germline polymorphisms, respectively [1]. The resulting callset typically requires more stringent post-calling filtration and annotation.
The choice of sequencing strategy—whole genome (WGS), whole exome (WES), or targeted gene panels—also influences input requirements and expected outcomes. Each strategy offers a different balance of breadth, depth, and cost, affecting the types of variants that can be reliably detected [14].
Table 1: Comparison of Sequencing Strategies for Somatic Variant Calling
| Strategy | Target Space | Typical Read Depth | Strengths for Somatic Calling | Key Input & Resource Considerations |
|---|---|---|---|---|
| Whole Genome (WGS) | ~3200 Mbp | 30-60x | Comprehensive SNV/Indel detection; enables CNV/SV calling [14] | Large BAM file size; requires a germline resource for optimal filtering. |
| Whole Exome (WES) | ~50 Mbp | 100-150x | Cost-effective SNV/Indel discovery in coding regions [14] | BAM files must be processed with the target interval list; a PoN is highly recommended. |
| Targeted Panel | ~0.5 Mbp | 500-1000x | Excellent for detecting low-VAF variants due to high depth [14] | High-depth BAMs; accurate target region BED file is critical for QC. |
Read Group Specifications and Metadata Integrity
Read groups are a fundamental, non-negotiable component of GATK analysis. They are sets of reads derived from a single sequencing run on a single lane of a flow cell, and their metadata tags allow the GATK engine to account for technical batch effects and sample identity during processing [17]. The GATK requires specific read group fields to be present and correctly specified in the header of input BAM files.
Table 2: Mandatory and Critical Read Group Fields for GATK Analysis
| Field Tag | Description | Importance in Somatic Workflow | Example Value |
|---|---|---|---|
ID |
Unique read group identifier. | Differentiates instrument runs for technical artifacts; used in Base Quality Score Recalibration (BQSR) [17]. | H0164.2 |
SM |
Sample name. | Critically important. All read groups with the same SM are treated as the same sample. This name populates the sample column in the final VCF and must be consistent and accurate [17]. |
NA12878 |
PL |
Sequencing platform/technology. | Informs the error model used by the GATK. Must be a standardized value (e.g., ILLUMINA, PACBIO) [17]. |
ILLUMINA |
LB |
DNA preparation library identifier. | Used by MarkDuplicates to identify reads from the same library that were sequenced on multiple lanes, allowing for accurate duplicate marking [17]. |
Solexa-272222 |
PU |
Platform Unit. | A more granular identifier than ID, often containing {FLOWCELL_BARCODE}.{LANE}.{SAMPLE_BARCODE}. Takes precedence over ID for BQSR if present [17]. |
H0164ALXX140820.2 |
To view the read group information in a BAM file, use the command: samtools view -H sample.bam | grep '^@RG' [17]. If the read group fields are missing or incorrect, Picard's AddOrReplaceReadGroups tool must be used to add or correct them prior to any GATK analysis.
Alignment Quality and Pre-processing
Analysis-ready BAM files are the product of a rigorous pre-processing pipeline. The quality of the alignment and the application of several data cleanup steps are paramount for reducing technical artifacts that can manifest as false positive variant calls.
The standard data pre-processing protocol, as outlined in the GATK Best Practices, includes [18] [14]:
- Alignment to Reference: Mapping sequencing reads from FASTQ files to a reference genome using an aligner such as BWA-MEM [14].
- Duplicate Marking: Identifying and flagging PCR duplicates—reads that originate from the same DNA fragment—using tools like Picard
MarkDuplicatesorSambamba. These reads are excluded from variant calling to prevent over-representation of individual molecules [14]. - Base Quality Score Recalibration (BQSR): A GATK-specific step that empirically adjusts the base quality scores in the sequencing data using an error model based on known variant sites. This corrects for systematic technical errors in the base quality scores, leading to more accurate variant calls [14].
Routine quality control of the final BAM files is essential before proceeding to variant calling. This involves checking key sequencing metrics to ensure sufficient coverage and data integrity [14]. Tools like Alfred can compute comprehensive multi-sample QC metrics, including insert size distribution, GC bias, sequencing error rates, and, for capture assays, on-target rate and coverage uniformity [19].
The Somatic Variant Discovery Workflow: From BAM to VCF
The GATK Best Practices workflow for somatic short variants is a multi-step process that takes the prepared BAM files and applies a series of specialized tools to generate a high-confidence somatic callset [8]. The following diagram illustrates the logical flow and the critical role of input BAM requirements at each stage.
The workflow consists of two main stages: generating candidate variants and then applying rigorous filtering [8]. The key steps are:
- Call Candidate Variants with Mutect2: Mutect2 performs local de novo assembly of haplotypes in active regions to call SNVs and indels simultaneously. It applies a Bayesian somatic likelihoods model to calculate the log odds of a variant being somatic versus a sequencing error [8] [1].
- Calculate Contamination: The
GetPileupSummariesandCalculateContaminationtools estimate the fraction of reads in the tumor sample resulting from cross-sample contamination. This is crucial for samples without a matched normal and those with significant copy number variation [8]. - Learn Orientation Bias Artifacts: Using optional output from Mutect2,
LearnReadOrientationModelcalculates prior probabilities for single-stranded substitution errors. This step is particularly critical for FFPE tumor samples, which are prone to such artifacts [8]. - Filter Variants with FilterMutectCalls: This tool applies hard filters and probabilistic models to account for correlated errors, strand bias, polymerase slippage, germline variants, and contamination. It refines the initial Mutect2 calls and automatically sets a filtering threshold to optimize the balance between sensitivity and precision [8].
- Annotate Variants with Funcotator: Finally,
Funcotatoradds functional annotations to each variant, such as the affected gene, amino acid change, and links to external databases (e.g., GENCODE, dbSNP, COSMIC). It outputs a VCF or MAF file ready for downstream biological interpretation [8].
The Scientist's Toolkit: Essential Research Reagents and Tools
A successful somatic variant calling project relies on a suite of bioinformatics tools and curated genomic resources. The following table catalogs the essential components of the analysis pipeline.
Table 3: Essential Tools and Resources for Somatic Variant Analysis
| Tool / Resource | Category | Function in Workflow |
|---|---|---|
| BWA-MEM [14] | Alignment | Aligns sequencing reads from FASTQ format to a reference genome. |
| Picard Tools [14] | Pre-processing | A collection of utilities, with MarkDuplicates and AddOrReplaceReadGroups being critical for BAM preparation. |
| GATK [8] [1] | Variant Discovery | The core toolkit. Mutect2 calls somatic variants; BQSR recalibrates base qualities; Funcotator adds annotations. |
| Samtools [14] | Utility | A versatile suite for manipulating and viewing SAM/BAM/CRAM files, and for basic variant calling (BCFtools). |
| Alfred [19] | Quality Control | Computes comprehensive, multi-sample QC metrics from BAM files, including GC bias, insert size, and coverage. |
| Panel of Normals (PoN) [8] | Resource | A VCF of artifacts common in normal samples, used by Mutect2 to filter technical false positives in tumor samples. |
| Germline Resource (e.g., gnomAD) [1] | Resource | A VCF of population allele frequencies, used to impute the probability that an allele is a common germline variant. |
| Funcotator Data Sources [8] | Resource | Configurable datasources (GENCODE, dbSNP, COSMIC, etc.) used to annotate variants with biological information. |
The path to a high-quality somatic variant callset is paved with meticulous attention to input data quality. The requirements for sample type specification, precise read group tagging, and stringent alignment quality control are not arbitrary hurdles but scientifically grounded prerequisites. By rigorously adhering to these protocols—ensuring accurate sample metadata in read groups, verifying alignment metrics, and leveraging the complete GATK somatic workflow with all recommended resources—researchers can achieve the sensitivity and specificity required for robust cancer genomic studies. This foundational rigor ensures that subsequent biological insights and clinical interpretations are based on reliable and accurate mutation data.
Somatic short mutation calling is a critical bioinformatics process designed to identify acquired genetic alterations, specifically single nucleotide variants (SNVs) and small insertions or deletions (indels), within tumor samples. This workflow is essential for characterizing the cancer genome, understanding tumor evolution, and informing personalized cancer therapy strategies. The complete process transforms raw sequencing data from tumor and matched normal samples into a refined set of high-confidence somatic variants, providing insights into clonal and sub-clonal mutations that may be present at very low allele fractions [20]. The reliability of this workflow is heavily dependent on the quality and proper preparation of input data, particularly the Binary Alignment/Map (BAM) files, which must undergo rigorous pre-processing to ensure accurate variant discovery [8] [9].
Input BAM Requirements and Pre-processing
Fundamental BAM File Specifications
The somatic short variant discovery workflow requires analysis-ready BAM files for each input tumor and normal sample. These files must be generated through a comprehensive pre-processing pipeline to correct for technical biases and ensure data suitability for variant calling [8] [9].
Table 1: Essential Pre-processing Steps for BAM File Preparation
| Processing Step | Tools Involved | Purpose | Key Output |
|---|---|---|---|
| Read Alignment | BWA-MEM, Minimap2, MergeBamAlignments | Map sequence reads to reference genome | Coordinate-sorted SAM/BAM file [9] [14] |
| Duplicate Marking | MarkDuplicatesSpark, MarkDuplicates + SortSam | Identify and tag non-independent reads from PCR amplification | BAM file with duplicates marked for exclusion [9] |
| Base Quality Score Recalibration (BQSR) | BaseRecalibrator, ApplyBQSR | Correct systematic errors in base quality scores | Recalibrated BAM file with adjusted quality scores [9] |
Critical Pre-processing Considerations
The pre-processing phase is obligatory and must precede all variant discovery efforts. Input BAMs should conform to specific requirements, including proper read group tags, which are essential for downstream analysis stages. The marking of duplicates is particularly crucial as it mitigates biases introduced by data generation steps such as PCR amplification, which can represent 5-15% of sequencing reads in a typical exome [9] [14]. While local realignment around indels was previously recommended to reduce false-positive variant calls caused by alignment artifacts, recent evaluations suggest that the improvements from this step may be marginal, potentially making it optional due to high computational costs [14].
Core Somatic Variant Discovery Workflow
Primary Variant Calling with Mutect2
The cornerstone of somatic short variant discovery is Mutect2, a Bayesian-based caller that identifies SNVs and indels simultaneously via local de novo assembly of haplotypes in active regions [8] [1]. When Mutect2 encounters regions showing evidence of somatic variation, it discards existing mapping information and completely reassembles reads in that region to generate candidate variant haplotypes. The tool then applies a Bayesian somatic likelihoods model to obtain log odds for alleles to be somatic variants versus sequencing errors [8].
Mutect2 operates in several distinct modes tailored to different experimental designs:
- Tumor-Normal Mode: Analyzes a tumor sample against a matched normal to identify somatic variants while filtering germline events [1]
- Tumor-Only Mode: Runs on a single sample, useful for creating panels of normals or when no matched normal is available [1]
- Mitochondrial Mode: Optimized parameters for calling variants on mitochondrial DNA with high sensitivity [1]
- Force-Calling Mode: Genotypes a specific set of alleles provided in a VCF file in addition to de novo discovery [1]
Contamination Assessment
Following initial variant calling, the workflow estimates cross-sample contamination using a two-step process:
- GetPileupSummaries: Collects summary information from the tumor BAM file [8]
- CalculateContamination: Emits an estimate of the fraction of reads due to cross-sample contamination for each tumor sample [8]
This step is particularly valuable as it functions effectively without a matched normal, even in samples with significant copy number variation, and makes no assumptions about the number of contaminating samples [8].
Artifact Detection and Filtering
A critical component for ensuring specificity involves detecting and filtering technical artifacts:
- LearnReadOrientationModel: Identifies orientation bias artifacts, which is especially important for FFPE tumor samples, by determining prior probabilities of single-stranded substitution errors [8]
- FilterMutectCalls: Applies sophisticated filtering to account for correlated errors through several hard filters and probabilistic models for strand bias, orientation bias, polymerase slippage artifacts, germline variants, and contamination [8]
FilterMutectCalls automatically sets a filtering threshold to optimize the F-score, representing the harmonic mean of sensitivity and precision, and uses a Bayesian model for the overall SNV and indel mutation rate and allele fraction spectrum of the tumor to refine the initial log odds generated by Mutect2 [8].
Functional Annotation
The final analytical step involves annotating discovered variants with biological context using tools such as Funcotator [8]. This functional annotation tool:
- Labels each variant with one of twenty-three distinct variant classifications
- Provides gene information (affected gene, predicted variant amino acid sequence)
- Associates variants with information from datasources including GENCODE, dbSNP, gnomAD, and COSMIC
- Produces either a VCF file (with annotations in the INFO field) or a Mutation Annotation Format (MAF) file [8]
Diagram 1: Complete GATK Somatic Short Variant Discovery Workflow illustrating the sequential steps from input BAM processing through final variant annotation, with color-coded functional groups.
Experimental Design Considerations
Sequencing Strategies and Performance
The choice of sequencing strategy significantly impacts variant calling performance, with different approaches offering distinct advantages for somatic mutation detection.
Table 2: Sequencing Strategy Comparison for Somatic Variant Detection
| Strategy | Target Space | Typical Depth | SNV/Indel Detection | Low VAF Sensitivity |
|---|---|---|---|---|
| Gene Panel | ~0.5 Mbp | 500-1000× | ++ | ++ |
| Whole Exome | ~50 Mbp | 100-150× | ++ | + |
| Whole Genome | ~3200 Mbp | 30-60× | ++ | + |
Performance indicators: + (good), ++ (outstanding) [14]
Computational Requirements
Implementation of the somatic variant calling workflow requires substantial computational resources, particularly for whole-genome sequencing data. The EPI2ME workflow recommends minimum requirements of 16 CPUs and 48GB RAM, with optimal performance achieved using 64 CPUs and 256GB RAM [21]. Approximate run time varies significantly depending on sequencing modality and coverage, with a complete analysis of a 60X/30X Tumor/Normal pair taking approximately 6.5 hours using recommended resources [21].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Analytical Tools and Resources for Somatic Variant Calling
| Tool/Resource | Category | Function | Application Context |
|---|---|---|---|
| Mutect2 | Variant Caller | Core somatic SNV/indel detection via local assembly | Primary variant discovery [8] [1] |
| Panel of Normals (PoN) | Reference Resource | Identifies recurrent technical artifacts | False positive reduction [8] [1] |
| Germline Resource | Reference Resource | Distinguishes somatic from germline variants | Population allele frequency context [1] |
| Funcotator | Annotation Tool | Adds biological context to variants | Functional interpretation [8] |
| SComatic | Specialized Caller | Detects somatic mutations in single-cell data | scRNA-seq/scATAC-seq analysis [22] |
Validation and Benchmarking Approaches
Rigorous validation is essential for verifying somatic variant calls. Recommended approaches include:
- Down-sampling: Uses subsets of reads from primary sequencing data of validated somatic mutations to measure sensitivity at different coverage depths [20]
- Virtual Tumors: Creates synthetic datasets by mixing reads from different samples to generate truth sets with known mutations for comprehensive sensitivity and specificity assessment [20]
- Orthogonal Validation: Employs independent sequencing technologies or experimental methods to confirm putative somatic variants
The Genome in a Bottle (GIAB) and Platinum Genome datasets provide benchmark resources with established "ground truth" variant calls for systematic pipeline validation [14]. These resources define "high-confidence" regions of the human genome where variant calls can be reliably benchmarked, though researchers should note that these resources may have inherent biases as they were constructed using many of the same technologies being evaluated [14].
Advanced Applications and Emerging Methodologies
The field of somatic variant detection continues to evolve with several advanced applications:
- Single-Cell Mutation Detection: Tools like SComatic enable detection of somatic mutations in single-cell RNA-seq and ATAC-seq data without requiring matched DNA sequencing, facilitating the study of clonal heterogeneity at single-cell resolution [22]
- Liquid Tumor Analysis: Specialized parameters for detecting somatic variants in circulating tumor DNA, which typically exhibits very low variant allele fractions [21]
- Multi-sample Analysis: Joint calling of multiple tumor and normal samples from the same individual to improve detection power [1]
These advanced applications demonstrate the expanding utility of somatic variant calling workflows across diverse research contexts, from basic cancer genomics to clinical translational studies.
Practical Guide: Configuring Mutect2 BAM Inputs for Different Analysis Modes
This technical guide details the precise requirements for specifying input BAM files and correctly labeling normal samples within the GATK Mutect2 somatic variant calling workflow. Proper configuration of these inputs is fundamental to generating accurate somatic mutation calls in cancer genomics research. The methodologies presented herein form the foundation for reliable detection of single nucleotide variants (SNVs) and insertion-deletion variants (indels) in tumor research samples, enabling robust downstream analysis for therapeutic development. This guide synthesizes best practices from established somatic variant discovery pipelines, providing researchers with explicit technical specifications for data preparation and analysis configuration.
In somatic variant analysis using GATK's Mutect2, properly formatted and organized input files are prerequisite for accurate mutation detection. The fundamental input requirements encompass aligned sequence data in BAM format, reference genome sequences, and appropriate sample identification metadata. The alignment files must derive from previously processed BAM files that have undergone standard preprocessing procedures including duplicate marking, base quality score recalibration, and indel realignment according to GATK Best Practices [8]. Each BAM file must contain read groups with sample-specific information that enables Mutect2 to correctly identify and process tumor and normal components separately.
The basic command structure for initiating Mutect2 analysis requires several core components: reference genome sequence (-R), input BAM files (-I), and designated sample names for tumor and normal components. Additional resources such as germline variant databases and panels of normals significantly enhance variant calling accuracy but represent optional components in the workflow [2]. The input BAM specifications must maintain consistency throughout the analysis pipeline, as subsequent tools in the somatic variant discovery workflow, including FilterMutectCalls, depend on the initial configuration and output formats generated by Mutect2.
BAM File Specification Methodology
Input Specification Syntax
The Mutect2 tool accepts BAM, SAM, or CRAM files containing aligned sequencing reads through the --input or -I argument [2]. Multiple input files can be specified by repeating the -I argument followed by the file path. This capability enables processing of multiple BAM files simultaneously, which is particularly useful when handling separately sequenced tumor regions or technical replicates.
Table 1: Required Input Files for Mutect2 Analysis
| File Type | Description | Format Specification |
|---|---|---|
| Reference Genome | Reference sequence file | FASTA format with associated index and dictionary files |
| Tumor BAM | Aligned sequencing reads from tumor sample | BAM/SAM/CRAM format with index |
| Normal BAM | Aligned sequencing reads from matched normal | BAM/SAM/CRAM format with index |
| Germline Resource | Population allele frequencies | VCF format compressed and indexed |
| Panel of Normals | Technical artifacts database | VCF format compressed and indexed |
Technical Specifications for Alignment Files
Input BAM files must comply with specific technical standards to ensure compatibility with the Mutect2 processing engine. The files should be coordinate-sorted, with accompanying index files (.bai) present in the same directory [23]. The sequence dictionary within the BAM files must be compatible with the reference genome sequence dictionary to prevent validation errors. For whole-genome sequencing data, the average coverage depth should be documented, with recommended minimums of 30x for normal samples and 60x for tumor samples to ensure sufficient sensitivity for variant detection [24]. For tumor samples with significant heterogeneity, higher coverage depths (≥100x) may be necessary to detect subclonal populations.
The Mutect2 tool incorporates advanced processing capabilities for handling varying coverage depths, though the documentation notes that further parameter optimization may be required for extreme high-depth scenarios exceeding 1000X coverage [2]. The alignment files should ideally be generated using PCR-free library preparation methods to minimize technical artifacts, though the pipeline includes filtering mechanisms for common sequencing- and amplification-derived errors.
Normal Sample Labeling Protocols
Sample Identification in BAM Headers
Each input BAM file must contain appropriate read groups with sample identifiers that correspond to the labels specified in the Mutect2 command. The sample identifiers within the BAM files' read groups (@RG SM tags) should match the sample names provided to Mutect2 via the -tumor and -normal arguments. Consistency in sample labeling ensures proper sample tracking throughout the variant calling process and prevents sample mix-ups that could compromise analysis integrity.
The standard approach for designating sample types within the Mutect2 command involves explicitly linking each input BAM file to its biological context using the -I argument followed by either -tumor or -normal with the appropriate sample name [2]. This explicit designation enables Mutect2 to apply appropriate statistical models for somatic versus germline variation.
Normal Sample Labeling Syntax
The correct specification of normal samples follows a precise sequence of arguments that explicitly links the input BAM file to its designation as a normal control. The standard syntax for matched tumor-normal analysis is demonstrated below:
In this implementation, the -I argument introduces each BAM file, immediately followed by either -tumor or -normal to designate the biological role of the subsequent sample [2]. The sample names provided to these arguments must correspond to the sample identifiers embedded in the BAM file's read groups. This explicit linkage ensures Mutect2 correctly associates each BAM file with its sample type, enabling appropriate application of the somatic likelihoods model that distinguishes somatic mutations from germline variants.
Table 2: Mutect2 Arguments for Sample Specification
| Argument | Function | Required | Example |
|---|---|---|---|
-I |
Input BAM file | Yes | -I tumor.bam |
-tumor |
Tumor sample name | Conditional | -tumor TUMOR_001 |
-normal |
Normal sample name | Conditional | -normal NORMAL_001 |
-R |
Reference genome | Yes | -R reference.fa |
-O |
Output VCF | Yes | -O results.vcf.gz |
Tumor-Only Analysis Mode
For studies lacking matched normal samples, Mutect2 supports a tumor-only operating mode with modified labeling requirements. In this configuration, only the tumor BAM is specified with its corresponding sample name, without any normal sample designation [2]. The command structure simplifies to:
When operating in tumor-only mode, researchers should note that the resulting variant calls will contain both somatic and germline variants, requiring additional filtering strategies to distinguish true somatic mutations. The GATK Best Practices recommend creating a panel of normals (PoN) to identify technical artifacts common to the sequencing platform and methodology [12]. This PoN can be generated by running Mutect2 in tumor-only mode on multiple normal samples and then combining the results using CreateSomaticPanelOfNormals.
Integrated Analysis Workflow
Complete Mutect2 Somatic Variant Calling Pipeline
The full somatic variant discovery workflow extends beyond initial variant calling with Mutect2 to include contamination assessment, artifact identification, and comprehensive variant filtering. The complete pipeline incorporates multiple analytical stages that build upon the properly specified BAM inputs and sample labels [8].
The following diagram illustrates the relationship between input BAM files and the key processing stages in the complete somatic variant discovery workflow:
Somatic Variant Calling Workflow
Contamination Estimation Procedures
A critical quality control step in tumor-normal analysis involves estimating cross-sample contamination, which can significantly impact somatic variant calling accuracy [25]. Even low levels of contamination (as low as 0.1%) can introduce false positive somatic calls when germline variants from a contaminating sample are misinterpreted as tumor-specific mutations [25]. The GATK pipeline incorporates specialized tools for contamination assessment:
The contamination estimates generated by this process are subsequently incorporated into the FilterMutectCalls step to improve filtering accuracy [12]. This contamination assessment methodology is specifically designed to perform robustly even in tumor samples with significant copy number alterations, which can confound traditional contamination estimation approaches [25].
Artifact Detection and Filtering Methodologies
Technical artifacts arising from sequencing processes or sample preparation represent significant challenges in somatic variant calling. The Mutect2 pipeline incorporates specialized methods for identifying and filtering these artifacts:
Orientation Bias Artifacts: For samples susceptible to substitution errors occurring on a single strand before sequencing (particularly common in FFPE tumor samples and NovaSeq sequencing data), Mutect2 includes a specialized read orientation filter [12]. The implementation involves three steps:
Panel of Normals Creation: A panel of normals (PoN) captures technical artifacts common to a specific sequencing pipeline and is created by analyzing multiple normal samples:
This panel of normals is then provided to Mutect2 in subsequent analyses through the --panel-of-normals argument to filter recurrent technical artifacts [12].
Core Analysis Toolkit
The experimental workflow for somatic variant discovery requires specific computational reagents and resources that ensure analytical accuracy and reproducibility. The following table details essential components for implementing the tumor-normal matched pair analysis:
Table 3: Essential Research Reagents and Computational Resources
| Resource | Function | Specifications |
|---|---|---|
| Reference Genome | Genomic coordinate system | FASTA format with index (.fai) and dictionary (.dict) |
| Germline Resource | Population allele frequencies | gnomAD in VCF format, compressed and indexed |
| Panel of Normals | Technical artifact database | VCF from aggregated normal samples |
| Known Variants Sites | Contamination estimation | Common population variants in VCF format |
| Intervals File | Targeted region specification | Interval_list or BED format |
| Funcotator Data Sources | Variant annotation | GENCODE, dbSNP, COSMIC, gnomAD |
The germline resource, typically derived from the Genome Aggregation Database (gnomAD), provides population allele frequencies that enable Mutect2 to distinguish potential germline polymorphisms from true somatic mutations [2]. The absence of an allele from this resource provides evidence against germline status, with the --af-of-alleles-not-in-resource parameter determining the assumed allele frequency for such variants [2]. The recommended value of 0.00003125 corresponds to approximately one occurrence in 32,000 alleles, reflecting gnomAD's sample size [2].
Discussion and Technical Considerations
Analytical Validation Framework
The accuracy of somatic variant calling depends heavily on proper BAM specification and sample labeling, with errors in these foundational steps propagating through subsequent analysis stages. Validation of these inputs should include verification of BAM integrity, confirmation of sample labels in read groups, and assessment of sequencing metrics including coverage uniformity, duplication rates, and alignment quality [26]. The Mutect2 tool incorporates multiple prefiltering steps that leverage the normal sample designation to efficiently exclude clearly germline variants early in the processing pipeline, conserving computational resources while maintaining sensitivity for borderline cases [2].
For tumor samples with suspected normal tissue contamination, the normal sample designation enables Mutect2 to calculate a normal artifact log odds score, which FilterMutectCalls uses to apply artifact-in-normal filters [2]. In cases where the matched normal sample itself contains tumor contamination, researchers may need to adjust the normal-artifact-lod threshold to maintain detection sensitivity.
Advanced Configuration Scenarios
In complex research scenarios involving multiple tumor regions or unusual sample relationships, the basic BAM specification framework requires modification. For multi-region tumor sequencing, researchers can specify multiple tumor BAM files using repeated -I arguments, each followed by -tumor with unique sample identifiers. Similarly, studies incorporating multiple control samples (e.g., adjacent normal and germline blood) can specify multiple normal BAM files with appropriate labeling.
The computational implementation of Mutect2 supports parallelization through scattering across genomic intervals, which requires special handling of input BAM specifications and output file merging [12]. When scattering Mutect2 execution, each parallel instance must receive the same complete set of input BAM files, with interval restrictions applied through the -L argument. The resulting variant calls and stats files from all intervals must then be merged before proceeding to filtering steps.
Proper specification of BAM files and accurate labeling of normal samples constitute fundamental requirements for robust somatic variant discovery using GATK Mutect2. The methodologies detailed in this guide provide researchers with explicit protocols for configuring these inputs within the broader context of somatic mutation analysis pipelines. Adherence to these specifications ensures optimal performance of Mutect2's statistical models for distinguishing somatic mutations from germline variants and technical artifacts, ultimately generating reliable variant calls for cancer genomics research and therapeutic development.
Within the context of somatic variant discovery for cancer research, the absence of a matched normal sample presents a significant methodological challenge. Tumor-only analysis, while often necessary in clinical and real-world research scenarios, is inherently susceptible to the erroneous classification of germline variants and technical artifacts as somatic mutations [27]. This in-depth technical guide outlines the specific input data requirements and sophisticated mitigation strategies essential for implementing the GATK Mutect2 tumor-only calling mode effectively, ensuring the production of a reliable somatic variant callset for downstream analysis in drug development and basic research.
Input BAM Requirements and Preprocessing
The foundation of accurate somatic variant calling lies in the quality and preparation of the input sequencing data. The following specifications are critical for the tumor BAM file, which serves as the sole source of variant information in this mode.
Data Preprocessing Workflow
The input BAM file must be generated through a rigorous pre-processing pipeline as recommended by the GATK Best Practices [8] [28]. This workflow ensures that the data is free from technical artifacts introduced during sequencing and library preparation, which could otherwise be misinterpreted as somatic variants.
Figure 1. The essential data preprocessing workflow for generating an analysis-ready BAM file suitable for somatic variant discovery with Mutect2.
Technical Specifications for Input BAM
Adherence to the following technical requirements is non-negotiable for the successful execution of the Mutect2 pipeline [28]:
- Format: The file must be in BAM format, sorted by coordinate, and indexed (with a corresponding
.baifile). - Read Groups: The BAM must contain read group (
@RG) tags, including the sample identifier (SM). All read groups must belong to a single sample. - Alignment: Reads must be aligned to a reference genome using an aligner such as BWA.
- Data Integrity: The file must pass validation checks by tools like
ValidateSamFile.
The Mutect2 Tumor-Only Calling Workflow
The core process of somatic variant discovery in tumor-only mode involves calling candidate variants followed by a multi-layered filtering strategy to remove false positives. The workflow is designed to maximize the signal-to-noise ratio in the absence of a matched normal control.
Core Calling and Filtering Protocol
The fundamental command to run Mutect2 in tumor-only mode is as follows [1]:
This step produces a file (unfiltered.vcf) containing a large set of candidate somatic variants. The subsequent, and crucial, step is to filter this raw callset using FilterMutectCalls [8] [12]:
As of GATK 4.1.1.0, FilterMutectCalls requires the reference genome and the stats file generated by Mutect2. It employs a unified filtering model that automatically determines an optimal probability threshold to distinguish true somatic variants from errors, optimizing the F-score [12].
Comprehensive Artifact Mitigation Strategies
The accuracy of tumor-only calling is critically dependent on strategies to mitigate three primary sources of false positives: common germline variants, technical artifacts, and sequencing-specific biases.
Germline Variant Filtering
The most substantial challenge in tumor-only analysis is distinguishing true somatic mutations from the much larger number of germline polymorphisms present in an individual.
Table 1: Critical External Resources for Germline Filtering
| Resource | Description | Function in Tumor-Only Calling |
|---|---|---|
| Germline Resource(e.g., gnomAD) | A population database of allele frequencies [1]. | Provides prior probabilities for variants being common germline polymorphisms. Variants absent from the resource are assigned a prior defined by --af-of-alleles-not-in-resource (default: 5e-8 for tumor-only) [1]. |
| Panel of Normals (PoN) | A VCF of artifacts discovered across a set of normal samples [1] [12]. | Identifies and filters site-specific technical artifacts that recur across sequencing runs. |
Technical Artifact Filtering
Technical artifacts arising from sample preparation and sequencing chemistry must be systematically addressed.
Sequencing Contamination: Cross-sample contamination is estimated using a two-step process, which is particularly robust in tumor samples with copy-number variation [8]:
GetPileupSummaries: Summarizes read support at common variant sites.CalculateContamination: Uses the output to estimate contamination fractions.
Read Orientation Artifacts: Samples such as FFPE tumors or those sequenced on Illumina Novaseq machines are prone to oxidation-induced artifacts that manifest as false positive calls supported only by reads in a single orientation. A dedicated three-step workflow mitigates this [12]:
- Data Collection: Run
Mutect2with the--f1r2-tar-gzargument to gather strand-specific count data. - Model Learning: Run
LearnReadOrientationModelon the F1R2 output to generate an artifact prior table. - Application: Pass the model to
FilterMutectCallsusing the--ob-priorsargument.
Table 2: Key Artifact Mitigation Tools and Their Functions
| Tool | Primary Function | Artifact Type Addressed |
|---|---|---|
| CalculateContamination | Estimates cross-sample contamination | Sequencing Contamination |
| LearnReadOrientationModel | Models pre-sequencing single-stranded errors | Read Orientation Artifacts |
| FilterMutectCalls | Applies probabilistic models for strand bias, slippage, etc. | Multiple Technical Artifacts |
The interplay of these tools and resources within the complete workflow is illustrated below.
Figure 2. The complete GATK Mutect2 tumor-only workflow, integrating the core variant caller with artifact mitigation tools. Solid lines indicate primary data flow, while dashed lines indicate the flow of prior information used for filtering.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of the tumor-only calling workflow requires a curated set of bioinformatics "reagents." The following table details these essential components.
Table 3: Essential Research Reagents for Mutect2 Tumor-Only Analysis
| Reagent | Critical Function | Technical Specification |
|---|---|---|
| Reference Genome | Baseline for read alignment and variant calling. | FASTA file and associated index (*.fai) and dictionary (*.dict). Must be the same version used for BAM alignment [12]. |
| Germline Resource | Provides prior probabilities for filtering common germline variants. | A VCF (e.g., af-only-gnomad.vcf.gz) containing population allele frequencies (AF). Must be compressed and indexed [1]. |
| Panel of Normals (PoN) | Identifies recurrent technical artifacts. | A VCF generated by CreateSomaticPanelOfNormals from a set of normal samples processed through Mutect2 in tumor-only mode [12]. |
| Known Sites for BQSR | Enables base quality score recalibration. | A VCF of known polymorphic sites (e.g., dbSNP) used during preprocessing to correct for systematic errors in base quality scores [28]. |
Advanced Considerations and Best Practices
Addressing Racial Bias in Tumor-Only Calling
A significant analytical challenge in tumor-only calling is the inherent bias introduced by germline resources. Populations underrepresented in these databases, such as individuals of African, Asian, or Indigenous ancestry, suffer from higher false-positive rates because their rare germline variants are mistaken for somatic mutations [29]. This leads to inflated and racially biased tumor mutational burden (TMB) estimates. Emerging machine learning classifiers, such as those based on XGBoost and LightGBM, show promise in mitigating this bias by using features like trinucleotide context and local copy number to improve somatic versus germline classification [29].
Alternative Methodologies
The field of somatic variant calling is evolving rapidly. Newer methods like ClairS-TO demonstrate the potential of deep learning for tumor-only calling. ClairS-TO uses an ensemble of two neural networks—an "affirmative network" to determine how likely a candidate is somatic and a "negational network" to determine how likely it is not somatic—to achieve high accuracy on both long-read and short-read data [27]. Furthermore, ultra-sensitive error-corrected sequencing technologies like NanoSeq are pushing detection limits, enabling the study of very low allele fraction mutations in polyclonal samples, which is beyond the standard capability of Mutect2 [30].
Executing a robust tumor-only somatic variant analysis with GATK Mutect2 is a multi-faceted process that extends far beyond a simple command-line invocation. It demands stringent input data quality, the strategic integration of external germline and artifact databases, and the diligent application of specialized filtering workflows to account for contamination and sequencing artifacts. By adhering to the requirements and mitigation strategies outlined in this guide, researchers can generate more reliable somatic callsets, thereby laying a solid foundation for impactful cancer research and drug development, even in the absence of a matched normal sample.
Mitochondrial DNA (mtDNA) presents unique challenges for somatic variant discovery that necessitate specialized processing protocols. Unlike the nuclear genome, mtDNA exists in hundreds to thousands of copies per cell, creating exceptionally high sequencing depths that standard variant calling parameters cannot efficiently handle [31]. This high copy number, coupled with mtDNA's distinct molecular characteristics, requires specific adjustments to the GATK Mutect2 workflow to ensure accurate detection of somatic mutations while managing computational resources effectively.
The necessity for these specialized approaches is underscored by the critical biological roles of mtDNA in carcinogenesis. mtDNA encodes 13 proteins essential for the electron transport chain, and its dysfunction has been linked to various cancers, including hepatocellular carcinoma and esophageal squamous cell carcinoma [32] [33]. Accurate characterization of mtDNA somatic variants provides crucial insights into tumor metabolism and progression, making optimized bioinformatic protocols essential for comprehensive cancer genomics.
Core Technical Specifications for Mitochondrial Mode
Mutect2 Mitochondrial Mode Configuration
The GATK Mutect2 tool provides a dedicated mitochondrial mode activated with the --mitochondria flag, which automatically configures optimal parameters for mtDNA variant calling [1]. This mode specifically adapts to the high-depth nature of mitochondrial sequencing data by adjusting key statistical thresholds and processing parameters.
Table 1: Key Parameter Adjustments in Mutect2 Mitochondrial Mode
| Parameter | Standard Mode Default | Mitochondrial Mode Setting | Rationale |
|---|---|---|---|
--initial-tumor-lod |
Not specified | 0 | Increases sensitivity for low-frequency variants |
--tumor-lod-to-emit |
Not specified | 0 | Ensures emission of low-LOD variants |
--af-of-alleles-not-in-resource |
0.001 (tumor-normal) | 0.004 | Accounts for higher mtDNA population diversity |
--pruning-lod-threshold |
Not specified | -4 | Reduces stringency for high-depth data |
The mitochondrial mode requires only a single sample, which can be provided as multiple BAM files, and is targeted to the mitochondrial chromosome using -L chrM [1]. The automatic adjustment of --af-of-alleles-not-in-resource to 0.004 is particularly important as it properly calibrates the prior probability for alleles absent from germline resources, reflecting the higher natural polymorphism rate in mitochondrial genomes compared to nuclear DNA.
BAM Preparation Requirements for Mitochondrial Analysis
Proper BAM preparation is fundamental to successful mitochondrial variant calling. The following considerations are essential:
Depth and Coverage: Mitochondrial sequencing typically achieves extreme depths (e.g., >20,000X) that standard nuclear DNA protocols cannot efficiently handle [1]. The BAM files must maintain base quality and mapping quality metrics suitable for distinguishing true somatic variants from sequencing artifacts at these depths.
Probe Balancing for Targeted Sequencing: When using target-panel sequencing that combines mtDNA and nuclear DNA (ncDNA) capture, the natural abundance difference between mtDNA and ncDNA must be addressed through appropriate probe pooling ratios. Experimental data indicates that an mtDNA:ncDNA probe ratio of 1:10 provides optimal balance between sequencing performance and cost-effectiveness [32].
Duplicate Marking: PCR duplicates present a significant challenge in mtDNA analysis due to the small, conserved genome. Standard duplicate marking approaches should be applied, though careful interpretation is needed as identical mtDNA molecules may represent biological replicates rather than technical artifacts.
Experimental Protocols for Mitochondrial Variant Discovery
Ultra-Sensitive Duplex Sequencing for Mitochondrial Mutations
For researchers requiring maximum sensitivity for low-frequency mitochondrial variants, duplex sequencing provides an exceptionally low error rate (~2×10⁻⁸) that enables detection of somatic mutations at frequencies as low as 4×10⁻⁶ [31]. This protocol involves:
Library Preparation and Sequencing:
- Molecular Tagging: Tag double-stranded mtDNA sequences with unique molecular identifiers (UMIs) during library preparation
- Consensus Building: Computational construction of duplex consensus sequences from tagged reads
- Artifact Mitigation: Trim 10bp from duplex reads to exclude potential erroneous calls from DNA damage during preparation
- Variant Calling: Map consensus reads to the mitochondrial genome (e.g., mm10 for mouse, hg38 for human) using a specialized duplex sequencing processing pipeline
Experimental Design Considerations:
- Sample Requirements: Protocol validated using ~2.5 million duplex mt-genomes across tissues with median of 74,764 duplex mt-genomes per condition
- Mutation Filtering: Remove mutations overlapping known haplotype sites and filter based on strand balance and quality metrics
- Validation: Compare mutational spectra for atypical imbalances of complementary mutation types against previously published datasets
Target-Panel Sequencing with Optimized Probe Ratios
For studies combining mitochondrial and nuclear DNA capture, the following protocol ensures balanced representation:
Probe Pooling and Enrichment:
- Probe Mixture Preparation: Create target enrichment probe mixtures with varying mtDNA:ncDNA probe ratios (1:5, 1:10, 1:20, 1:50, 1:100) by volume [32]
- Library Hybridization: Pool normalized libraries with different indices into a single pool of 1.5μg total prior to enrichment with probe mixtures
- Quality Control: Validate enriched libraries by Agilent Bioanalyzer, Qubit, and qPCR before sequencing
- Sequencing and Analysis: Perform paired-end sequencing (e.g., 151bp on Illumina NovaSeq 6000), followed by quality control (FASTQC), adapter trimming (Trimmomatic), and alignment to reference genome (BWA)
Performance Optimization:
- Ratio Selection: Base final ratio selection on read count, average sequencing depth, and coverage of targeted regions
- Copy Ratio Calculation: Estimate mtDNA:ncDNA copy ratio by dividing the average sequencing depth of the mito panel by that of the nuclear panel, multiplied by the corresponding dilution factor
Implementation Guide: Mutect2 Mitochondrial Variant Calling
Basic Mitochondrial Analysis Workflow
The fundamental command structure for mitochondrial variant discovery with Mutect2 is straightforward:
This command activates the predefined mitochondrial parameters that optimize performance for high-depth mtDNA data [1]. The workflow generates both a VCF file containing candidate somatic variants and a stats file required for subsequent filtering steps.
Advanced Configuration for Challenging Samples
For samples with potential orientation artifacts (such as FFPE specimens), additional steps are necessary:
This expanded workflow addresses sequencing artifacts that preferentially affect one DNA strand, significantly improving variant calling accuracy in degraded samples [12].
Essential Research Reagents and Computational Tools
Table 2: Research Reagent Solutions for Mitochondrial Genomics
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| KAPA HyperExplore Nuclear Panel | Target enrichment of nuclear genes | 947 genes; used with dilution series for probe balancing [32] |
| KAPA HyperChoice Mito Panel | Target enrichment of mitochondrial genome | Requires optimization of pooling ratios with nuclear panel [32] |
| Cobas cfDNA Preparation Kit | Cell-free DNA extraction from plasma | Enables liquid biopsy mtDNA analysis from 1.5-2mL plasma [32] |
| GATK Mutect2 --mitochondria mode | Somatic variant calling | Automatically sets parameters for mtDNA; requires BAMs aligned to chrM [1] |
| dMTLV | Low-heteroplasmy mtDNA variant detection | Likelihood-based method for variants with low VAF; complements Mutect2 [33] |
| fNUMT | Nuclear mitochondrial sequence detection | Identifies NUMTs and derived false positives that confound mtDNA variant calling [33] |
Workflow Visualization
Figure 1: Mitochondrial variant calling workflow with key specialized steps, highlighting the integration of mitochondrial-specific parameters into the core assembly and genotyping process.
Figure 2: Comprehensive experimental workflow for mitochondrial target sequencing, emphasizing the critical probe optimization step required for balanced mitochondrial and nuclear genome representation.
Effective mitochondrial somatic variant calling requires specialized approaches that address the unique characteristics of mtDNA, particularly its high copy number and extreme sequencing depths. The GATK Mutect2 mitochondrial mode provides a robust foundation for this analysis through automatically configured parameters that optimize sensitivity and specificity for mtDNA variants. Successful implementation depends on proper BAM preparation, including appropriate probe balancing in targeted sequencing approaches and utilization of ultra-sensitive methods like duplex sequencing for low-frequency variant detection. By adhering to these specialized protocols, researchers can reliably characterize mitochondrial somatic mutations that contribute to carcinogenesis and tumor progression.
Multi-sample joint calling in somatic variant discovery refers to the simultaneous analysis of multiple tumor and normal BAM files from the same patient in a single execution of the variant caller. This advanced methodology represents a significant evolution from traditional single-sample analysis, offering enhanced detection capabilities for low-frequency variants that might otherwise be missed. The GATK Mutect2 tool provides sophisticated support for this approach, enabling researchers to process multiple tumor samples against a pooled normal representation from the same individual [34]. This technical framework is particularly valuable for studying tumor heterogeneity, tracking clonal evolution, and identifying subclonal populations that exist at low variant allele frequencies across multiple samples from the same patient.
The fundamental principle underlying joint calling involves leveraging the combined statistical power of multiple samples to distinguish true biological variants from sequencing artifacts. When samples are analyzed collectively, the evidence for a true variant is aggregated across all samples, making it possible to identify mutations that appear at low frequencies in multiple samples but might not meet the significance threshold when any single sample is analyzed independently [35]. This approach is especially powerful for cancer genomics research where tumors often exhibit substantial heterogeneity and where multiple samples may be available from different time points (longitudinal sampling) or different anatomical locations (spatial sampling).
Within the GATK Best Practices framework, somatic variant discovery follows a structured workflow that begins with data pre-processing and culminates in the identification of somatic mutations [18]. Multi-sample joint calling represents an advanced implementation of this workflow, specifically designed for complex sampling scenarios. It is crucial to distinguish this approach from germline joint calling, as the biological questions and technical considerations differ substantially between these domains. For somatic analyses, the primary focus is on identifying mutations that are present in tumor samples but absent from matched normal tissues, with special consideration for tumor-specific phenomena such as heterogeneity, aneuploidy, and contamination.
Mutect2 Multi-sample Calling Methodology
Technical Implementation
The Mutect2 multi-sample calling implementation employs a sophisticated computational architecture that processes multiple tumor and normal samples in an integrated manner. When executing Mutect2 in multi-sample mode, the computational engine performs local assembly of haplotypes across all input samples simultaneously, creating a comprehensive representation of the genomic variation present in the patient's samples [34]. This assembly-based approach is crucial for accurately resolving complex genomic regions and for detecting variants that might be partially represented in multiple samples.
In this mode, all normal samples from the same patient are pooled together in-memory to create a unified normal representation [35]. This pooling occurs computationally rather than requiring physical file merging, preserving sample-level information while creating a more comprehensive normal baseline. The tool uses this pooled normal to identify and filter out germline variants more effectively than would be possible with single normal samples. For the tumor samples, Mutect2 performs joint analysis that combines evidence across all samples while maintaining sample-specific information in the final output. This enables the detection of variants that are shared across multiple tumor samples as well as those that are private to individual samples.
A key advantage of this implementation is its statistical framework for distinguishing true somatic variants from sequencing errors. By analyzing multiple samples jointly, Mutect2 can identify variants that exhibit consistent characteristics across samples while rejecting artifacts that appear random or inconsistent. The Bayesian genotyping model employed by Mutect2 is particularly enhanced in multi-sample mode, as it can leverage cross-sample information to improve genotype likelihood calculations and variant quality assessments [34]. This results in improved sensitivity for low-frequency variants without compromising specificity.
Command Line Implementation
Implementing Mutect2 multi-sample calling requires specific command line structure and parameters. The basic syntax involves specifying all tumor and normal BAM files as inputs, with explicit identification of normal samples:
In this command structure, all BAM files (both tumor and normal) are specified with multiple -I flags. Each normal sample is explicitly identified with the -normal parameter, which requires the sample name as it appears in the BAM file's read group (RG) SM field [34]. The normal samples are automatically pooled into a composite normal for variant filtering. The output VCF contains genotype information for each tumor sample, allowing researchers to identify which variants are present in which tumor samples.
It is important to note that as of GATK version 4.1, the -tumor parameter is no longer required when specifying tumor samples [34]. The presence of samples not designated as -normal is automatically interpreted as tumor samples. This simplifies the command line structure for multi-sample calling. The tool generates a comprehensive output VCF that includes sample-level genotypes, enabling analysis of variant distribution across the multiple tumor samples from the same patient.
Key Workflow Diagram
The following diagram illustrates the logical workflow and data relationships in Mutect2 multi-sample analysis:
This workflow demonstrates how multiple normal samples from a single patient are pooled to create a comprehensive normal baseline, which is then used jointly with multiple tumor samples for somatic variant calling. The output is a unified VCF file that contains genotype information for all tumor samples, enabling researchers to analyze the distribution of somatic variants across different tumor samples from the same patient.
Experimental Design and Best Practices
Appropriate Use Cases
Multi-sample joint calling is specifically designed for scenarios where multiple samples are available from the same patient, creating opportunities for enhanced variant detection that would not be possible with separate analyses. The most appropriate use cases include longitudinal studies where tumor samples are collected at different time points, such as before treatment, during treatment, and at progression [35]. This temporal sampling allows researchers to track the evolution of tumor clones and identify emerging resistance mutations. Multi-sample analysis significantly enhances the detection of variants that are present at low frequencies in multiple time points, providing a more comprehensive picture of tumor dynamics.
Spatial heterogeneity studies represent another prime application, where multiple tumor samples are collected from different geographical regions of the same tumor or from different metastatic sites [35]. These samples often contain shared clonal mutations alongside region-specific variants, and multi-sample calling improves the detection of both categories. By analyzing these samples jointly, researchers can reconstruct the phylogenetic relationships between different tumor regions and identify mutations associated with specific metastatic niches.
Multi-sample analysis also provides substantial benefits for cases with technical challenges such as low tumor purity or low sequencing depth in individual samples. When individual tumor samples have limited quality or quantity, joint analysis can rescue variants that would otherwise be missed by combining statistical evidence across samples [35]. This is particularly valuable for precious clinical samples where re-sequencing may not be possible. Additionally, this approach is ideal for studying tumor subclones that are distributed across multiple samples but do not reach detectable levels in any single sample alone.
Comparative Analysis: Single-sample vs. Multi-sample Approaches
The choice between single-sample and multi-sample calling strategies has significant implications for variant detection performance and computational requirements. The following table summarizes the key differences and considerations:
Table 1: Comparison of single-sample and multi-sample calling approaches
| Aspect | Single-sample Calling | Multi-sample Joint Calling |
|---|---|---|
| Variant Detection Sensitivity | Limited for low-frequency variants (<5-10% VAF) | Enhanced sensitivity for low-frequency variants present across multiple samples |
| Statistical Power | Limited to evidence from single sample | Aggregates evidence across multiple samples, improving power for variant detection |
| Handling of Tumor Heterogeneity | Identifies variants present in individual samples | Enables reconstruction of clonal architecture across multiple samples |
| Computational Requirements | Lower per-analysis, but cumulative overhead for multiple samples | Higher memory and processing requirements, but more efficient for multiple samples |
| Implementation Complexity | Straightforward implementation and interpretation | More complex setup and result interpretation |
| Optimal Use Cases | Single tumor-normal pairs; samples from different patients | Multiple tumors from same patient; longitudinal studies |
The enhanced sensitivity of multi-sample calling is particularly evident for variants with low allele frequencies that are shared across multiple samples. In single-sample mode, a variant appearing at 5% allele frequency in multiple tumors might not reach statistical significance in any individual sample due to limited evidence. However, in multi-sample mode, the combined evidence across samples can make this variant clearly detectable [35]. This sensitivity improvement comes with increased computational requirements, as the joint analysis involves more complex statistical calculations and larger memory footprints to process multiple samples simultaneously.
Practical Implementation Guidelines
Successful implementation of multi-sample joint calling requires careful attention to sample preparation, computational resources, and parameter configuration. The following experimental protocol outlines the key steps:
Sample Preparation and Sequencing:
- Collect multiple tumor samples and, if possible, multiple normal samples from the same patient
- Ensure consistent library preparation and sequencing protocols across all samples
- Sequence all samples to sufficient depth (typically >100x for tumors, >60x for normals)
- Use the same reference genome build for all alignments
Computational Requirements:
- Allocate sufficient memory (typically 32GB or more depending on sample count)
- Ensure adequate temporary storage space for intermediate files
- Plan for longer runtime compared to single-sample analyses
Data Preprocessing:
- Process all BAM files through standardized preprocessing pipelines
- Follow GATK Best Practices for data pre-processing including duplicate marking, base quality score recalibration, and alignment [18]
- Verify that sample names in BAM read groups are unique and meaningful
- Ensure all BAM files are coordinate-sorted and indexed
Mutect2 Execution:
- Specify all tumor and normal BAM files using multiple
-Iparameters - Identify each normal sample using the
-normalparameter with the sample name from the read group - Include appropriate germline resources (e.g., gnomAD) and panels of normals
- Use the
--af-of-alleles-not-in-resourceparameter appropriate for your organism and sample count
Downstream Analysis:
- Process the raw Mutect2 output with FilterMutectCalls
- Annotate variants using tools like Funcotator
- Analyze the sample-specific genotype information in the output VCF
- Perform validation studies to confirm low-frequency variants detected through joint calling
Technical Considerations and Limitations
Critical Parameter Configuration
Multi-sample joint calling requires careful parameter configuration to optimize performance while maintaining specificity. The --af-of-alleles-not-in-resource parameter is particularly important, as it determines the prior probability for alleles not found in the germline resource. In multi-sample mode, this parameter should be adjusted based on the number of samples being analyzed [34]. For tumor-only calling, the default is automatically set to 5e-8, while for tumor-normal calling it defaults to 1e-6. The --initial-tumor-lod and --tumor-lod-to-emit parameters control the sensitivity thresholds for variant detection and may require adjustment based on the specific characteristics of the sample set.
The --germline-resource parameter is essential for effective filtering of common germline variants. In multi-sample mode, the germline resource helps distinguish true somatic variants from rare germline polymorphisms that might be present in the pooled normal. The --panel-of-normals (PoN) parameter provides additional filtering power against sequencing artifacts and common technical artifacts. When analyzing multiple samples from the same patient, it is particularly important to use a comprehensive PoN to avoid false positives from systematic errors that might affect all samples similarly.
For the filtering step with FilterMutectCalls, several parameters may require optimization for multi-sample analyses. The --normal-artifact-lod threshold may need adjustment when using pooled normals, as the combined evidence from multiple normal samples can affect the artifact detection logic. The --tumor-lod threshold controls the final sensitivity of the filtered callset and should be validated using known positive controls when possible. The statistical clustering model in FilterMutectCalls assumes all tumors are from the same patient, which is appropriate for multi-sample analyses but requires verification that this assumption holds for the specific experimental design.
Analysis Scope and Limitations
Mutect2 multi-sample calling is specifically designed for samples derived from the same patient and should not be used for combining samples from different patients [35]. When samples from different patients are combined, several problems arise: the normal samples become ineffective for filtering patient-specific germline variants, the somatic likelihoods model behaves unpredictably, and computational performance degrades significantly due to excessive genetic diversity. For multi-patient analyses, separate single-sample calls should be generated and combined in downstream analyses.
The computational requirements of multi-sample calling increase with the number of samples analyzed. Runtime and memory usage scale with the total sequencing data across all samples, and very large sample sets may require partitioning or specialized computational resources. Additionally, the current implementation assumes that all tumor samples are independent biopsies from the same patient. If multiple samples are simply technical replicates or different sequencing runs of the same biological sample, they should be merged before analysis rather than analyzed as separate samples in multi-sample mode.
Another important limitation concerns the handling of copy number variations and complex structural variations. While Mutect2 excels at detecting small somatic variants (SNVs and indels), it is not designed for comprehensive identification of larger structural variants or copy number alterations. These variant types require specialized tools and approaches that are beyond the scope of Mutect2's capabilities. Researchers working with highly aneuploid tumors or complex genomic rearrangements should complement Mutect2 analysis with dedicated structural variant callers.
Table 2: Key research reagents and computational resources for multi-sample joint calling
| Resource Category | Specific Examples | Function in Workflow |
|---|---|---|
| Reference Genome | GRCh38, GRCh37 | Standardized genomic coordinate system for alignment and variant calling |
| Germline Resource | gnomAD (af-only-gnomad.vcf.gz) | Filtering of common germline polymorphisms; provides population allele frequencies |
| Panel of Normals | Custom-generated from normal samples | Filtering of recurring technical artifacts and sequencing errors |
| Variant Databases | dbSNP, COSMIC, ClinVar | Variant annotation and functional interpretation |
| Alignment Tools | BWA-MEM, minimap2 | Mapping of sequencing reads to reference genome |
| BAM Processing Tools | Picard, Sambamba | Duplicate marking, BAM file manipulation and quality control |
| Variant Filtering | FilterMutectCalls | Application of statistical filters to remove false positive calls |
| Variant Annotation | Funcotator, VEP | Functional annotation of variants for biological interpretation |
The germline resource, typically derived from the Genome Aggregation Database (gnomAD), is particularly important for distinguishing true somatic variants from rare germline polymorphisms [34]. The panel of normals (PoN) is a critical resource for filtering technical artifacts that recur across multiple normal samples. Creating an effective PoN requires sequencing data from multiple normal samples processed through the same experimental and computational pipelines as the tumor samples. For multi-sample analyses, both of these resources enhance specificity without compromising sensitivity for true somatic variants.
The reference genome choice has implications for alignment efficiency and variant accuracy. The current best practice is to use the GRCh38 reference genome with decoy sequences and alternate contigs. All samples in a multi-sample analysis must be aligned to the same reference genome build to ensure coordinate consistency. Additionally, the use of standardized variant annotation databases enables consistent biological interpretation of variants across multiple samples, facilitating the identification of clinically relevant mutations and potential therapeutic targets.
Multi-sample joint calling with Mutect2 represents a powerful advancement in somatic variant detection, enabling researchers to leverage the full potential of multiple samples from the same patient. This approach provides enhanced sensitivity for low-frequency variants, improved statistical power for variant detection, and unique insights into tumor heterogeneity and evolution. The technical implementation requires careful attention to sample preparation, computational resources, and parameter configuration, but offers substantial rewards in terms of variant detection capability.
As cancer genomics continues to evolve toward more complex sampling strategies, including longitudinal monitoring and multi-region sequencing, the importance of sophisticated analysis methods like multi-sample joint calling will only increase. By implementing the best practices and considerations outlined in this guide, researchers can maximize the biological insights gained from their precious sample collections and contribute to advancing our understanding of cancer genomics. The integration of multi-sample calling into standardized analysis workflows represents an important step toward more comprehensive and accurate characterization of somatic variation in cancer.
In somatic variant analysis, distinguishing true somatic mutations from germline variants and technical artifacts is a fundamental challenge. Two specialized resources are essential for this task: the germline resource and the Panel of Normals (PON). When used with a matched normal sample, these resources enable the GATK Mutect2 pipeline to achieve high specificity and sensitivity, forming a multi-layered filtration system critical for accurate variant calling in cancer research and drug development [36] [1].
Core Concepts and Definitions
Germline Reference Resource
A germline resource is a population-scale catalog of germline variations, such as gnomAD, provided in a VCF file. This resource contains population allele frequencies (AF) that inform the Bayesian statistical model of Mutect2 about the prior probability of a variant being common in the population. Variants with high population allele frequency are more likely to be germline and are thus filtered out of the final somatic callset [1] [37].
Panel of Normals (PON)
A Panel of Normals (PON) is a custom-built resource derived from the sequencing data of multiple normal (healthy) tissue samples. Its primary purpose is to capture recurrent technical artifacts present in a specific sequencing pipeline. Artifacts may arise from mapping errors, sequencing errors, or other platform-specific biases. By identifying sites that are recurrently noisy across many normal samples, the PON allows Mutect2 to pre-emptively filter these artifacts from tumor samples [36].
Table 1: Key Characteristics of Supporting Resources
| Feature | Germline Resource | Panel of Normals (PON) |
|---|---|---|
| Primary Purpose | Filter common germline variants [1] | Filter recurrent technical artifacts [36] |
| Data Source | Population databases (e.g., gnomAD) [1] | In-house normal samples from healthy tissue [36] |
| Content | Known germline variants with allele frequencies [1] | Somatic-like artifactual sites found in multiple normals [36] |
| File Format | VCF [1] | Sites-only VCF [36] |
| Role in Analysis | Provides prior probabilities for germline status [37] | Identifies and flags unreliable, noisy genomic sites [36] |
The Scientist's Toolkit: Essential Research Reagents
The following table details the key materials and computational reagents required to implement this somatic calling workflow effectively.
Table 2: Essential Research Reagents and Resources
| Item | Function / Purpose | Technical Notes |
|---|---|---|
| Reference Genome (FASTA) | Baseline for read alignment and variant calling; must be the same for all steps [1]. | Use a consistent version (e.g., hg38) for all data. |
| Normal Sample BAMs | Input for creating a Panel of Normals; should be derived from healthy tissue [36]. | Technically matched to tumor samples; minimum of 40 recommended [36]. |
| Tumor and Matched Normal BAMs | Primary data for somatic variant discovery. | The input BAMs must be coordinate-sorted and indexed. |
| Germline Resource (e.g., gnomAD VCF) | Provides allele frequencies for population-based germline filtering [1]. | Must contain AF INFO field; should be matched to reference genome build [1]. |
| Panel of Normals (PON VCF) | A sites-only VCF used to filter recurrent technical artifacts [36]. | Can be created in-house or use public resources [36]. |
| af-only-gnomad.vcf.gz | A specific version of the germline resource without genotypes, only allele frequencies [12]. | Commonly used in GATK best practices workflows [12]. |
Quantitative Guidelines and Best Practices
Panel of Normals Construction
The selection of normal samples for a PON is critical. The most important criteria are technical properties; the normals should be as technically similar as possible to the tumor samples, encompassing the same exome or genome preparation methods, sequencing technology, and analysis software [36]. To minimize the risk of including an undiagnosed tumor, samples should ideally come from young, healthy subjects and are typically derived from blood [36].
- Minimum Sample Count: While there is no definitive rule, a minimum of 40 samples is recommended for creating a PON. In practice, larger cohorts of hundreds of samples are used at large production centers like the Broad Institute [36].
- Variant Frequency Threshold: During PON creation, the
--min-sample-countargument (default: 2) determines how many normal samples must exhibit a variant for it to be included in the PON. This targets artifacts that are recurrent but not necessarily present in every sample [38].
Germline Resource Configuration
When using a germline resource, the --af-of-alleles-not-in-resource parameter becomes crucial. This value represents the imputed allele frequency for any variant not found in the germline resource. Mutect2 dynamically sets appropriate defaults based on the analysis mode [1]:
- Tumor-only mode: Default is 5e-8
- Tumor-normal mode: Default is 1e-6
- Mitochondrial mode: Default is 4e-3
For a resource like the gnomAD exomes, which represents ~200,000 samples, a value of 0.0000025 (1/(2 * number of exome samples)) is appropriate [37].
Experimental Protocol: Creating a Panel of Normals
Creating a robust PON is a multi-step process that involves running Mutect2 on each normal sample individually and then combining the results [12] [38].
Step-by-Step Methodology
Run Mutect2 on Individual Normal Samples: Execute Mutect2 in tumor-only mode for each normal BAM file. The
--max-mnp-distance 0parameter is included to avoid a known bug in GenomicsDBImport [38].Import VCFs into a GenomicsDB: Use GenomicsDBImport to consolidate the individual VCFs into a single, queryable database. This tool efficiently handles the genomic intervals and multiple input files [12].
Create the Combined PON: Use CreateSomaticPanelOfNormals to process the database and generate the final panel. The
--min-sample-countargument can be adjusted to change the recurrence threshold [38].
Integrated Analysis Workflow in Mutect2
The germline resource and PON work in concert within the Mutect2 pipeline to filter out false positives before statistical genotyping and filtering.
A typical Mutect2 command incorporating both resources is structured as follows [1]:
In this command:
- The
--germline-resourceprovides population allele frequencies that Mutect2 uses to calculate the prior probability of a variant being germline. - The
--panel-of-normalsimmediately excludes sites that are listed in the PON from further analysis, as these are known artifact-prone loci.
Publicly available Panels of Normals and germline resources can be accessed to begin analysis without constructing a custom PON. The GATK resource bundle provides the following PONs for common use cases [36]:
- hg38:
gs://gatk-best-practices/somatic-hg38/1000g_pon.hg38.vcf.gz - hg19/b37 (WGS):
gs://gatk-best-practices/somatic-b37/Mutect2-WGS-panel-b37.vcf - hg19/b37 (Exome):
gs://gatk-best-practices/somatic-b37/Mutect2-exome-panel.vcf
For the germline resource, the af-only-gnomad VCFs for the appropriate reference genome build are standard in the GATK best practices workflows [12] [37].
Best Practices for Input BAM Specifications in Command-line Implementation
In somatic variant discovery research, the Binary Alignment/Map (BAM) file format serves as the fundamental data structure containing aligned sequencing reads for both tumor and normal samples. The BAM format represents the compressed binary version of the Sequence Alignment/Map (SAM) format, providing a compact and indexable representation of nucleotide sequence alignments [39]. For somatic calling pipelines, particularly those utilizing the GATK Mutect2 workflow, properly formatted and processed BAM files are critical inputs that directly impact variant calling accuracy, sensitivity, and specificity. The BAM file structure consists of two primary components: a header section containing information about the entire file (sample name, sample length, and alignment method), and an alignment section containing detailed information for each read or read pair [39].
The GATK Best Practices for somatic short variant discovery provide a standardized framework for processing high-throughput sequencing data, with specific requirements for input BAM files at different stages of the analysis workflow [18] [8]. These specifications ensure that the biological signals of somatic variation can be reliably distinguished from technical artifacts introduced during sample preparation, sequencing, and alignment processes. For drug development professionals and translational researchers, adherence to these specifications is essential for generating clinically actionable insights from cancer genomic data.
Core BAM File Requirements for GATK Somatic Calling
Structural and Content Specifications
Input BAM files for GATK somatic variant discovery must meet specific structural requirements to ensure compatibility with the Mutect2 pipeline and downstream filtering tools. The alignment section of each BAM file must contain complete information for each read, including read name, read sequence, read quality, alignment information, and custom tags [39]. The read name includes the chromosome, start coordinate, alignment quality, and the CIGAR (Concise Idiosyncratic Gapped Alignment Report) string, which details the alignment structure through operators that indicate matches, insertions, deletions, and other discontinuities in the alignment [40].
Table 1: Essential BAM File Components for GATK Somatic Analysis
| Component | Description | Requirements for Somatic Calling |
|---|---|---|
| Header Section | Contains metadata about the entire file | Must include proper @SQ lines for all reference contigs and @RG lines with complete read group information |
| Alignment Section | Contains the aligned read records | Must be coordinate-sorted and include all mandatory fields (QNAME, FLAG, RNAME, POS, MAPQ, CIGAR, etc.) |
| Read Groups | Sample identification and processing metadata | Must contain unique ID, sample name (SM), sequencing platform (PL), and library (LB) information |
| Quality Scores | Per-base sequencing quality values | Must be encoded in standard Phred scale and properly calibrated |
| Alignment Quality | MAPQ field indicating mapping confidence | Values should reflect genuine mapping uncertainty; 255 indicates unknown quality |
| CIGAR String | Alignment structure representation | Must accurately represent matches, insertions, deletions, and splicing events |
BAM files must be coordinate-sorted, with alignments ordered by their leftmost position on the reference genome, and must be accompanied by a proper index file (BAI) that enables efficient random access to genomic regions [41]. The index file serves as a "table of contents" for the BAM file, indicating where specific reads or sets of reads can be found, which is particularly important for the regional processing approach used by Mutect2 [40] [41].
Sample Identification and Read Group Specifications
Proper sample identification within BAM files is crucial for somatic variant discovery, especially when analyzing multiple tumor-normal pairs or incorporating panel of normal (PoN) resources. Each read group must contain specific metadata fields that unambiguously identify the sample and experimental conditions. The GATK-SV framework imposes specific restrictions on sample names to avoid parsing errors, requiring that sample IDs must be unique within the cohort and contain only alphanumeric characters and underscores (no dashes, whitespace, or special characters) [42].
For somatic analysis with Mutect2, the -tumor and -normal parameters must match the read group sample names (the SM field values) in their respective BAM files [37]. To look up the read group SM field, researchers can use GetSampleName or samtools view -H tumor.bam | grep '@RG' [37]. Sample IDs should not contain only numeric characters (e.g., "10004928"), be a substring of another sample ID in the same cohort, or contain any of the following substrings: chr, name, DEL, DUP, CPX, CHROM [42].
Table 2: Read Group Requirements for Somatic Variant Discovery
| Field | Description | Criticality for Somatic Analysis |
|---|---|---|
| ID | Read group identifier | Must be unique for each flow cell lane; differentiates technical batch effects |
| SM | Sample identifier | Must match the -tumor and -normal arguments in Mutect2; consistent across lanes for same sample |
| PL | Sequencing platform | Required for platform-specific processing; e.g., "ILLUMINA" for Illumina instruments |
| LB | Library identifier | Important for marking duplicates; should be unique for each library preparation |
| PU | Platform unit | Flow cell lane identifier; helps identify lane-specific artifacts |
| CN | Sequencing center | Useful for multi-center studies and metadata tracking |
Samples with a high percentage of improperly paired or chimeric reads should be filtered out as technical outliers prior to running GATK-SV's GatherSampleEvidence, as they may indicate issues with library preparation, degradation, or contamination and can lead to poor variant set quality [42].
BAM Preprocessing Workflows for Somatic Analysis
Data Preprocessing Phase
The GATK Best Practices workflow for data pre-processing involves converting raw sequence data (provided in FASTQ or uBAM format) to produce analysis-ready BAM files through alignment to a reference genome and data cleanup operations to correct for technical biases [18]. The parsimonious operating procedures outlined in the three-step preprocessing workflow maximize data quality, storage, and processing efficiency to produce a mapped and clean BAM file ready for analysis workflows that start with MarkDuplicates [43].
The preprocessing workflow emphasizes cleaning up prior meta information while providing the opportunity to assign proper read group fields, marking adapter sequences using MarkIlluminaAdapters such that they contribute minimally to alignments, and piping three processes (SamToFastq, BWA-MEM, and MergeBamAlignment) to produce the final BAM while avoiding storage of larger intermediate FASTQ and SAM files [43]. The resulting clean BAM is coordinate sorted, indexed, and retains original sequencing read information within the final BAM file such that data is amenable to reversion and analysis by different means [43].
BAM Preprocessing Workflow for Somatic Analysis
Quality Control and Validation Procedures
Prior to somatic variant calling, BAM files must undergo rigorous quality control to identify potential issues that could compromise variant calling accuracy. The MarkIlluminaAdapters tool adds the XT tag to read records to mark the 5' start position of specified adapter sequences and produces a metrics file [43]. This step is crucial for identifying reads with adapter sequences arising from read-through of short inserts, which can affect mapping and variant calling.
For large files, Java memory settings (-Xmx) and temporary directory specifications (TMP_DIR) should be used to prevent out-of-memory errors and ensure efficient processing [43]. The preprocessing workflow produces BAM files where reads with adapter sequences are marked with a tag in XT:i:# format, where # denotes the 5' starting position of the adapter sequence, while reads without adapter sequence remain untagged [43].
Experimental Protocols for BAM-Based Somatic Variant Discovery
Mutect2 Somatic Variant Calling Workflow
The GATK Mutect2 workflow for somatic short variant discovery identifies somatic single nucleotide variants (SNVs) and indels in one or more tumor samples from a single individual, with or without a matched normal sample [8]. The workflow requires pre-processed BAM files as input, prepared according to GATK Best Practices for data pre-processing [8]. The main steps involve generating a large set of candidate somatic variants using Mutect2, then filtering them to obtain a more confident set of somatic variant calls [8].
The basic command structure for Mutect2 follows the GATK4 command-line syntax, using the gatk wrapper script with the tool name as a positional argument [44]. A typical Mutect2 command for somatic variant calling includes:
This command produces a raw unfiltered somatic callset, which then undergoes additional processing including contamination estimation, orientation bias modeling, and filtering [8] [12]. The BAM input files must be properly formatted with correct read groups to ensure the -tumor and -normal arguments correctly identify the respective samples [37].
Post-Calling Filtering and Annotation
Following variant calling with Mutect2, several additional processing steps are required to generate a high-confidence somatic callset. FilterMutectCalls performs probabilistic filtering based on the probability that a variant is a somatic mutation, automatically determining the threshold that optimizes the F score (the harmonic mean of sensitivity and precision) [12]. The tool requires the same reference FASTA file used with Mutect2 and a stats file generated by Mutect2 [12].
For samples with potential substitution errors occurring on a single strand before sequencing (such as FFPE tumor samples or samples sequenced on Illumina Novaseq machines), Mutect2's orientation bias filter should be applied [12]. This three-step process involves:
- Running Mutect2 with the
--f1r2-tar-gzargument to create raw data for orientation bias modeling - Passing this data to LearnReadOrientationModel to generate an orientation model
- Providing the learned model to FilterMutectCalls with the
--ob-priorsargument
Functional annotation with Funcotator adds gene-level information to each variant, labeling each with one of twenty-three distinct variant classifications and producing gene information (affected gene, predicted variant amino acid sequence) and associations to datasources including GENCODE, dbSNP, gnomAD, and COSMIC [8].
Table 3: Essential Research Reagent Solutions for BAM-Based Somatic Analysis
| Resource Category | Specific Tools/Resources | Function in Somatic Workflow |
|---|---|---|
| Alignment Tools | BWA-MEM, Samtools, Picard | Generate coordinate-sorted BAM files from raw sequencing data |
| Variant Callers | GATK Mutect2 | Perform somatic short variant discovery from BAM inputs |
| Filtering Tools | FilterMutectCalls, CalculateContamination, LearnReadOrientationModel | Filter raw variant calls to produce high-confidence somatic variants |
| Reference Sequences | GRCh38, GRCh37 | Provide reference genome for alignment and variant calling |
| Germline Resources | gnomAD, 1000 Genomes | Filter common germline variants from somatic callsets |
| Panel of Normals | Custom-generated from normal samples | Identify and filter technical artifacts and common germline variants |
| Annotation Databases | Funcotator, dbSNP, COSMIC, GENCODE | Add functional and biological context to variant calls |
| Quality Control Metrics | GetPileupSummaries, CollectMultipleMetrics | Assess BAM file quality and identify potential issues |
The GATK Best Practices emphasize that several key steps make use of external resources, including validated databases of known variants [18]. If few or no such resources are available for non-human organisms, researchers may need to bootstrap their own or use alternative methods [18]. The Mutect2 workflow is extensively validated on data from human whole-genome or whole-exome samples sequenced with Illumina technology, and adaptations may be necessary for different data types, organisms, or experimental designs [18].
Properly formatted and processed BAM files are foundational to successful somatic variant discovery using the GATK Mutect2 pipeline. Adherence to the Best Practices for BAM specifications—including proper read group assignments, coordinate sorting, indexing, adapter marking, and quality control—ensures that biological signals of somatic variation can be accurately distinguished from technical artifacts. For researchers and drug development professionals, rigorous attention to these input specifications enables the generation of reliable, reproducible somatic variant callsets that can inform biological insights and clinical applications.
The experimental protocols and toolkit resources outlined in this technical guide provide a comprehensive framework for implementing GATK somatic calling workflows with appropriate BAM file inputs. As somatic variant discovery continues to evolve with new sequencing technologies and analysis methods, these foundational BAM specifications will remain critical for maintaining analytical accuracy and reliability in cancer genomics research.
Troubleshooting Common BAM File Issues and Optimizing Performance
Resolving Expected Variant Missingness Due to Alignment Artifacts
Variant missingness, the failure to detect true somatic variants in next-generation sequencing data, is a significant challenge in cancer genomics. A predominant source of this missingness stems from alignment artifacts, which occur during the process of mapping sequencing reads to a reference genome. These artifacts systematically obscure true biological variation, compromising the sensitivity and accuracy of somatic variant calling pipelines, particularly within the context of GATK-based research. In clinical drug development and research settings, such omissions can lead to missed therapeutic targets and inaccurate patient stratification.
Alignment artifacts arise primarily from two major sources: erroneous realignment in low-complexity regions and an incomplete reference genome with respect to the sample [45]. When sequence reads originate from highly repetitive genomic regions or regions poorly represented in the reference, aligners may incorrectly map them to an incorrect locus. This results in apparent variants that are false positives, or more problematically, the obscuring of true variants, leading to false negatives (variant missingness). The GATK Best Practices workflow acknowledges these challenges and emphasizes the importance of data pre-processing to produce analysis-ready BAM files, as the quality of the input BAM is foundational for all subsequent variant discovery [18].
This technical guide provides a comprehensive framework for understanding, identifying, and resolving alignment artifacts to minimize variant missingness, with a specific focus on meeting the input BAM requirements for robust GATK somatic short variant discovery.
The table below summarizes the primary categories of alignment artifacts, their causes, and their direct impact on variant calling.
Table 1: Characterization of Major Alignment Artifact Types
| Artifact Type | Primary Cause | Impact on Variant Calling | Common Genomic Context |
|---|---|---|---|
| Misalignment in Low-Complexity Regions [45] | Ambiguous mapping due to repetitive sequences (e.g., homopolymers, short tandem repeats). | False positives and false negatives due to reads being clustered incorrectly. | Mononucleotide and dinucleotide repeats [46]. |
| Reference Genome Incompleteness [45] | Sample-specific sequences absent from the reference genome are forced to align to incorrect, homologous loci. | Erosion of true variants and introduction of artifactual variants at incorrect sites. | Highly polymorphic or poorly characterized regions. |
| Poor Mapping Quality | Reads with high sequence similarity to multiple genomic locations are assigned inappropriately high mapping quality scores by the aligner. | Introduction of false positive calls, which may be filtered out, but can obscure true variants during caller processing [47]. | Segmental duplications and repetitive elements. |
The error rate of raw variant calls can be as high as 1 error per 10-15 kilobases, primarily driven by these artifacts. While post-filtering can reduce this rate to 1 error per 100-200 kilobases, this process often eliminates genuine variants as well, contributing to variant missingness [45].
Experimental Evidence of Prevalence
Studies utilizing haploid human cell lines (CHM1) have been instrumental in quantifying artifacts, as any heterozygous call is unequivocally an error. This approach has revealed that erroneous realignment and an incomplete reference genome are two major sources of false heterozygous calls [45]. Furthermore, consensus approaches from real tumor cohorts demonstrate that while individual somatic callers are susceptible to these artifacts, full consensus predictions can achieve validation rates exceeding 98% [48]. This highlights that artifact-induced missingness is a systematic issue affecting most callers, but it can be mitigated through robust bioinformatic protocols.
Methodologies for Artifact Detection and Resolution
Computational Filtering with GATK
The GATK suite provides specialized tools for the removal of alignment-induced false positives within a somatic variant callset.
Tool:
FilterAlignmentArtifacts: This experimental tool is designed specifically to filter false positives caused by reads being mapped to the wrong genomic locus [47].- Mechanism: It leverages a highly sensitive realignment process using a BWA-MEM index of the best available reference genome (e.g., hg38 for human), independent of the reference used for the input BAM's original alignment. By comparing the original alignment to this more sensitive realignment, it can identify and remove variants that are likely due to mapping errors [47].
- Usage Context: It is a core component of the Somatic Short Mutation Calling Best Practice Workflow, applied after the initial variant calling with Mutect2 and before further filtering with
FilterMutectCalls[47] [8].
Tool:
FilterMutectCalls: This tool employs a broader set of probabilistic models and hard filters to detect various error types, including alignment artifacts, strand bias, and polymerase slippage [8]. It works in concert withFilterAlignmentArtifactsto produce a high-confidence callset.
The Consensus Calling Approach
Employing multiple, diverse somatic callers and requiring variants to be detected by a consensus of them is a highly effective strategy for improving specificity.
- Protocol: A standard protocol involves running at least two or three callers (e.g., MuTect2, MuSE, Strelka2) on the same tumor-normal BAM pair and taking the intersection of their calls [49] [48].
- Performance: Full consensus calls have been shown to achieve a validation rate of >98% via Sanger sequencing [48]. This method effectively bypasses the unique error profiles of individual callers, though it may slightly reduce sensitivity.
- Implementation: For large-scale studies, accelerated callers like MuSE 2 and Strelka2 are recommended for their combination of speed and accuracy, making a consensus approach computationally feasible for large cohorts [49].
Manual Review with a Standardized SOP
Despite computational advances, manual review remains a critical step for the final refinement of somatic variants, especially in clinical settings.
- Standardized Protocol: A detailed Standard Operating Procedure (SOP) for manual review using the Integrative Genomics Viewer (IGV) exists to reduce inter-reviewer variability [46].
- Annotation System: The SOP defines a set of calls (
Somatic,Germline,Ambiguous,Fail) and 19 standardized tags to annotate the rationale for a decision. - Key Artifact Tags: During review, specific patterns indicative of alignment artifacts should be tagged [46]:
AI(Adjacent Indel): For variants attributable to misalignment caused by a nearby insertion or deletion.MN/DN(Mono-/Dinucleotide Repeat): For variants in or adjacent to homopolymer or short tandem repeats.LM(Low Mapping Quality): For variants mostly supported by reads with low mapping quality.HDR(High Discrepancy Region): For variants in regions where supporting reads show recurrent mismatches.
The following workflow diagram integrates these methodologies into a cohesive strategy for resolving alignment artifacts.
The Scientist's Toolkit: Essential Research Reagents and Tools
The following table details key resources required for experiments aimed at characterizing and mitigating alignment artifacts.
Table 2: Essential Research Toolkit for Artifact Investigation
| Tool / Resource | Function | Use-Case in Artifact Resolution |
|---|---|---|
| Haploid Cell Line (CHM1) [45] | A genetically haploid human cell line. | Serves as a ground truth set; any heterozygous call is an alignment artifact, enabling precise quantification and study. |
| BWA-MEM Aligner [45] [47] | A widely-used read alignment algorithm. | The standard aligner for initial mapping; also used internally by GATK's FilterAlignmentArtifacts for sensitive realignment. |
| IGV & IGVNav [46] | Genomic visualization tools for manual variant inspection. | Critical for manual review. IGVNav is a plugin that standardizes the process with predefined calls and tags for artifacts. |
| Synthetic Tumor/Normal Pairs (e.g., via BAMSurgeon) [50] | In silico generated datasets with known truth variants. | Provides a controlled training set with known positives/negatives to benchmark artifact detection methods and train models. |
| NeuSomatic [50] | A somatic mutation detector based on Deep Convolutional Neural Networks (CNNs). | Can be trained to recognize complex patterns of alignment artifacts from data, offering a universal and accurate detection method. |
| Consensus Truth Sets (e.g., from TCGA, PCAWG) [49] | High-confidence somatic variant callsets from large consortia. | Used as a gold standard benchmark to evaluate the sensitivity and precision of artifact filtration methods. |
Advanced and Emerging Strategies
Deep Learning Approaches
Tools like NeuSomatic represent the cutting edge in artifact detection. By using deep convolutional neural networks, these models can learn complex, multi-dimensional features from sequence alignments that are indicative of true variants versus artifacts [50]. NeuSomatic can operate as a stand-alone caller or in an ensemble mode, where it integrates the outputs of multiple traditional callers (e.g., MuTect2, Strelka2) to make a final, more accurate prediction. This ensemble approach is particularly powerful for suppressing the false positives unique to each individual caller, many of which are alignment artifacts.
Multi-Platform Sequencing for Comprehensive Resolution
For the most challenging regions of the genome, short-read sequencing may be inherently limited. Long-read sequencing technologies (PacBio, Oxford Nanopore) and optical mapping (Bionano Genomics) can traverse repetitive low-complexity regions and detect large SVs that confound short-read aligners [51]. Integrating data from these multiple platforms has been shown to yield a 3 to 7-fold increase in structural variant detection compared to standard short-read studies [51]. This multi-platform approach is the ultimate solution for resolving variant missingness in complex genomic regions, providing a haplotype-resolved view of the genome that dramatically reduces alignment ambiguity.
The following diagram illustrates the decision process for selecting the appropriate resolution strategy based on the biological context and available data.
Addressing High-Depth Sequencing Challenges and Computational Limits
High-depth sequencing has become a cornerstone of somatic variant discovery in cancer genomics and therapeutic development, enabling researchers to detect low-frequency subclonal populations that are critical for understanding tumor evolution and therapeutic resistance. In the context of GATK somatic calling workflows, high-depth data (often exceeding 200x coverage) presents both unprecedented opportunities and substantial computational challenges. The Genome Analysis Toolkit Best Practices for somatic short variant discovery provide a comprehensive framework for identifying SNVs and indels from tumor samples with or without matched normals, but efficient implementation requires careful consideration of input BAM requirements and computational constraints [8]. As sequencing technologies advance, with platforms like Illumina NovaSeq producing exceptionally high yields and long-read technologies from Oxford Nanopore and PacBio achieving increasing adoption, the bioinformatics community must develop strategies to manage the resulting data deluge while maintaining analytical precision [52] [53].
The fundamental challenge lies in balancing sensitivity for detecting low-allele-fraction variants against computational feasibility, especially in large-scale studies or clinical settings where turnaround time is critical. This technical guide examines the specific bottlenecks encountered when processing high-depth BAM files through GATK somatic calling pipelines and provides evidence-based solutions for researchers, scientists, and drug development professionals working under computational constraints.
Computational Bottlenecks in High-Depth BAM Processing
Performance Limitations in Variant Calling
Processing high-depth BAM files through GATK's Mutect2 reveals several specific computational bottlenecks that scale non-linearly with sequencing depth. The local assembly of haplotypes in active regions, while superior for sensitivity, becomes increasingly resource-intensive as depth exceeds 500x, with memory requirements growing substantially in complex genomic regions [1]. Benchmarking studies indicate that Mutect2 running time increases approximately 1.8-fold when depth doubles from 200x to 400x, with even more pronounced slowdowns in regions with high variant density [8].
The Bayesian genotyping model employed by Mutect2 must evaluate evidence from all reads at each potential variant site, creating significant computational overhead at extreme depths (e.g., 20,000X as mentioned in GATK documentation) [1]. This is particularly challenging in cancer genomes with aneuploidy and copy-number variations, where the assumptions of diploid genotyping break down and additional computational cycles are required to properly evaluate somatic evidence. For large panels or whole genomes at high depth, these bottlenecks can extend processing time from hours to days without proper optimization.
Memory and Storage Constraints
The memory footprint for high-depth BAM processing constitutes another major constraint, particularly during the variant calling and filtering stages. BAM files from 30x whole genomes typically require ~80-100GB of storage, but this increases substantially for high-depth targeted sequencing, where depths of 1000x can produce BAM files exceeding 50GB for a single sample [54]. When multiple such files are processed simultaneously in joint calling mode (supported in Mutect2 v4.1.0.0+), memory requirements can exceed 64GB of RAM, creating infrastructure barriers for many research settings [1].
Storage bottlenecks extend beyond the BAM files themselves to intermediate files generated during processing. The Mutect2 workflow produces additional data products including F1R2 tar files for orientation bias analysis, contamination tables, and statistical summaries that collectively can approach the size of the input BAMs [8]. Without careful management of temporary files and output compression, storage requirements can easily double during active processing, creating challenges for shared computational environments.
Table 1: Computational Requirements for High-Depth BAM Processing in GATK Somatic Workflow
| Processing Stage | CPU Intensity | Memory Requirements | Scaling with Depth | Intermediate Storage |
|---|---|---|---|---|
| BAM Pre-processing | Medium | 8-16GB | Linear | Low |
| Mutect2 Calling | Very High | 32-64GB | Super-linear | High |
| Contamination Calculation | Low | 4-8GB | Linear | Minimal |
| Read Orientation | Medium | 16-32GB | Linear | Medium |
| FilterMutectCalls | Medium | 8-16GB | Linear | Minimal |
Strategic Approaches for Computational Efficiency
BAM Pre-processing and Optimization
Proper BAM pre-processing represents the most effective first step in managing computational load for high-depth sequencing data. The GATK Best Practices emphasize that "input BAMs should be pre-processed as described in the GATK Best Practices for data pre-processing" before somatic variant discovery [8]. This includes coordinate sorting, duplicate marking, and proper read group specification, all of which contribute significantly to computational efficiency.
Duplicate marking is particularly important for high-depth data, as PCR duplicates can constitute 10-30% of reads in amplified libraries, unnecessarily increasing computational overhead without adding informative content [55]. While the GATK documentation notes that "marking duplicates is less relevant to targeted amplicon sequencing and RNA-Seq analysis," it remains critical for most somatic calling applications to avoid false positives from over-amplified fragments [55]. For large BAM files, the MarkDuplicates tool in Picard can be accelerated through careful management of temporary directory space and increasing the memory allocation to avoid disk thrashing.
Read group information, while often overlooked, enables more efficient processing by providing metadata about sample origin, library preparation, and sequencing run [56]. Properly formatted read groups with correct SM, LB, and PU fields allow tools to optimize memory usage and implement sample-specific processing logic. The Picard AddOrReplaceReadGroups tool provides a mechanism to fix missing or incorrect read group information in existing BAM files, which should be addressed before variant calling [55].
Workflow Optimizations for High-Depth Data
Several specific workflow optimizations can dramatically improve performance with high-depth BAM files without sacrificing analytical sensitivity:
Interval Scattering: By breaking the genome into smaller intervals (typically 1-10Mb) and processing them in parallel, overall runtime can be reduced by 70-90% on multi-core systems. This approach distributes memory load across jobs and enables recovery from failures without restarting complete analyses. The GATK workflow scripts provided as reference implementations demonstrate effective interval scattering patterns optimized for human genomes [18].
Resource-Aware Configuration: Mutect2 parameters can be tuned for high-depth data by adjusting assembly-related parameters such as --max-reads-per-alignment-start and --max-assembly-region-size to prevent excessive resource consumption in complex regions. The GATK documentation notes that Mutect2 "accommodates extreme high depths, e.g. 20,000X" through internal optimizations, but explicit parameter tuning may still be beneficial for specific applications [1].
Strategic Normalization: For tumor-only analyses, creating and using a panel of normals (PoN) significantly reduces false positives and computational overhead during filtering. The PoN captures common technical artifacts and low-frequency germline variants that would otherwise require extensive computational resources to evaluate and filter in each sample individually [8]. Similarly, proper use of germline resources like gnomAD enables more efficient filtering of common polymorphisms.
Diagram 1: Computational optimization workflow for high-depth BAM processing
Experimental Protocols and Benchmarking
Benchmarking Methodologies for Performance Validation
Rigorous benchmarking using established standards ensures that computational optimizations do not compromise analytical sensitivity. The Global Alliance for Genomics and Health (GA4GH) has developed benchmarking protocols that utilize Genome in a Bottle (GIAB) reference samples with established ground-truth variant calls [53]. These resources enable quantitative assessment of pipeline performance through precision (positive predictive value) and recall (sensitivity) metrics calculated as follows:
Precision = True Positives / (True Positives + False Positives) Recall = True Positives / (True Positives + False Negatives) [53]
In recent benchmarking of a nanopore sequencing pipeline using GIAB samples, researchers achieved a precision of 0.997 and recall of 0.992 for SNVs, while small indel identification approached a precision of 0.922 and recall of 0.838 [53]. Similar methodologies should be applied when optimizing GATK workflows for high-depth data to ensure variant detection sensitivity remains acceptable after computational optimizations.
For somatic-specific benchmarking, titration experiments using tumor-normal cell line admixtures with known variant profiles provide the most relevant performance assessment. By creating mixtures with varying allele fractions (1-25%) and processing them through optimized workflows, researchers can establish the relationship between sequencing depth, computational parameters, and detection sensitivity for low-frequency variants.
Platform-Specific Considerations
Different sequencing platforms present distinct computational challenges for high-depth analyses. Illumina short-read platforms typically produce the "highest number of reads and bases, depth of coverage, completeness of consensus genomes," creating substantial processing demands but with relatively straightforward error profiles [52]. By comparison, long-read technologies from Oxford Nanopore and PacBio have "lower yields, sequencing depth, and mapping coverage" per run but produce longer reads that require different computational approaches during alignment and variant calling [52].
The GATK Best Practices were developed primarily using "data from human whole-genome or whole-exome samples sequenced with Illumina technology," meaning researchers working with other platforms or organisms may need to adapt certain parameters [18]. For all platforms, base quality score recalibration remains a critical preprocessing step that improves downstream variant calling accuracy, though the specific covariates may differ between technologies.
Table 2: Platform-Specific Computational Considerations for High-Depth Sequencing
| Sequencing Platform | Typical Max Depth | Primary Computational Challenge | Recommended GATK Adjustments |
|---|---|---|---|
| Illumina NovaSeq | 1000x+ | Memory usage in complex regions | Increase assembly region size |
| Illumina HiSeq | 500x | General processing throughput | Standard parameters |
| PacBio Sequel II | 200x | Long read handling | Adjust min pruning, assembly parameters |
| ONT PromethION | 200x | High error rate processing | Modify quality thresholds, use mitochondrial mode |
Essential Research Reagents and Computational Tools
Table 3: Research Reagent Solutions for High-Depth Sequencing Studies
| Reagent/Tool | Function | Application in GATK Workflow |
|---|---|---|
| GIAB Reference Standards | Benchmarking variant calls | Performance validation |
| Coriell CNV Reference Panel | Large variant detection | SV/CNV benchmarking |
| gnomAD Resource | Germline variant filtering | Contamination estimation |
| Picard Tools | BAM preprocessing | Read group addition, duplicate marking |
| SAMtools | BAM manipulation | File sorting, indexing, format conversion |
| BAMscale | Peak quantification | Coverage analysis for targeted sequencing |
| deepTools | Coverage visualization | BigWig track generation |
| Funcotator | Variant annotation | Functional consequence prediction |
Implementation Framework for Resource-Constrained Environments
Practical Implementation Guidelines
For research groups operating within computational constraints, a tiered approach to high-depth sequencing analysis balances resource limitations with analytical requirements:
Targeted Processing: When whole-genome sequencing at high depth is computationally prohibitive, targeted sequencing of cancer gene panels (1-5Mb) at ultra-high depth (500-1000x) provides a cost-effective alternative for detecting low-frequency variants. The GATK workflows "can be adapted to others with some modifications" for targeted designs [18]. Tools like BAMscale provide efficient "quantification of sequencing peaks from diverse experimental sources" for such targeted approaches [54].
Progressive Filtering: Implementing multiple filtering stages reduces the computational load at each step. Initial quality-based filtering of reads, followed by region-specific processing (e.g., excluding low-complexity regions in initial passes), and finally comprehensive analysis of target regions dramatically improves throughput without significantly compromising results.
Cloud-Based Scaling: The WDL workflows provided as GATK reference implementations are designed to run on Cromwell, an execution engine that supports cloud-based scaling [18]. This approach allows researchers to maintain standard analytical pipelines while leveraging elastic cloud resources during peak processing demands, particularly important for high-depth BAM files that exceed local computational capacity.
Validation and Quality Control
Comprehensive quality control metrics specific to high-depth data ensure that computational optimizations do not introduce analytical artifacts:
Contamination Monitoring: The CalculateContamination tool in GATK is specifically "designed to work well without a matched normal even in samples with significant copy number variation" [8]. For high-depth data, contamination estimates should be reviewed alongside mapping quality statistics to identify potential sample swaps or cross-individual contamination that might be masked by high coverage.
Orientation Bias Assessment: FFPE-derived samples and other degraded DNA sources exhibit specific artifacts that become more pronounced at high depth. The LearnReadOrientationModel tool "finds prior probabilities of single-stranded substitution errors prior to sequencing for each trinucleotide context," which is "extremely important for FFPE tumor samples" [8]. This step prevents false positives from strand-biased artifacts that might otherwise pass filters in high-depth data.
Coverage Uniformity: While total sequencing depth receives significant attention, the uniformity of coverage critically impacts variant detection sensitivity. Tools like deepTools bamCoverage enable assessment of coverage distribution across regions of interest, with normalization options including RPKM, CPM, BPM, and RPGC [57]. For cancer genomes with copy-number alterations, careful interpretation of coverage metrics is essential, as expected ploidy variations can be confused with technical coverage biases.
Diagram 2: Quality control framework for high-depth variant calling
High-depth sequencing represents a powerful approach for detecting low-frequency somatic variants, but requires thoughtful computational strategies to implement efficiently within GATK somatic calling workflows. By combining proper BAM pre-processing, workflow optimizations like interval scattering, and rigorous quality control, researchers can overcome the computational barriers associated with high-depth data while maintaining analytical sensitivity. As sequencing technologies continue to evolve toward higher throughput and longer reads, these optimization strategies will become increasingly critical for cancer researchers and drug development professionals working to translate genomic findings into clinical insights.
The GATK Best Practices provide a solid foundation for somatic variant discovery, but researchers should view them as a starting point rather than a rigid protocol, particularly when working with high-depth data or non-standard experimental designs. As noted in the GATK documentation, "any workflow that has been significantly adapted or customized, whether for performance reasons or to fit a use case that we do not explicitly cover, should not be called 'GATK Best Practices'" [18]. This adaptive approach ensures that computational limitations do not constrain biological discovery in the era of high-depth genomic analysis.
Managing Contamination Estimation with GetPileupSummaries and CalculateContamination
In next-generation sequencing (NGS) analysis, particularly for cancer genomics, cross-sample contamination is a pervasive technical challenge that can significantly compromise the integrity of variant calls. Even low levels of contamination can lead to both false-positive and false-negative somatic variant calls, ultimately affecting downstream clinical and research conclusions [58]. Within the GATK Best Practices workflow for somatic short mutation discovery, the tools GetPileupSummaries and CalculateContamination form an essential quality control module specifically designed to quantify this contamination [59] [60]. This technical guide details the implementation of this contamination estimation pipeline, framing it within the broader context of BAM file preparation requirements essential for robust somatic variant analysis. The methodology represents an evolution from earlier tools like ContEst, offering improved performance in the presence of copy number variations and without requiring matched normal samples [59] [61].
GetPileupSummaries: Data Collection Module
GetPileupSummaries functions as the data collection front-end of the contamination estimation pipeline. This tool processes a BAM file and a reference set of known germline variants to produce a compact table summarizing allele counts at specific genomic positions [62]. The tool is designed to tabulate counts of reads supporting reference, alternate, and other alleles for a predefined set of common biallelic SNP sites, typically derived from population frequency databases like gnomAD. Critically, these sites must contain population allele frequency (AF) annotations in their INFO field [62].
CalculateContamination: Estimation Algorithm Module
CalculateContamination serves as the computational engine that transforms the pileup summary data into a quantitative contamination estimate. The tool implements a statistical method that estimates contamination by analyzing the fraction of reference alleles observed at homozygous alternate sites [59]. In uncontaminated samples, such sites should exhibit nearly 100% alternate alleles; the presence of reference reads provides evidence of foreign DNA mixture. This approach works with copy number variations and multiple contaminating sources, representing a significant improvement over earlier probabilistic models that assumed diploid genotypes with no CNVs [59] [61].
Table 1: Core Tool Functions within the Somatic Variant Calling Workflow
| Tool Name | Primary Function | Input Requirements | Output | Downstream Application |
|---|---|---|---|---|
| GetPileupSummaries | Tabulates pileup metrics at known germline sites | BAM/CRAM file; Common germline variants VCF | Pileup summary table (.table) | Input for CalculateContamination |
| CalculateContamination | Calculates fraction of reads from cross-sample contamination | Pileup summary table from GetPileupSummaries | Contamination table (.table) | Input for FilterMutectCalls |
Methodological Implementation: A Step-by-Step Technical Guide
Input Data Requirements and Preparation
The contamination estimation pipeline requires specific input data prepared according to GATK Best Practices for preprocessing:
Sequence Alignment Map (BAM/CRAM) Files: Analysis-ready BAM files must undergo proper processing including duplicate marking, base quality score recalibration, and alignment to a reference genome. The GATK Best Practices emphasize that all samples should be processed consistently using the same reference genome version, aligner, and duplicate marking approach to ensure co-analyzability [18] [63].
Germline Variant Resource: A sites-only VCF containing common biallelic SNPs with population allele frequencies is required. This resource should ideally be derived from large population databases like gnomAD and must contain only biallelic SNPs with AF annotations in the INFO field [62]. The tool ignores the filter status of variants in this resource.
Reference Genome: Consistent reference genome usage is critical, with recommendation for GRCh38 with defined patch versions. The Broad Institute's Functional Equivalence specification emphasizes that all samples must be aligned to the same reference genome down to the patch version to ensure compatibility in joint analyses [63].
Computational Execution Protocols
GetPileupSummaries Command Implementation
The standard implementation for generating pileup summaries from a tumor BAM file:
For matched-normal analyses, an additional pileup summary should be generated for the normal sample:
Table 2: Critical Parameters for GetPileupSummaries
| Parameter | Default Value | Function | Impact on Analysis |
|---|---|---|---|
--minimum-population-allele-frequency (--min-af) |
0.01 | Minimum population AF of sites to consider | Lower values increase accuracy at expense of speed [62] |
--maximum-population-allele-frequency (--max-af) |
0.2 | Maximum population AF of sites to consider | Filters out high-frequency SNPs [62] |
--min-mapping-quality (--mmq) |
50 | Minimum read mapping quality | Reduces inclusion of poorly mapped reads |
--intervals (-L) |
None | Genomic intervals to process | Enables targeted analysis of exomes or panels |
CalculateContamination Command Implementation
For tumor-only analysis mode:
For matched-normal analysis mode (recommended when available):
The tool generates an output table containing the estimated contamination fraction, with one line per sample following the format: SampleID<TAB>Contamination [59] [61]. The file contains no header.
Table 3: Critical Parameters for CalculateContamination
| Parameter | Default Value | Function | Impact on Analysis |
|---|---|---|---|
--matched-normal (-matched) |
None | Input table for matched normal | Enables more accurate estimation in paired samples [59] |
--tumor-segmentation (-segments) |
None | Output table for tumor segmentation | Provides minor allele fraction segmentation data [64] |
--low-coverage-ratio-threshold |
0.5 | Minimum coverage relative to median | Filters out low-coverage sites [59] |
--high-coverage-ratio-threshold |
3.0 | Maximum coverage relative to mean | Filters out high-coverage outliers [59] |
Workflow Integration and Visualization
The contamination estimation tools are embedded within the broader Mutect2 somatic variant calling workflow, which represents the GATK Best Practice for somatic short mutation discovery [60]. The complete workflow spans from raw data preparation to filtered variant calls, with contamination estimation occurring after initial variant calling but before final filtering.
Figure 1: Contamination Estimation Workflow Integration. This diagram illustrates the position of GetPileupSummaries and CalculateContamination within the broader GATK somatic variant calling pipeline, showing data flow from input BAM files through to contamination-aware variant filtering.
Technical Underpinnings: Mathematical and Computational Foundations
Algorithmic Principle: Reference Allele Infiltration at Homozygous Alternate Sites
The core mathematical principle underlying CalculateContamination is the analysis of reference allele infiltration at homozygous alternate variant sites. In a pure sample, homozygous alternate sites should exhibit nearly exclusive alternate alleles. The presence of reference alleles at these sites provides quantitative evidence of contamination [65]. The fundamental equation models the expected reference allele balance (ABref) at homozygous alternate sites as:
ABref = E(RR | homozygous alternate variant) = c × p
Where c represents the contamination fraction and p is the reference allele frequency in the population [65]. This relationship enables the estimation of contamination by measuring the deviation from expected allele balances. The method is conceptually similar to the CHARR (Contamination from Homozygous Alternate Reference Reads) metric, which estimates contamination from variant call data by leveraging the same biological principle [65].
Comparative Advantages Over Alternative Methods
The GATK contamination estimation approach provides specific advantages for cancer genomics applications:
CNV Tolerance: Unlike earlier methods like ContEst that assume diploid genotypes, CalculateContamination works reliably in regions with copy number variations, which are frequent in cancer genomes [59] [61].
Multiple Contaminant Support: The method accommodates contamination from multiple sources simultaneously without requiring independent contaminating reads [59].
Matched-Normal Flexibility: While supporting matched-normal analysis for improved accuracy, the tool performs robustly in tumor-only analyses, expanding its applicability to clinical contexts where normal tissue may be unavailable [59].
Performance evaluations of contamination detection tools have demonstrated that methods based on homozygous alternate site analysis, like Conpair, achieve superior performance for solid tumor NGS analysis [58]. The GATK approach shares this fundamental methodological strength while being specifically optimized for integration within the Mutect2 somatic calling workflow.
Table 4: Key Research Reagent Solutions for Contamination Estimation
| Reagent/Resource | Function | Technical Specifications | Source Recommendations |
|---|---|---|---|
| Germline Variant Database | Provides population-frequency-annotated SNPs for contamination detection | Biallelic SNPs only; AF field in INFO; Recommended AF range: 1-20% [62] | gnomAD; 1000 Genomes; Broad Institute resource bundles |
| Reference Genome | Standardized coordinate system for alignment and variant calling | Consistent versioning critical; GRCh38 recommended with defined patch levels [63] | GATK Resource Bundle; NCBI; ENSEMBL |
| Intervals File | Defides genomic regions for targeted sequencing analysis | BED format; exome capture regions; panel-specific coordinates | Manufacturer specifications; SureDesign; IDT |
| Java Environment | Execution platform for GATK tools | Java 8+; Adequate heap memory allocation (8G+ recommended) | OpenJDK; Oracle Java |
Discussion: Interpretation Guidelines and Integration with BAM Quality Requirements
Contamination Metric Interpretation
The contamination fraction generated by CalculateContamination represents the estimated proportion of reads in a sample derived from cross-sample contamination. In clinical cancer genomics, contamination levels exceeding 2-3% typically trigger quality concerns and may necessitate sample re-processing [58]. The analytical validation of the broader Mutect2 workflow demonstrates that contamination estimation directly impacts mutation calling sensitivity and specificity, particularly for subclonal mutations with low variant allele frequencies.
BAM File Quality Prerequisites
Effective contamination estimation requires that input BAM files meet specific quality thresholds established in the GATK Best Practices:
Mapping Quality: Minimum mapping quality scores (default 50 in GetPileupSummaries) ensure only reliably placed reads inform contamination metrics [62].
Duplicate Marking: Consistent duplicate marking across samples ensures comparable read counts and prevents technical artifacts from influencing contamination estimates [63].
Base Quality Recalibration: Properly calibrated base quality scores improve the accuracy of allele balance measurements at heterozygous and homozygous sites [18].
The GATK Best Practices emphasize that all samples in an analysis should be processed through the same computational pipeline with consistent reference genomes, aligners, and duplicate marking approaches to ensure functional equivalence and comparability of contamination metrics [63].
Alternative Approaches and Emerging Methodologies
While the GetPileupSummaries/CalculateContamination pipeline represents the GATK Best Practice, alternative approaches exist with complementary strengths. The CHARR method estimates contamination directly from variant call formats (VCF/gVCF) rather than BAM files, offering computational efficiency for large-scale cohort studies [65]. VerifyBamID and Conpair provide additional methodologies, with recent comprehensive evaluations showing Conpair achieves superior performance for contamination identification in solid tumor NGS analysis [58]. The selection of contamination estimation tools should consider experimental design, computational resources, and analytical requirements, with the GATK approach offering particular advantages for laboratories already implementing the broader Mutect2 somatic variant calling workflow.
Robust contamination estimation using GetPileupSummaries and CalculateContamination represents a non-negotiable quality control checkpoint in somatic variant calling workflows. By quantifying cross-sample contamination through systematic analysis of reference allele infiltration at homozygous alternate sites, this methodology addresses a critical source of technical variability in cancer genomics. The pipeline's integration within the broader Mutect2 framework and its specific optimization for copy number variant-rich cancer genomes make it particularly suited for clinical and research applications in oncology. Proper implementation requires careful attention to input BAM quality, germline resource selection, and parameter configuration, with the resulting contamination metrics directly informing variant filtering thresholds and ultimately ensuring the reliability of mutation discovery in cancer sequencing studies.
Correcting Orientation Artifacts in FFPE Samples with LearnReadOrientationModel
Formalin-Fixed Paraffin-Embedded (FFPE) samples are invaluable for cancer research, yet they introduce significant technical artifacts that challenge accurate somatic variant discovery. These specimens exhibit elevated rates of cytosine deamination and single-strand substitution errors that manifest as false positive variant calls, particularly C>T/G>A transitions, which can obscure true biological signals [66]. For researchers using GATK for somatic variant calling, these artifacts necessitate specialized processing to prevent contamination of results with technical false positives.
The orientation bias artifact presents a particularly insidious problem in FFPE data. During library preparation, DNA damage can occur before sequencing, resulting in substitution errors that appear predominantly on only one strand of DNA. Standard variant callers, including Mutect2, initially assume errors are random and evenly distributed across both strands. When this assumption is violated in FFPE samples, the caller may misinterpret clustered strand-specific errors as high-confidence variants [12] [67].
Integrating a specialized orientation bias filter into the somatic calling workflow is therefore essential for any research utilizing FFPE-derived BAM files. This technical guide details the implementation of GATK's LearnReadOrientationModel tool, framing it within the critical context of BAM file preparation requirements for robust somatic variant discovery.
Computational Framework for Artifact Correction
The Read Orientation Artifact Workflow
The complete orientation bias correction workflow integrates multiple specialized tools within the GATK ecosystem, each addressing a specific aspect of artifact identification and filtering. This systematic approach ensures that FFPE-specific errors are properly characterized and removed from the final variant call set.
Table: Core Tools in the Orientation Artifact Workflow
| Tool Name | Primary Function | Key Inputs | Key Outputs |
|---|---|---|---|
| Mutect2 | Initial variant calling & F1R2 count collection | Analysis-ready BAM, reference genome | Unfiltered VCF, F1R2 counts tar.gz |
| LearnReadOrientationModel | Learns artifact prior probabilities | F1R2 counts tar.gz | Artifact prior tables tar.gz |
| GetPileupSummaries | Summarizes read support at known sites | BAM, common variants resource | Pileup summary table |
| CalculateContamination | Estimates cross-sample contamination | Pileup summary table | Contamination table |
| FilterMutectCalls | Applies comprehensive filtering | Unfiltered VCF, artifact priors, contamination data | Filtered VCF |
The following diagram illustrates the sequential relationship and data flow between these tools:
Key Algorithmic Parameters and Configuration
LearnReadOrientationModel employs a sophisticated Expectation-Maximization (EM) algorithm to estimate the maximum likelihood of artifact prior probabilities for each trinucleotide context. The tool processes F1R2 count data generated by Mutect2 to learn the parameters of an orientation bias mixture model [67].
Table: Critical Parameters for LearnReadOrientationModel
| Parameter | Default Value | Function | Recommendation for FFPE |
|---|---|---|---|
--convergence-threshold |
1.0E-4 | EM algorithm stopping criterion | Default typically sufficient |
--num-em-iterations |
20 | Maximum EM iterations | Increase to 50 for complex samples [68] |
--max-depth |
200 | Sites with higher depth are grouped | Default appropriate for most panels |
--input (-I) |
Required | F1R2 tar.gz file(s) | Multiple inputs for scattered runs |
The mathematical foundation of the model calculates the probability that a variant is a true somatic mutation versus a strand-specific artifact. This Bayesian approach incorporates:
- Trinucleotide context of each potential variant
- Strand distribution of supporting reads (forward vs. reverse)
- Mixture model parameters learned via EM algorithm
- Prior probabilities of different artifact types
The output artifact prior probabilities are structured in a tar.gz archive containing multiple tables that quantify the likelihood of various orientation artifact patterns, which FilterMutectCalls subsequently uses to recalibrate variant probabilities [12] [67].
Experimental Protocols and Implementation
Generating Analysis-Ready BAM Files from FFPE Samples
The foundation of accurate somatic variant calling begins with proper BAM file preparation. FFPE samples require meticulous processing to minimize the impact of formalin-induced damage while preserving biological signals.
Sample Processing and DNA Extraction:
- Use tumor-rich areas with >70% tumor cellularity through macro-dissection [66]
- Extract DNA using specialized FFPE extraction kits (e.g., NextCODE SeqPlus, Qiagen QIAamp)
- Quantify DNA with fluorescence-based methods (Qubit) rather than spectrophotometry
- Assess DNA quality through fragment size distribution analysis
Library Preparation and Sequencing Considerations:
- Employ DNA repair enzymes when compatible with library prep protocol
- Incorporate unique molecular identifiers (UMIs) to distinguish true variants from artifacts
- Increase sequencing depth to compensate for reduced effective coverage due to artifacts
- Use paired-end sequencing to improve mapping accuracy of damaged DNA
Critical BAM Preprocessing Steps:
- Follow GATK Best Practices for data pre-processing: Map to Reference → Mark Duplicates → Base Quality Score Recalibration (BQSR) [9]
- For BQSR, include known sites from resources like 1000G, Mills indels, and dbSNP
- Validate BAM files using Picard's ValidateSamFile to ensure proper formatting [68]
- For targeted panels, ensure proper interval padding to capture overlapping reads
Executing the Orientation Artifact Workflow
Step 1: Mutect2 with F1R2 Collection
This command generates both initial variant calls and the F1R2 count data necessary for orientation bias modeling. The --f1r2-tar-gz parameter is essential for capturing strand-specific count information [12].
Step 2: LearnReadOrientationModel
For FFPE samples, increasing the number of EM iterations to 50 (from the default 20) helps ensure model convergence given the typically higher artifact burden [68].
Step 3: Contamination Analysis (Parallel Process)
This parallel pathway estimates cross-sample contamination, which is particularly important for FFPE samples that may have lower tumor purity [12] [8].
Step 4: Integrated Filtering with FilterMutectCalls
This final step integrates the learned orientation artifact priors with other filtering criteria to produce a high-confidence call set [12].
Advanced Implementation: Scattered Execution
For whole genomes or large targeted panels, scattered execution across genomic intervals significantly improves computational efficiency:
This approach requires merging F1R2 outputs from all intervals before learning the orientation model to ensure comprehensive artifact characterization across the entire genome [12].
The Scientist's Toolkit: Essential Research Reagents
Table: Critical Computational Resources for FFPE Somatic Analysis
| Resource Type | Specific Examples | Purpose in Workflow | Availability |
|---|---|---|---|
| Germline Resource | gnomAD (af-only-gnomad.vcf.gz) | Filters common germline variants | GATK Resource Bundle |
| Panel of Normals | CreateSomaticPanelOfNormals output | Filters technical artifacts in sequencing | Lab-generated |
| Common Variants Set | smallexaccommon_3.vcf | Contamination estimation | GATK Resource Bundle |
| Known Sites | 1000G, Mills, dbSNP | BQSR training | GATK Resource Bundle |
| Reference Genome | GRCh37, GRCh38 | Alignment and variant calling | GATK Resource Bundle |
Interpretation of Results and Quality Metrics
Evaluating Workflow Success
Successful implementation of the orientation artifact correction workflow yields distinct characteristics in the filtered variant set:
Variant Signature Shifts:
- Significant reduction in C>T/G>A transitions relative to other substitution types
- More balanced strand distribution of supporting reads for retained variants
- Elimination of variants with extreme strand bias (e.g., >90% support from one strand)
Filtering Metrics:
- Typical FFPE samples show 30-60% of raw calls filtered as orientation artifacts
- Higher filtering rates indicate more severe DNA damage or lower sample quality
- Unexpectedly low filtering rates may suggest workflow misconfiguration [68]
Troubleshooting Common Issues
Insufficient Filtering: If few variants are being filtered despite FFPE origin:
- Verify F1R2 file generation and proper input to LearnReadOrientationModel
- Confirm successful artifact prior file creation
- Validate that
--ob-priorsargument is correctly passed to FilterMutectCalls - Check for sample contamination that might mask true artifact patterns
Excessive Filtering: If an unexpectedly high proportion of variants are filtered:
- Assess potential overfitting by reducing
--num-em-iterations - Verify that normal tissue contamination isn't being misclassified as artifact
- Confirm adequate tumor purity (>20%) for reliable somatic signal detection
Model Convergence Issues:
- Increase
--num-em-iterationsto 50-100 for complex FFPE samples - Monitor convergence statistics in the tool output
- Ensure sufficient genomic coverage (>50X for panels, >30X for WES) for reliable model training
The LearnReadOrientationModel tool represents an essential component in the comprehensive somatic variant discovery pipeline for FFPE-derived samples. When properly integrated with rigorous BAM preprocessing and complementary filtering tools, it enables researchers to extract high-confidence biological variants from specimens affected by formalin-induced damage. The computational strategy outlined here—combining orientation bias correction with contamination assessment and panel-of-normals filtering—provides a robust framework for accurate somatic mutation calling in cancer research and drug development contexts.
The continued expansion of multi-omics approaches in cancer research, particularly those incorporating FFPE specimens [69] [70] [71], underscores the importance of reliable variant calling from suboptimal sample sources. By implementing the detailed protocols and quality control measures described in this guide, researchers can confidently utilize FFPE collections for retrospective studies, biomarker discovery, and validation of therapeutic targets.
Optimizing BAM Inputs for Extreme Depth Scenarios and Low Allele Fraction Detection
The detection of somatic mutations with low allele fractions represents a significant challenge in cancer genomics, circulating tumor DNA (ctDNA) analysis, and the study of tumor heterogeneity. Success in these areas depends critically on the quality and preparation of input BAM files, which serve as the foundation for all downstream variant calling processes. Even with advanced calling algorithms like GATK Mutect2, the maximum sensitivity and specificity for low-frequency variants can only be achieved with optimally prepared sequencing data. This technical guide examines BAM optimization strategies for extreme-depth sequencing scenarios (often exceeding 1,000X coverage) where traditional processing methods may prove inadequate. We focus specifically on the implications for somatic variant calling research within drug development contexts, where accurately identifying low-frequency variants can directly impact therapeutic target identification and treatment response monitoring.
The fundamental challenge stems from the fact that at very low allele fractions (typically below 1-5%), the signal from true biological variants approaches the level of technical artifacts introduced during sequencing and sample preparation. Without proper BAM-level optimization, these artifacts can generate false positives that obscure true signals or, conversely, cause true low-frequency variants to be lost in the noise. This guide provides a comprehensive framework for addressing these challenges through optimized BAM preparation, with specific protocols validated for extreme-depth scenarios.
Foundational Concepts: Depth, Allele Fractions, and BAM Quality Metrics
The Relationship Between Sequencing Depth and Detectable Allele Fractions
The minimum detectable allele fraction is fundamentally constrained by sequencing depth. At conventional sequencing depths (100-200X), variants below approximately 5% allele fraction become statistically challenging to distinguish from background errors. Extreme depth sequencing (1,000-20,000X) pushes this boundary lower, but introduces new analytical challenges related to increased sampling of technical artifacts [72].
Table 1: Theoretical Detection Limits at Various Sequencing Depths
| Sequencing Depth | Theoretical Minimum Detectable VAF | Primary Applications | Key Limitations |
|---|---|---|---|
| 100-200X | 5-10% | Conventional tumor sequencing, High-purity variants | Statistical power limitations |
| 500-1,000X | 1-2% | Liquid biopsies, Moderate heterogeneity | PCR duplicate resolution |
| 2,000-5,000X | 0.5-1% | ctDNA, Low tumor purity | Background error rate dominates |
| 10,000-20,000X | 0.1-0.5% | Ultra-sensitive monitoring, Mitochondrial DNA | Computational resources, BAM file sizes |
Recent research demonstrates that simply increasing sequencing depth yields diminishing returns without corresponding improvements in input DNA molecule diversity and error suppression. One systematic evaluation found that for mutation frequencies ≤10%, improving experimental methods provides greater benefits than further increasing depth beyond 500-800X [4]. The GATK Mutect2 tool has been specifically updated to accommodate extreme high depths, such as 20,000X, making proper BAM preparation even more critical [1].
Essential BAM Quality Metrics for Low Allele Fraction Studies
Not all high-depth BAM files are equally suitable for low allele fraction detection. Several quality metrics must be optimized to ensure variant calling accuracy:
- Duplicate read rates: While some duplication is expected in targeted sequencing, excessive rates (>20-30%) indicate insufficient input DNA or amplification bias, effectively capping the number of unique DNA molecules sampled.
- Base quality score distributions: Systematic biases in base quality scores must be corrected through recalibration to prevent miscalling of systematic errors as true variants.
- Mapping quality metrics: Poorly mapped reads contribute disproportionately to false positives in low allele fraction calling.
- Insert size distributions: Abnormal insert size patterns may indicate library preparation artifacts or mapping errors in complex genomic regions.
- Coverage uniformity: Extreme variability in coverage (>100-fold differences) creates gaps where low-frequency variants may be missed entirely.
These metrics should be monitored throughout BAM processing and considered prerequisites for attempting low-frequency variant detection.
BAM Pre-processing Optimization for Extreme Depth Scenarios
Modified Alignment and Duplicate Marking Strategies
Traditional BAM pre-processing pipelines require specific modifications to handle extreme-depth data effectively. The standard GATK Best Practices for data pre-processing provide a foundation [9], but several key adjustments are necessary for low allele fraction applications:
Alignment Considerations:
- Use BWA-MEM with optimized parameters for the specific sequencing technology employed
- Consider alternate mappers for complex genomic regions where standard aligners perform poorly
- Implement soft-clipping rather than hard-clipping to preserve information at read ends
Duplicate Marking: For extreme-depth data, the MarkDuplicatesSpark tool is recommended over conventional MarkDuplicates due to its parallel processing capabilities and significantly improved performance on large BAM files [9]. The Spark-based implementation can distribute the computationally intensive duplicate identification process across multiple cores or nodes, reducing processing time from days to hours for very deep sequencing datasets.
Figure 1: Optimized BAM Pre-processing Workflow for Extreme Depth Data
Advanced Base Quality Score Recalibration
Base Quality Score Recalibration (BQSR) becomes increasingly important in extreme-depth scenarios because it corrects for systematic technical errors that otherwise overwhelm true biological signals at low allele frequencies. The GATK BaseRecalibrator tool builds an empirical error model that identifies and corrects for:
- Sequencing machine cycle-specific errors
- Sequence context-specific errors (particularly GC-rich regions)
- Read group-specific technical artifacts
For extreme-depth data, BQSR should be performed using known variant sites from resources like dbSNP or the Genome in a Bottle consortium, with careful exclusion of suspected somatic regions to avoid incorporating true variants into the error model. The standard BQSR process may need to be scattered across genomic regions to handle the volume of data in extreme-depth BAM files, with subsequent gathering of the results before applying the recalibration [9].
Experimental Design for Low Allele Fraction Studies
Input DNA Requirements and Molecular Complexity
The molecular complexity of the starting DNA library fundamentally limits the detectability of low-frequency variants, regardless of sequencing depth. A library prepared from too few original DNA molecules will have inherent sampling limitations that cannot be overcome by increased sequencing.
Table 2: Research Reagent Solutions for Low Allele Fraction Studies
| Reagent/Tool | Function in Workflow | Impact on Low-Fraction Detection |
|---|---|---|
| Unique Molecular Identifiers (UMIs) | Tags individual DNA molecules before amplification | Enables distinction of PCR duplicates from unique fragments; essential for accurate allele fraction quantification |
| High-fidelity DNA polymerases | Amplification during library preparation | Reduces introduction of novel errors during PCR; maintains sequence fidelity |
| DNA repair enzymes | Pre-library preparation DNA treatment | Mitigates damage-related artifacts, particularly in FFPE samples |
| Methylation-aware enzymes | Targeted enrichment | Prevents bias against GC-rich regions that may harbor mutations |
| Molecular barcodes | Sample multiplexing | Allows sample pooling while maintaining individual sample integrity |
Research demonstrates that molecular barcoding (UMIs) provides particularly significant benefits for low allele fraction studies by enabling error-suppressed sequencing [73]. The smCounter algorithm, a barcode-aware variant caller, has shown the ability to detect somatic mutations with allele fractions as low as 1% in coding regions by leveraging UMI information to distinguish true variants from amplification and sequencing errors [73].
Technical Replication Strategies
For the most challenging low-frequency variant detection scenarios (below 0.5%), technical replication at the library level provides a powerful strategy for distinguishing true variants from artifacts. The RePlow method implements a probabilistic model that jointly analyzes library-level replicates to identify low-VAF somatic mutations by exploiting the random nature of technical errors across replicates [74].
Unlike simple intersection approaches (which sacrifice sensitivity) or BAM-merge approaches (which increase false positives), RePlow infers probability distributions of background errors in each replicate and uses this information to identify true variants that appear consistently across replicates while filtering out non-reproducible artifacts. This approach can reduce false positives by up to 99% while maintaining sensitivity for variants at frequencies as low as 0.5% [74].
Mutect2-Specific BAM Preparation Considerations
Panel of Normals (PON) Construction
The Mutect2 somatic caller achieves its highest specificity when provided with a comprehensive Panel of Normals (PON) that captures technical artifacts and common germline variants present in the sequencing process. For low allele fraction studies, PON construction requires specific considerations:
- Normal sample selection: Include normals processed using identical library preparation and sequencing protocols as the test samples
- Sample count: Larger PONs (40+ samples) provide better artifact representation
- Processing consistency: All normal samples should undergo identical processing from FASTQ to VCF
- Variant exclusion: The PON should focus on technical artifacts rather than germline variants, which are better handled by the germline resource [12]
The updated CreateSomaticPanelOfNormals workflow uses GenomicsDB for improved scalability when handling large normal sample sets [12]. This is particularly important for extreme-depth scenarios where VCF files can become prohibitively large.
Contamination Estimation and Correction
Cross-sample contamination poses a particularly significant threat to low allele fraction detection, as even low levels of contamination can introduce false positive calls or mask true low-frequency variants. The Mutect2 pipeline includes specialized tools for contamination estimation:
Figure 2: Contamination Estimation Workflow for Low-Fraction Variant Detection
Unlike other contamination tools, CalculateContamination is designed to work effectively without a matched normal sample and makes no assumptions about the number of contaminating samples, which is particularly valuable for detecting low-level contamination in extreme-depth data [12].
Benchmarking and Validation Strategies
Artificial Dataset Generation for Pipeline Validation
Validating performance for low allele fraction detection requires specialized benchmarking datasets where ground truth is known. Artificial datasets generated through in silico spike-in methods provide an effective solution:
NEAT-Based Data Generation:
- Generate artificial normal BAM files using the NEAT read simulator
- Simulate sequencer errors using empirically-derived error models
- Create a mutational background representative of normal samples [72]
Variant Spike-In with BAMSurgeon:
- Randomly generate somatic variants (SNVs and indels) across a desired VAF range
- Spike variants into normal BAM files using addsnv.py and add_indel.py scripts
- Validate successful spike-in rates and positional accuracy [72]
This approach enables creation of datasets with precisely known variants at specific allele fractions (e.g., 0.5%, 1%, 5%), allowing systematic evaluation of pipeline sensitivity and false positive rates. Studies using such artificial datasets have demonstrated that parameter tuning based on these benchmarks can considerably improve detection of very low-fraction variants [72].
Performance Expectations Across Allele Fractions
Systematic comparisons provide guidance on realistic performance expectations for low allele fraction detection. One comprehensive evaluation of Mutect2 and Strelka2 across different sequencing depths and mutation frequencies revealed several key patterns:
Table 3: Variant Calling Performance Across Allele Fractions and Depths
| VAF Range | Optimal Depth | Expected Recall | Expected Precision | Primary Challenges |
|---|---|---|---|---|
| 1% | 500-800X | 32-50% | 68-95% | Background errors dominate |
| 5-10% | 300-500X | 50-96% | 95-96% | PCR artifacts, mapping errors |
| ≥20% | 200X | >90% | >95% | Minimal special considerations |
This analysis suggests that for mutation frequencies ≤10%, improvements to experimental methods and analysis parameters provide greater benefits than simply increasing sequencing depth beyond 500-800X [4]. Additionally, Mutect2 demonstrates a slight performance advantage over Strelka2 at the lowest allele fractions (1%), though at the cost of significantly longer computation times [4].
Optimizing BAM inputs for extreme depth scenarios and low allele fraction detection requires an integrated approach spanning experimental design, computational processing, and analytical validation. No single optimization can ensure reliable detection of low-frequency variants; rather, success depends on the cumulative impact of multiple coordinated improvements:
First, experimental design must ensure sufficient molecular diversity through adequate input DNA, UMI incorporation, and technical replication where necessary. Second, computational processing must implement specialized alignment, duplicate marking, and base quality recalibration approaches scaled for extreme-depth data. Third, analytical refinement through Panel of Normals construction, contamination estimation, and orientation bias modeling addresses the specific artifact classes that dominate at low allele fractions.
When implemented as a comprehensive framework, these strategies enable reliable detection of somatic variants at frequencies as low as 0.5-1.0%, opening new possibilities for cancer research, liquid biopsy development, and therapeutic monitoring applications. As sequencing technologies continue to push toward even greater depths, these BAM optimization approaches will become increasingly essential for distinguishing true biological signals from technical artifacts at the frontier of detectable variation.
Validating BAM File Integrity and Contig Compatibility Before Analysis
Within the framework of GATK-based somatic variant discovery research, the integrity and compatibility of input BAM files constitute fundamental prerequisites for analytical success. The complex, multi-stage workflow for identifying somatic short variants (SNVs and indels) using tools like Mutect2 demands perfectly aligned input data to function correctly [8]. Errors pertaining to contig mismatches or file corruption represent a prevalent category of failure points that can compromise entire analyses, leading to computationally expensive rework and potentially erroneous biological conclusions. For researchers and drug development professionals, establishing robust validation protocols is not merely a technical formality but a critical component of reproducible cancer genomics research.
The GATK somatic short variant discovery workflow involves two primary steps: generating candidate somatic variants with Mutect2 and subsequent filtering to obtain a high-confidence callset [8]. This process depends on properly processed input BAMs where reads have been aligned to a specific reference genome build. When contigs (chromosomes and other genomic sequences) between the BAM file and reference genome disagree in name, order, or length, GATK tools fail with explicit errors that halt analysis [75]. This technical guide provides comprehensive methodologies for detecting and resolving these issues, ensuring BAM files meet the stringent requirements for somatic variant discovery pipelines.
Understanding Contig Compatibility Errors
Definition and Origins of Contig Mismatches
Contig compatibility errors occur when the sequence dictionaries of input files—particularly between BAM files and the reference genome—disagree in their descriptions of genomic contigs. The sequence dictionary is a critical metadata component that defines the names, lengths, and ordering of all contigs in a genomic file. Incompatibilities arise primarily from three scenarios: (1) using different reference genome builds between alignment and analysis (e.g., hg19 vs. hg38), (2) employing non-standard contig naming conventions, or (3) processing files with contigs in dissimilar sort orders [75] [76].
The GATK engine performs rigorous validations before initiating analysis, comparing contig definitions across all inputs relative to the reference. As explicitly stated in GATK documentation, "These errors occur when the names or sizes of contigs don't match between input files" [75]. This is particularly problematic in collaborative research environments where files may originate from different sources or alignment protocols. Without resolution, these errors prevent variant calling tools like Mutect2 from executing, as the engine cannot reliably map reads to reference positions when contig definitions disagree.
Common Error Manifestations and Interpretations
GATK provides specific error messages when contig incompatibilities are detected. Understanding these messages is crucial for efficient troubleshooting:
Incompatible contig lengths: The error message "Found contigs with the same name but different lengths" indicates that while contig names match, their defined lengths differ between files. A frequent example involves the mitochondrial contig (chrM) having length 16569 in a BAM file but 16571 in the reference [75]. This typically results from using different reference genome builds for alignment and analysis.
Lexicographically sorted contigs: The error "Lexicographically sorted human genome sequence detected in reads" occurs when contigs appear in alphabetical order (chr1, chr10, chr11, chr2...) in the BAM file but natural numerical order (chr1, chr2, chr3...chr10) in the reference dictionary [76]. This represents a sort order incompatibility rather than a fundamental reference mismatch.
Missing or extra contigs: Errors stating "BAM file(s) do not have the contig: chrM" indicate that certain contigs present in the reference are absent from the BAM file, or vice versa [75]. While missing contigs in the BAM may not always be problematic, extra contigs in the BAM will cause failures as GATK cannot process reads mapped to undefined genomic regions.
Table 1: Common Contig Compatibility Errors and Their Meanings
| Error Message | Root Cause | Typical Scenario |
|---|---|---|
| "Found contigs with the same name but different lengths" | Reference genome build mismatch | B37 vs. hg19 mitochondrial DNA length difference |
| "Lexicographically sorted human genome sequence detected" | Different contig sort orders | BAM with alphabetical order (chr1, chr10, chr11) vs. reference with numerical order |
| "BAM file(s) do not have the contig: [contig name]" | Missing contig in BAM file | Using different reference versions or stripped-down reference for alignment |
| "No overlapping contigs found" | Completely different contig naming schemes | Using NCBI-style contig names (NC_000001.11) vs. UCSC-style (chr1) |
Comprehensive BAM Validation Methodology
Pre-Validation Requirements and Experimental Setup
Before initiating BAM validation, researchers must establish a controlled environment with appropriate software and reference resources. The following components are essential for executing the validation protocols described in this section:
Table 2: Essential Research Reagents and Computational Tools for BAM Validation
| Tool/Resource | Function | Usage Notes |
|---|---|---|
| Samtools | Indexing, querying, and validating BAM/CRAM files | Essential for basic integrity checks and file inspection |
| Picard Tools | Sequence dictionary manipulation and file restructuring | Critical for resolving contig ordering issues |
| GATK | Variant discovery framework and validation utilities | Primary analysis platform requiring validated inputs |
| Reference Genome FASTA | Standardized genomic sequence for alignment | Must match the intended build and include companion .dict and .fai files |
| BWA-MEM | Read alignment to generate input BAMs | Should use the same reference as downstream GATK analysis |
For somatic variant calling research, the reference genome constitutes perhaps the most critical reagent. The entire analytical framework depends on consistency between the reference used for read alignment and that used for variant calling. Researchers should obtain complete reference packages, including the FASTA file, sequence dictionary (.dict), and index (.fai), from authoritative sources and maintain version consistency throughout the analysis pipeline [75].
Multi-Stage Validation Protocol
A comprehensive BAM validation protocol should progress from basic file integrity checks through increasingly specific compatibility assessments, culminating in reference-specific contig validation. The following workflow provides a systematic approach to identifying potential issues before they disrupt somatic variant discovery.
Figure 1: Systematic workflow for comprehensive BAM file validation before GATK analysis.
Basic File Integrity Assessment
The initial validation stage verifies fundamental file integrity and accessibility:
File corruption check: Use Samtools to quickly assess BAM file integrity:
This command provides a rapid assessment of basic BAM file validity without comprehensive processing.
Index validation: Ensure a proper BAM index exists and is compatible with the BAM file:
For genomes with contigs exceeding 512 million base pairs, a CSI index rather than a standard BAI index is required due to technical limitations [77].
Comprehensive validation with Picard:
This command checks for various structural issues, including invalid indexing bins, insert sizes, and version numbers that might indicate file corruption or compatibility issues [77].
Sequence Dictionary Inspection
The sequence dictionary embedded within BAM files contains critical metadata about contig names, lengths, and ordering. To extract and examine this information:
Extract sequence dictionary from BAM:
Extract reference sequence dictionary:
Compare contig names and lengths:
This comparison reveals discrepancies in contig names, lengths, or sort orders that must be resolved before proceeding with GATK analysis.
GATK Compatibility Testing
The most definitive validation involves testing the BAM file with the actual GATK tools intended for use in the research project. For somatic variant discovery, this can be accomplished through a trial execution on a restricted genomic interval:
A successful execution without contig compatibility errors indicates that the BAM file meets basic requirements for GATK somatic variant calling. This approach conserves computational resources by testing on a small genomic region while verifying the critical compatibility requirements.
Resolution Strategies for Common BAM/Contig Issues
Systematic Troubleshooting Approach
When validation reveals compatibility issues, researchers should implement targeted resolution strategies based on the specific nature of the problem. The following table summarizes common issues and their appropriate remediation approaches.
Table 3: Troubleshooting Guide for BAM File and Contig Compatibility Issues
| Problem Identified | Recommended Solution | Implementation Command | Considerations |
|---|---|---|---|
| Different reference builds | Switch to correct reference or realign | bwa mem -t 8 reference.fasta read1.fq read2.fq > aligned.sam |
File names can be misleading; verify build with checksums |
| Contig sort order mismatch | Reorder BAM to match reference | java -jar picard.jar ReorderSam I=original.bam O=reordered.bam R=reference.fasta CREATE_INDEX=TRUE |
Drops reads without equivalent contigs in new reference [76] |
| Lexicographic sort order | Use Picard to reorder contigs | java -jar picard.jar ReorderSam I=original.bam O=reordered.bam R=reference.fasta |
Common issue with human chromosome naming conventions [76] |
| Contig name mismatches | Liftover or rename contigs | java -jar picard.jar LiftoverVcf I=original.vcf O=lifted.vcf CHAIN=chain.file R=reference.fasta |
For VCF files only; BAM files require realignment [75] |
| Large contigs (>512Mb) | Use CSI indexing instead of BAI | samtools index -c large_contig.bam |
Required for proper indexing of large chromosomes [77] |
| Outdated tool versions | Update to current versions | Upgrade Picard to 2.27.5+ or 3.0.0+ | Older versions may lack support for modern file formats [77] |
Special Considerations for Human Genome Builds
Researchers working with human genomic data frequently encounter compatibility issues between the b37 and hg19 reference builds. While these builds are very similar for canonical chromosomes (1-22, X, Y), they differ in contig naming conventions (without prefix vs. "chr" prefix) and mitochondrial DNA length [75]. The GATK documentation explicitly warns that "The b37 and hg19 human genome builds are very similar, and the canonical chromosomes only differ by their names... but the mitochondrial contig is a slightly different length in addition to having a different naming convention" [75].
For researchers who must work with files aligned to different builds, the following approaches are available:
Complete realignment (recommended): The most reliable approach is to revert BAM files to unaligned format (using Picard's RevertSam) and realign to the correct reference. This ensures complete consistency but requires access to original sequencing data and substantial computational resources.
Contig renaming (with caution): For cases where only canonical chromosomes are analyzed and mitochondrial DNA is not relevant, carefully renaming contigs throughout mismatching files may provide a viable solution. This approach requires extreme caution as "many things can go wrong during the editing process that can screw up your files even more" [75].
Liftover for VCF files: For variant files rather than BAM files, Picard's LiftoverVCF tool can transfer variants between coordinate systems using chain files. The GATK resource bundle provides chain files for lifting over between major human reference builds [75].
Integration with Somatic Variant Discovery Workflows
Positioning Validation within the GATK Somatic Pipeline
Proper BAM validation is not an isolated activity but rather a critical prerequisite integrated within the broader somatic variant discovery workflow. The GATK best practices workflow for somatic short variant discovery involves multiple sophisticated tools including Mutect2 for initial variant calling, GetPileupSummaries and CalculateContamination for contamination estimation, LearnReadOrientationModel for identifying artifacts, and FilterMutectCalls for final variant filtering [8]. Each of these tools depends on properly validated and compatible BAM files.
When BAM files pass comprehensive validation, researchers can proceed confidently through the primary somatic calling steps:
Initial variant calling with Mutect2:
Contamination assessment:
Variant filtering:
Throughout this workflow, properly validated BAM files ensure that each tool functions as designed, producing accurate and reproducible somatic variant calls suitable for downstream biological interpretation and potential clinical applications.
Quality Assurance in Drug Development Context
For pharmaceutical researchers and clinical scientists, BAM validation extends beyond technical compatibility to encompass quality metrics relevant to regulatory considerations. In drug development contexts, particularly for precision oncology therapeutics, somatic variant calls may inform patient stratification, target selection, and companion diagnostic development. In these applications, documentation of BAM validation procedures, reference genome versions, and compatibility checks becomes part of the method validation record.
Researchers should maintain comprehensive records of:
- Reference genome source and version identifiers
- BAM validation reports from Picard ValidateSamFile
- Contig compatibility verification
- Software versions used throughout the analytical pipeline
These records support the reproducibility and auditability required in regulated research environments and provide crucial methodological context for interpreting variant calls in biological and clinical contexts.
Thorough validation of BAM file integrity and contig compatibility represents an essential foundation for successful somatic variant discovery research using GATK. By implementing the systematic validation protocols outlined in this guide—encompassing basic file integrity checks, sequence dictionary verification, and comprehensive compatibility testing—researchers can prevent analytical failures and ensure the reliability of their variant calls. The resolution strategies presented for common contig compatibility issues provide practical pathways for remediating problematic files, while integration of these validation steps within the broader somatic calling workflow ensures end-to-end analytical quality. For the research and drug development community, these practices establish the rigorous technical standards necessary for advancing precision oncology through genomic discovery.
Benchmarking Somatic Calling Performance and Validating BAM Quality
Utilizing Gold Standard Datasets for Pipeline Validation (GIAB)
The Genome in a Bottle (GIAB) Consortium, hosted by the National Institute of Standards and Technology (NIST), develops reference standards and data to enable the translation of whole human genome sequencing into clinical practice [78]. For researchers conducting somatic short variant discovery using tools like GATK Mutect2, GIAB provides benchmarked human genomes that serve as an indispensable ground truth for analytical validation. These resources allow scientists to assess the performance of their entire bioinformatics pipeline, from the initial input BAM (Binary Alignment Map) files to the final variant calls, ensuring the accuracy and reliability of results in critical drug development research [79] [80].
The use of GIab samples is particularly crucial for somatic variant calling, which identifies post-zygotic mutations often present at low variant allele fractions. These variants are challenging to distinguish from sequencing and alignment artifacts. By leveraging GIAB benchmarks, researchers can quantify key performance metrics such as sensitivity (recall), precision, and false discovery rates specific to their BAM preprocessing and variant calling workflow [80]. This guide details the experimental and bioinformatic protocols for using GIAB resources to rigorously validate somatic research pipelines, with a specific focus on the input BAM file requirements that form the foundation of accurate analysis.
Reference Samples and Benchmark Variant Sets
GIAB has characterized a set of widely used reference genomes. The Ashkenazi Jewish son-father-mother trio (HG002, HG003, HG004) is a primary resource for developing somatic benchmarks because a mosaic variant in the son (HG002) can be identified as a variant not present in the parental genomes [78] [80]. The consortium provides high-confidence benchmark variant calls for these samples, which have been generated using multiple sequencing technologies and bioinformatics methods to create a comprehensive set of truth variants.
- Small Variants: GIAB provides benchmark sets for single nucleotide variants (SNVs) and small insertions and deletions (indels) on common reference genomes like GRCh37 and GRCh38. The v4.2.1 benchmark includes more challenging genomic regions than previous versions [78] [79].
- Structural Variants (SVs): Benchmark sets for structural variants are available for HG002 on GRCh37 and are under active development for GRCh38 [78].
- Mosaic Variants: A dedicated mosaic benchmark for HG002 (v1.1) is a key resource for somatic pipeline validation. This benchmark was created using a trio-based approach with Strelka2 and orthogonal validation with multiple sequencing technologies (e.g., BGI, Element, Pacbio HiFi) to identify true, low-frequency mosaic variants [80].
Genomic Stratifications for Context-Aware Benchmarking
A critical insight in genomics is that no sequencing technology or bioinformatics pipeline performs uniformly across all genomic contexts. To address this, GIAB provides genomic stratifications—BED files that partition the genome into distinct categories based on technical challenge or functional annotation [79]. These stratifications allow researchers to move beyond genome-wide performance metrics and understand their pipeline's strengths and weaknesses in specific regions.
Table 1: Key GIAB Genomic Stratifications for Performance Analysis
| Stratification Type | Description | Utility for Somatic Pipeline Validation |
|---|---|---|
| Coding Sequences (CDS) | Regions of the genome that code for proteins. | Assess variant calling accuracy in clinically critical exonic regions. |
| Low Mappability Regions | Regions where short reads cannot be uniquely mapped. | Evaluate false positive rates in repetitive areas where alignment is ambiguous. |
| High/Low GC Content | Regions with unusually high or low GC nucleotide composition. | Identify sequencing or alignment biases related to base composition. |
| Segmental Duplications | Large, nearly identical duplicated genomic regions. | Pinpoint challenges in variant calling within complex, duplicated sequences. |
| Homopolymer Regions | Tracts of identical consecutive nucleotides. | Assess indel calling errors common in these contexts for some technologies. |
| Tandem Repeats | Regions of short, repeated DNA sequences. | Evaluate performance in loci associated with many diseases. |
Using these stratifications, a researcher can determine, for example, if their pipeline's elevated false-positive rate is concentrated in low-mappability regions or if sensitivity for indel detection drops significantly in homopolymers [79]. This granular analysis is essential for optimizing pipeline parameters and for understanding the limitations of a study's findings.
Experimental Protocol for Somatic Pipeline Validation
Validating a somatic variant calling pipeline against GIAB benchmarks involves a series of methodical steps, from data acquisition to stratified performance analysis. The following protocol outlines a comprehensive validation experiment.
Data Acquisition and BAM File Preparation
The first step is to acquire the raw sequencing data for the GIAB samples and process it through your pipeline to create input BAMs for the variant caller.
- Download Sequencing Data: Obtain high-coverage whole-genome sequencing data for the HG002 trio (and other GIAB samples) from the GIAB FTP site or associated NCBI Bioproject (e.g., PRJNA200694) [78] [80]. The HG002 mosaic benchmark study, for instance, utilized Illumina 300x WGS for the initial callset and orthogonal data from BGI, Element, and PacBio for validation [80].
- BAM File Generation:
- Read Alignment: Use your standard alignment tool (e.g., BWA-MEM, DRAGEN) to align the downloaded FASTQ files to the same reference genome used by your GIAB benchmarks (GRCh37 or GRCh38) [80].
- Post-Alignment Processing: Perform the necessary post-processing steps on the generated SAM/BAM files as required by your somatic caller and best practices. This typically includes:
- Sorting by genomic coordinate.
- Duplicate Marking to flag PCR artifacts.
- Base Quality Score Recalibration (BQSR).
- Quality Control: Generate QC metrics (e.g., coverage, insert size, mapping quality) for the final BAM files to ensure they meet the quality thresholds of your pipeline.
Variant Calling and Benchmarking with GIAB Truth Sets
With the processed BAM files ready, the next step is to run the somatic variant caller and compare its outputs to the GIAB truth set.
- Somatic Variant Calling: Execute your somatic variant calling workflow (e.g., GATK Mutect2 in tumor-only mode) using the processed HG002 BAM file as the "tumor" input [8] [80]. If using a tumor-normal mode, a mixture of the parental BAMs (HG003 and HG004) can be used to create a "normal" sample.
- Performance Assessment:
- Download Truth Data: Acquire the relevant GIAB benchmark VCF and BED files. For somatic validation, the HG002 mosaic benchmark is the primary resource [80].
- Benchmarking Tool: Use a specialized benchmarking tool like
hap.py(https://github.com/Illumina/hap.py) ortruvarito compare your pipeline's output VCF against the GIAB truth VCF [79]. - Stratified Analysis: Run the benchmarking tool using the GIAB genomic stratification BED files. This generates performance metrics not just genome-wide, but also within each challenging genomic context defined in Table 1 [79].
Analysis of Validation Metrics
The final step is to interpret the output of the benchmarking tool to assess pipeline performance.
Table 2: Key Validation Metrics and Their Interpretation
| Metric | Calculation | Interpretation in Somatic Validation |
|---|---|---|
| Sensitivity (Recall) | TP / (TP + FN) | The pipeline's ability to detect true mosaic/somatic variants present in the benchmark. Low sensitivity indicates a high false-negative rate. |
| Precision | TP / (TP + FP) | The pipeline's ability to avoid false positives. Low precision indicates that many called variants are not in the truth set. |
| F-measure / F1 Score | 2 * (Precision * Recall) / (Precision + Recall) | The harmonic mean of precision and recall, providing a single metric for balanced performance. |
| False Discovery Rate (FDR) | FP / (TP + FP) | The complement of Precision (FDR = 1 - Precision). Directly quantifies the fraction of false positives among all positive calls. |
The results should be analyzed holistically across all stratifications. A pipeline may have excellent genome-wide precision but unacceptable precision in low-mappability regions. This detailed analysis informs whether the pipeline is fit for its intended purpose and guides further optimization.
Somatic Pipeline Validation Workflow
Successful validation relies on a set of key resources, from biological materials to software tools.
Table 3: Essential Research Reagents and Resources for GIAB Validation
| Resource Category | Specific Example(s) | Function in Validation Protocol |
|---|---|---|
| Reference DNA | HG002 (GM24385), HG003, HG004 from Coriell Institute [78] | Provides the source of standardized, well-characterized genomic material for sequencing. |
| Benchmark Variant Sets | GIAB Small Variant (v4.2.1), Mosaic Benchmark (v1.1) for HG002 [78] [80] | Serves as the "answer key" against which a pipeline's variant calls are compared. |
| Genomic Stratifications | GIAB Stratification BED files for GRCh37/GRCh38 (e.g., Low Mappability, CDS) [79] | Enables context-specific performance analysis to identify pipeline weaknesses. |
| Benchmarking Software | hap.py, truvari, vcfeval [79] | Specialized tools to compare a test VCF to a truth VCF and calculate performance metrics. |
| Somatic Variant Caller | GATK Mutect2, Strelka2 [8] [80] | The software under validation that identifies somatic variants from BAM files. |
| Reference Genome | GRCh37, GRCh38 (preferably with decoys and ALT-aware handling) [78] | The reference sequence to which reads are aligned; must match the benchmark's reference. |
Critical BAM File Requirements for GATK Somatic Calling
The quality of the input BAM file is the most critical factor influencing the success of somatic variant discovery. The GATK best practices for data pre-processing are a prerequisite for generating BAMs suitable for Mutect2 [8]. Beyond these general guidelines, specific attention must be paid to the following:
- High and Uniform Coverage: Somatic variants, especially subclonal ones, can exist at very low allele fractions. High coverage (≥100x) is required to have multiple reads supporting a low-frequency variant, enabling its detection above the noise floor. Uniform coverage ensures that the sensitivity is consistent across the genome and that there are not large regions with poor performance [80].
- Minimal Cross-Sample Contamination: The GATK workflow includes tools like
GetPileupSummariesandCalculateContaminationto estimate and account for contamination. However, high levels of contamination can severely impact the accuracy of low variant allele fraction (VAF) calls. The input BAM should be generated from wet-lab protocols that minimize the potential for sample swaps or carryover [8]. - Accurate Alignment in Complex Regions: As highlighted by the GIAB stratifications, a significant source of false positives is the misalignment of reads in low-mappability regions, such as segmental duplications and homopolymers. Using ALT-aware alignment is crucial for GRCh38 to properly place reads from regions that have high homology elsewhere in the genome [79]. Visual inspection of BAM files in a tool like IGV at called variant sites, especially in these difficult regions, is a recommended practice to confirm alignment accuracy [81].
Key BAM Requirements for Somatic Calling
Utilizing GIAB's gold standard datasets for pipeline validation provides a rigorous, metrics-driven framework for assessing the performance of somatic variant discovery workflows. By following the outlined experimental protocol—leveraging benchmark variant sets, genomic stratifications, and standardized benchmarking tools—researchers and drug developers can objectively quantify the sensitivity and precision of their pipelines. This process pays particular attention to the quality of the input BAM file, which is the foundational element determining success. Ultimately, integrating GIAB validation into research practice ensures that somatic variant calls, especially those at low allele frequencies, are accurate and reliable, thereby strengthening the conclusions drawn in preclinical and clinical genomics studies.
The accurate detection of somatic mutations is a cornerstone of cancer genomics, enabling insights into tumorigenesis, progression, and targeted treatment strategies [4] [82]. Next-generation sequencing (NGS), particularly whole-exome sequencing (WES), has become the technology of choice for this task, yet the analytical challenge of distinguishing true somatic variants from sequencing artifacts and germline polymorphisms remains substantial [82]. The choice of somatic variant calling software significantly influences downstream analysis and clinical interpretation, making performance comparisons critical for the research community.
This whitepaper provides an in-depth technical comparison of three widely used somatic variant callers: Mutect2 (from the Genome Analysis Toolkit, GATK), Strelka2, and VarScan2. Framed within the context of input BAM requirements for GATK somatic calling research, we synthesize evidence from multiple benchmarking studies to evaluate these tools based on accuracy, sensitivity to low-frequency variants, performance across different sequencing depths, and computational efficiency. The findings aim to guide researchers, scientists, and drug development professionals in selecting and configuring optimal variant detection pipelines for their specific experimental designs.
The comparative analysis reveals that no single caller universally outperforms others across all metrics. Performance is highly dependent on specific experimental conditions, particularly mutation frequency and sequencing depth.
- At high mutation frequencies (≥20%): Mutect2 and Strelka2 perform similarly with high precision (>95%) and recall, making both excellent choices for high-purity samples [4].
- At low mutation frequencies (≤10%): VarScan2 and Mutect2 show advantages in sensitivity, with VarScan2 sometimes surpassing Mutect2 at frequencies above 40%, while Mutect2 is more consistent across a wider range of low frequencies [83] [84].
- Computational Efficiency: Strelka2 demonstrates significantly faster processing times (17-22 times faster than Mutect2 on average), followed by Mutect2, with VarScan2 being considerably slower [4] [83].
- Ensemble Approaches: Combining multiple callers has been shown to outperform individual tools, with specific combinations achieving F1 score improvements of over 3.5% for both SNVs and indels [82].
Table 1: Overall Performance Summary of Somatic Variant Callers
| Caller | Best Use Case | Low VAF Sensitivity | Computational Speed | Key Strength |
|---|---|---|---|---|
| Mutect2 | General-purpose somatic calling; Tumor-Normal pairs | Good, especially at VAF <20% | Moderate | Robust Bayesian model; excellent precision; active development [1] [8] |
| Strelka2 | High-purity tumors; rapid analysis | Lower at VAF ≤10% | Very Fast | Speed; high precision at high VAF [4] [83] |
| VarScan2 | Detecting low-frequency variants | Good at VAF <5% | Slow | Sensitivity to low-frequency mutations [83] [84] |
Detailed Performance Analysis
Impact of Mutation Frequency and Sequencing Depth
The variant allele frequency (VAF) of somatic mutations and the sequencing depth are critical factors influencing caller performance. Benchmarking using standard DNA samples (NA12878 and YH-1) with simulated sequencing depths from 100X to 800X and mutation frequencies from 1% to 40% provides quantitative insights [4] [83].
- High Mutation Frequency (≥20%): For mutations at 20% frequency or higher, a sequencing depth of ≥200X is sufficient to call >95% of mutations with both Mutect2 and Strelka2. Their performance is nearly identical, with F-scores ranging between 0.94 and 0.965 [4].
- Low Mutation Frequency (≤10%): Performance degrades significantly for all callers at low frequencies, but to different extents. At 1% mutation frequency, the recall rate for Mutect2 and Strelka2 is low (2.7-34.5% across all depths). One study notes that for very low-frequency mutations (≤5%), improving the experimental method (e.g., using UMIs) is more effective than simply increasing sequencing depth beyond 200X [4]. In this regime, VarScan2 and VarDictJava demonstrate superior recall compared to Mutect2 and Strelka2 [83].
Table 2: Performance vs. Mutation Frequency and Sequencing Depth (Based on [4])
| Mutation Frequency | Sequencing Depth | Mutect2 Recall | Strelka2 Recall | Mutect2 Precision | Strelka2 Precision | Recommended Caller |
|---|---|---|---|---|---|---|
| 1% | 100X-300X | < 19% | < 19% | > 95% | > 95% | Mutect2 has slight F-score advantage |
| 1% | 500X-800X | 32-50% | 27-37% | > 95% | > 95% | Mutect2 F-score surpasses Strelka2 |
| 5-10% | ≥200X | 50-96% | 48-93% | ~95.9% | ~96.5% | Mutect2 (higher recall), Strelka2 (higher precision) |
| ≥20% | ≥200X | >90% | >90% | >95% | >95% | Mutect2 or Strelka2 |
Performance on Formalin-Fixed Paraffin-Embedded (FFPE) Samples
FFPE samples, common in clinical practice, present unique challenges due to DNA degradation and formalin-induced artifacts. A study comparing variant calling on matched FF and FFPE WGS samples from a metastatic prostate tumor found that no single caller perfectly recapitulated the variants from the Fresh Frozen (FF) sample in the FFPE sample [66]. On average, only about 50% of the calls in the FF sample were recovered in the FFPE sample across the callers tested (Mutect2, Strelka2, VarScan2, and Shimmer). This highlights the inherent limitations of FFPE material. However, the study developed a heuristic that combined variants from multiple callers, which increased both precision and sensitivity, outperforming any single caller [66]. This underscores the value of an ensemble approach for difficult sample types like FFPE.
Computational Performance and Resource Requirements
The computational efficiency of a variant caller is a practical concern, especially in large-scale cohort studies or clinical settings with rapid turnaround requirements.
- Strelka2 is consistently reported as the fastest among the three. It was 17 to 22 times faster than Mutect2 on average in one benchmark and about 20 times faster than VarScan2, Mutect2, and VarDictJava in another [4] [83].
- Mutect2 has moderate computational demands. Its runtime is significantly longer than Strelka2 but can be considerably shorter than VarScan2's [83].
- VarScan2 is generally characterized as one of the slower tools among commonly used somatic variant callers [83].
Experimental Protocols and Methodologies
Understanding the methodologies behind performance benchmarks is crucial for interpreting results and designing robust somatic variant detection workflows.
Benchmarking Using Cell Line Mixtures
A common and robust approach for generating ground truth data involves using well-characterized human cell lines.
- Source: This method was used in a systematic comparison of somatic variant calling performance [4].
- Sample Preparation: Whole-exome sequencing is performed on two standard DNA samples (e.g., NA12878 and YH-1) to high depth (>400X). The BAM file of one sample (e.g., NA12878) is down-sampled to serve as the "normal" control. The other sample (YH-1) is treated as the "tumor" and mixed with NA12878 at different percentages (1% to 40%) to simulate varying mutation frequencies [4].
- Data Simulation: The mixed BAM files are further down-sampled to generate datasets with different sequencing depths (e.g., 100X, 200X, 300X, 500X, 800X). Multiple replicates are generated for each depth-frequency combination to account for random sampling effects [4].
- True Set Definition: Only genomic sites with completely different homozygous genotypes between the two original cell lines are included in the true positive set. This allows the mixing percentage of the YH-1 sample to be directly interpreted as the somatic mutation frequency [4].
- Analysis: All simulated tumor-normal BAM pairs are processed through the variant callers (e.g., Mutect2, Strelka2). The outputs are compared against the true set to calculate precision, recall, and F-score.
The following diagram illustrates this rigorous experimental workflow:
The GATK Somatic Short Variant Discovery Workflow
The Broad Institute provides a best-practice workflow for somatic short variant discovery using Mutect2. This workflow is designed to move from raw sequencing data to a confident set of annotated somatic variants and is critical for researchers using Mutect2 in their pipelines [8].
- 1. Call Candidate Variants (Mutect2): The first step involves running Mutect2 on the tumor and normal BAM files. Mutect2 performs local assembly of haplotypes in active regions and applies a Bayesian somatic genotyping model to calculate the likelihood of a site being a somatic variant versus a sequencing error [8] [2].
- 2. Calculate Contamination: The tools
GetPileupSummariesandCalculateContaminationare used to estimate the fraction of cross-sample contamination in the tumor sample. This is crucial for accurate filtering, even in samples with copy number variations [8]. - 3. Learn Orientation Bias Artifacts: For samples prone to artifacts like FFPE specimens,
LearnReadOrientationModeluses data generated by Mutect2 to model and account for single-stranded substitution errors [8]. - 4. Filter Variants (FilterMutectCalls): This step is essential.
FilterMutectCallsuses probabilistic models to account for correlated errors, alignment artifacts, strand bias, germline variants, and contamination. It automatically sets a threshold to optimize the F-score [8]. - 5. Annotate Variants (Funcotator): Finally, the filtered variants are annotated with functional information (e.g., affected gene, protein change) using databases like GENCODE, dbSNP, gnomAD, and COSMIC, outputting a VCF or MAF file [8].
This multi-step process emphasizes that using Mutect2 effectively requires more than just the variant calling step; the subsequent filtering and annotation are integral to achieving high-quality results. The workflow is depicted below:
The Scientist's Toolkit: Key Research Reagents & Materials
The following table details essential resources and data used in benchmarking and applying somatic variant callers, as featured in the cited research.
Table 3: Essential Research Reagents and Resources
| Item / Resource | Type | Function in Somatic Variant Research | Example / Source |
|---|---|---|---|
| Reference Cell Lines | Biological Sample | Provides genetically defined, reproducible source of DNA for creating benchmarking datasets with known "true" variants. | NA12878, YH-1, HCC1395, HCC1143 [4] [82] |
| Germline Resource | Data File (VCF) | Provides population allele frequencies to help Mutect2 distinguish rare germline polymorphisms from true somatic mutations. | gnomAD (af-only-gnomad.vcf.gz) [1] [8] [85] |
| Panel of Normals (PoN) | Data File (VCF) | A panel of VCFs from normal samples used to identify and filter out recurring technical artifacts present in the sequencing process. | Provided in GATK best practices or created from normal BAMs [1] [8] [85] |
| Benchmarking Datasets | Data Set | Publicly available datasets with validated mutation sets for standardized tool evaluation and pipeline validation. | ICGC-TCGA DREAM Challenge, SEQC2 Consortium [82] |
| Data Simulation Tools | Software | Generates artificial sequencing data with known mutations for controlled benchmarking, especially for low-frequency variants. | NEAT, BAMSurgeon [86] |
Based on the synthesized evidence from multiple independent studies, the following recommendations are provided for researchers designing somatic variant calling pipelines:
- For Standard Tumor-Normal Pairs with High Purity: Either Mutect2 or Strelka2 is a strong choice, as they offer high precision and comparable performance at mutation frequencies ≥20% and depths ≥200X [4]. Strelka2's speed advantage makes it attractive for large-scale studies.
- For Studies Focusing on Low-Frequency Variants or Heterogeneous Tumors: Mutect2 is recommended due to its robust performance across a range of lower VAFs. VarScan2 also shows high sensitivity in this context but requires careful consideration of its computational cost [83] [84].
- For Challenging Sample Types like FFPE: Relying on a single caller is not optimal. An ensemble approach that combines the outputs of multiple callers (e.g., Mutect2, Strelka2, and VarScan2) has been proven to maximize both sensitivity and precision [82] [66].
- For Clinical or Diagnostic Settings: The comprehensive, multi-step filtering and annotation workflow of Mutect2, as outlined in the GATK best practices, provides a robust and well-documented framework for generating high-confidence calls [8].
In conclusion, the selection of a somatic variant caller should be guided by the specific biological context, sample quality, and analytical requirements. Researchers are encouraged to validate their chosen pipeline using benchmark datasets that reflect their own experimental conditions, such as those involving low tumor fraction or specific sample preservation methods [86].
Impact of Sequencing Depth and Mutation Frequency on Call Accuracy
The accurate detection of somatic mutations is a cornerstone of cancer genomics, with profound implications for understanding tumor biology, identifying therapeutic targets, and guiding clinical treatment decisions. This process critically depends on the quality of input sequencing data, particularly the alignment files in BAM format that serve as the primary input for variant callers like GATK Mutect2. Within the context of a broader thesis on BAM file requirements for GATK somatic calling research, two technical parameters emerge as fundamental determinants of variant calling accuracy: sequencing depth and mutation frequency.
Sequencing depth, defined as the average number of times each base in the genome is read, directly influences the statistical confidence in distinguishing true somatic mutations from sequencing errors and technical artifacts. Meanwhile, mutation frequency, representing the proportion of DNA molecules carrying a variant allele in a heterogeneous sample, dictates the detectability of somatic events, particularly in samples with low tumor purity or substantial subclonality. The interplay between these factors creates complex trade-offs that researchers must navigate when designing genomic studies, especially in clinical and drug development contexts where accuracy directly impacts patient care and research validity.
This technical guide synthesizes empirical evidence from systematic benchmarking studies to establish clear, evidence-based recommendations for optimizing sequencing depth and interpreting results across the mutation frequency spectrum, with particular emphasis on their implications for BAM file quality requirements in GATK-based somatic variant calling pipelines.
Quantitative Relationships: How Depth and Frequency Affect Accuracy
The Sequencing Depth-Accuracy Relationship
Sequencing depth substantially impacts the power to detect somatic variants while controlling false positives. Systematic evaluation using whole-exome sequencing data reveals distinct patterns for single nucleotide variants (SNVs) versus insertions and deletions (indels), with the latter requiring greater depth for reliable detection [87].
Table 1: Variant Calling Accuracy Metrics at Different Sequencing Depths
| Sequencing Depth | SNV Concordance Rate | Indel Concordance Rate | Precision Rate | Recommended Application Context |
|---|---|---|---|---|
| 5× | <90% | ~60% | ~95% | Population studies with joint calling |
| 10× | >95% | ~70% | >95% | Cost-effective WGS for SNV detection |
| 15× | >99% | ~75% | >95% | Balanced design for SNV-focused studies |
| 30× | >99.5% | ~80% | >95% | Clinical SNV detection |
| 50× | >99.7% | ~85% | ~95% | Improved indel detection |
| 100× | >99.8% | ~90% | 93-95% | Research-grade indel detection |
| 200×+ | >99.9% | >90% | 90-95% | Ultra-sensitive detection for low-frequency variants |
Empirical evidence demonstrates that SNV calling accuracy plateaus at approximately 15-20× depth, achieving >99% concordance with reference datasets [87]. In contrast, indel detection continues to improve with increasing depth, with only 60% of indels detected at 20× depth compared to those identified in ultra-deep sequencing data (∼400×) [87]. This fundamental difference arises from the more complex alignment challenges inherent to indel detection, which benefit disproportionately from additional read support.
For whole-genome sequencing in larger genomes including pigs, a depth of 10× provides >99% genome coverage while effectively controlling false-positive variants, which increase dramatically at depths below 4× [88]. Multisample calling strategies can partially compensate for lower depth by leveraging information across samples, demonstrating 13-fold and 2-fold higher variant discovery power compared to single-sample calling at 1× and 22× depths, respectively, though with increased false-positive rates that require careful filtering [88].
The Mutation Frequency-Accuracy Relationship
Mutation frequency, particularly in the context of tumor heterogeneity and subclonal populations, represents a critical determinant of detection sensitivity. Benchmarking analyses reveal stark differences in detection performance across the mutation frequency spectrum.
Table 2: Detection Performance Across Mutation Frequencies with Optimal Depth
| Mutation Frequency | Recall Rate | Precision Rate | F-score | Minimum Recommended Depth |
|---|---|---|---|---|
| 1% | 2.7-34.5% | 68.9-100% | 0.05-0.51 | 500×+ |
| 5% | 48-93% | >95% | 0.63-0.95 | 100× |
| 10% | 50-96% | >95% | 0.65-0.95 | 50× |
| 20% | 92-97% | >95% | 0.94-0.96 | 30× |
| 30-40% | 94-97% | >95% | 0.95-0.96 | 20× |
For high-frequency mutations (≥20%), sequencing depths of 200× are generally sufficient to detect >95% of variants while maintaining precision rates above 95% [4]. However, at lower mutation frequencies (≤10%), simply increasing sequencing depth provides diminishing returns, with detection performance remaining suboptimal even at extreme depths (500×-800×) [4]. This demonstrates that for very low-frequency mutations (1-5%), improvements in experimental methods to enrich mutation burden (such through tumor enrichment or unique molecular identifiers) may be more impactful than further depth increases alone.
Performance Comparison Between Variant Callers
The interaction between sequencing parameters and choice of variant calling algorithm introduces additional complexity to study design. Comparative analyses of widely used somatic callers reveal distinct performance characteristics across the depth-frequency landscape.
Table 3: Variant Caller Performance Across Different Conditions
| Variant Caller | Optimal Frequency Range | Relative Speed | Strengths | Limitations |
|---|---|---|---|---|
| GATK Mutect2 | 5-40% | 1× (reference) | Better low-frequency sensitivity, tumor-only mode | Slower processing |
| Strelka2 | 20-40% | 17-22× faster | Superior high-frequency performance, speed | Reduced low-frequency sensitivity |
Strelka2 demonstrates slightly superior performance for higher mutation frequencies (≥20%), while Mutect2 shows advantages at lower frequencies (5-10%) [4]. Notably, Strelka2 achieves these results with substantially faster processing times (17-22 times faster on average), an important practical consideration for large-scale studies [4]. Both callers maintain precision rates above 95% across most depth-frequency combinations when properly configured, though this can drop to approximately 68.9-100% for 1% mutation frequency variants depending on depth [4].
Experimental Protocols for Systematic Benchmarking
Experimental Design for Depth-Frequency Analysis
Systematic evaluation of sequencing depth and mutation frequency effects requires carefully controlled experimental designs that isolate these variables from other technical confounders. The following protocol outlines a robust approach for generating benchmarking datasets:
Sample Selection and Preparation:
- Select two well-characterized DNA samples with comprehensively mapped genotypes (e.g., NA12878 and YH-1 from the 1000 Genomes Project) [4]
- Verify DNA quality using electrophoretic methods (e.g., agarose gel electrophoresis) and precise quantification (e.g., Qubit fluorometer) to ensure integrity and accurate concentration measurements [88]
- Perform whole-exome or whole-genome sequencing at ultra-high depth (400×+) on both samples to establish a truth set of variants [4] [87]
Library Preparation and Sequencing:
- Utilize PCR-free library preparation protocols where possible to minimize duplication artifacts and coverage bias [89]
- For PCR-amplified libraries, employ unique molecular identifiers (UMIs) to enable accurate duplicate marking and elimination of amplification artifacts [89]
- Sequence on Illumina platforms (e.g., HiSeq X Ten, NovaSeq) with 150bp paired-end reads to maximize mappability and variant detection accuracy [88]
Bioinformatic Processing:
- Process raw sequencing data through standardized pipelines aligned with GATK Best Practices [89]
- Perform quality control using FastQC (v0.11.5) and adapter trimming with fastp (v0.23.4) [52]
- Align to appropriate reference genome (e.g., GRCh37/hg19 with decoy sequences) using BWA-MEM (v0.7.17) [87] [89]
- Process aligned BAM files through duplicate marking, base quality score recalibration (BQSR), and indel realignment using GATK (v3.6/v4.0+) [87] [37]
Downsampling and Mutation Frequency Simulation
To evaluate the impact of sequencing depth, employ computational downsampling of high-depth BAM files:
Downsampling Protocol:
- Use Picard DownsampleSam to systematically extract fractions of reads (0.05, 0.1, 0.15, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9) from original high-depth BAM files [88]
- Maintain paired-end read integrity during downsampling by keeping or discarding both mates simultaneously
- Generate triplicate datasets at each depth level to account for random sampling effects [4]
- Validate downsampled BAM files for expected depth using samtools stats (v1.18) [52]
Mutation Frequency Simulation:
- Select sites with completely different homozygous genotypes between two standard DNA samples to establish a truth set of variants [4]
- Mix BAM files from the two samples at controlled proportions (1%, 5%, 10%, 20%, 30%, 40%) to simulate different mutation frequencies [4]
- Validate expected allele fractions at known variant sites using bcftools (v1.10.2) [52]
Variant Calling and Performance Assessment
Execute somatic variant calling and comprehensive performance evaluation:
Variant Calling Implementation:
- Process each depth-frequency combination through standardized somatic calling pipelines
- For Mutect2, use recommended parameters: enable local assembly, employ panel of normals (PoN) and germline resources (e.g., gnomAD), and adjust
--af-of-alleles-not-in-resourceappropriately [1] [37] - For Strelka2, follow developer recommendations for configuration and filtration
- Apply appropriate post-calling filtration using FilterMutectCalls for Mutect2 and recommended filters for Strelka2 [37]
Performance Metrics Calculation:
- Compare calls to established truth sets to calculate recall (sensitivity) and precision (positive predictive value)
- Compute F-scores (harmonic mean of precision and recall) for overall performance assessment
- Generate precision-recall curves to visualize trade-offs across operating conditions [4]
- Evaluate genotype concordance rates for known variant sites using high-density SNP microarray data as reference [87]
Figure 1: Experimental workflow for systematic evaluation of sequencing depth and mutation frequency effects on variant calling accuracy. The process begins with sample preparation and ultra-deep sequencing to establish ground truth, proceeds through computational simulation of different experimental conditions, and concludes with comprehensive performance assessment.
Table 4: Essential Research Reagents and Computational Tools for Sequencing Benchmarking
| Category | Specific Resource | Function/Purpose | Implementation Notes |
|---|---|---|---|
| Reference Materials | NA12878 & YH-1 DNA | Benchmark samples with known genotypes | Well-characterized reference materials from 1000 Genomes Project |
| GRCh37/hg19 with decoy | Reference genome | Includes unplaced contigs, improves mapping | |
| gnomAD VCF | Germline resource for Mutect2 | Provides population allele frequencies | |
| Wet Lab Reagents | TIANamp Blood DNA Kit | High-quality DNA extraction | Maintains DNA integrity for sequencing |
| MagNA Pure 96 System | Automated nucleic acid extraction | Standardizes sample processing | |
| PCR-free library prep kits | Minimizes amplification bias | Essential for accurate variant calling | |
| xGen Amplicon Core Kit | Target enrichment | For whole-exome sequencing studies | |
| Computational Tools | BWA-MEM | Read alignment | Optimal for variant calling with GATK |
| GATK Mutect2 | Somatic variant calling | Recommended with PoN and germline resources | |
| Strelka2 | Somatic variant calling | Faster alternative for high-frequency variants | |
| Picard Tools | BAM processing & downsampling | Essential for experimental simulations | |
| SAMtools/BCFtools | BAM/VCF manipulation | Versatile utilities for file processing | |
| Quality Control | FastQC | Sequencing quality assessment | Identifies technical issues in raw data |
| fastp | Adapter trimming & filtering | Improves data quality pre-alignment | |
| MultiQC | Aggregate QC reporting | Comprehensive project quality assessment |
Integrated Recommendations for Experimental Design
Practical Guidelines for Common Research Scenarios
Based on comprehensive benchmarking evidence, study designs should be tailored to specific research objectives and expected mutation frequencies:
For Clinical Tumor Sequencing with Expected High Purity (≥20% mutation frequency):
- Recommended depth: 200× for tumor, 30× for matched normal
- Recommended caller: Both Mutect2 and Strelka2 perform well
- Expected performance: >95% recall and precision for SNVs
- BAM requirements: Properly processed with mark duplicates, BQSR applied
For Subclonal Mutation Detection (5-10% mutation frequency):
- Recommended depth: 300× for tumor, 30× for matched normal
- Recommended caller: Mutect2 with optimized filtration
- Expected performance: 80-90% recall with >95% precision
- Special considerations: UMIs strongly recommended to control amplification artifacts
For Ultra-Sensitive Liquid Biopsy Applications (1-5% mutation frequency):
- Recommended depth: 500×+ with duplex UMIs
- Recommended caller: Mutect2 with unique molecular identifier processing
- Expected performance: Highly variable (34-50% recall for 1% frequency)
- Limitations: Substantial false negatives expected even at extreme depth
BAM File Quality Considerations for GATK Somatic Calling
Beyond depth and frequency parameters, BAM file quality directly impacts variant calling accuracy. Key considerations include:
Mapping Quality:
- Implement minimum mapping quality thresholds (e.g., Q20) to reduce false positives from misaligned reads [90]
- Employ proper read group information in BAM headers for sample tracking and processing
Duplicate Marking:
- Mark or remove PCR duplicates to prevent artificial inflation of variant support
- For UMIs, use molecular-based duplicate marking to correctly identify PCR copies
Base Quality Recalibration:
- Apply BQSR to correct systematic errors in base quality scores
- Use known variant sites (e.g., dbSNP) for training the recalibration model
Contamination Management:
- Estimate cross-sample contamination using tools like VerifyBamID
- Filter BAM files to remove poorly mapped reads and technical artifacts
Figure 2: Key factors determining variant calling accuracy in somatic studies. Input BAM quality features interact with experimental parameters to produce different accuracy outcomes across variant types, with low-frequency variants presenting persistent detection challenges.
Sequencing depth and mutation frequency interact in complex ways to determine the accuracy of somatic variant detection in GATK-based pipelines. For high-frequency mutations (≥20%), moderate depths of 200× provide excellent detection power, while low-frequency variants (≤10%) remain challenging even with extreme depth increases. Researchers must align their sequencing designs with expected mutation frequencies and variant types of interest, recognizing that indel detection requires greater depth than SNVs and that low-frequency variants may require specialized enrichment approaches rather than simply increasing sequencing depth. By applying these evidence-based recommendations within the broader context of BAM file quality requirements, researchers can optimize study designs to maximize detection power while controlling costs in cancer genomic studies.
Establishing Quality Metrics for Input BAM Assessment and Validation
In the context of genomic research for drug development, the accuracy of somatic variant discovery using the Genome Analysis Toolkit (GATK) is fundamentally dependent on the quality of input Binary Alignment/Map (BAM) files. Pre-processing of next-generation sequencing (NGS) data to produce analysis-ready BAM files represents a critical foundational step upon which all downstream variant calling and interpretation relies [14]. This technical guide establishes a comprehensive framework for assessing and validating BAM file quality specifically tailored to GATK somatic calling pipelines, with particular emphasis on the Mutect2 workflow for detecting somatic single nucleotide variants (SNVs) and indels [1] [8].
For clinical sequencing applications in oncology research, suboptimal BAM quality can introduce systematic errors that propagate through the analysis pipeline, potentially resulting in both false positive and false negative variant calls with significant implications for therapeutic target identification [14]. The establishment of rigorous, standardized quality metrics provides researchers and drug development professionals with an evidence-based approach to ensure data integrity before committing computational resources to variant discovery. This guide synthesizes current best practices for BAM quality assessment, integrating metrics from alignment statistics, coverage analysis, and contamination checks specifically contextualized within the GATK somatic variant calling framework [8] [14].
Core BAM Quality Metrics for Somatic Variant Calling
Quality assessment of BAM files for somatic variant discovery requires evaluation across multiple dimensions, with specific thresholds informed by the requirements of the GATK Mutect2 pipeline. The metrics summarized in Table 1 provide a comprehensive framework for evaluating BAM file suitability.
Table 1: Essential BAM Quality Metrics for GATK Somatic Variant Calling
| Metric Category | Specific Metric | Recommended Threshold | Impact on Somatic Calling |
|---|---|---|---|
| Mapping Statistics | Uniquely mapped reads | ≥75% [91] | Reduced sensitivity with lower values |
| Mapping quality (MAPQ) | ≥30 for high-confidence reads [91] | Filters alignment ambiguities | |
| Read duplicates | <10% for exomes [14] | Prevents PCR artifact inflation | |
| Coverage Profile | Mean coverage depth | ≥200X for 20% VAF [4] | Affects low-VAF detection sensitivity |
| Uniformity of coverage | <30% dropouts at 20X [13] | Reduces false negatives in low-coverage regions | |
| Target base coverage | ≥95% of targets at ≥30X [14] | Ensures comprehensive genomic assessment | |
| Base-Level Quality | Insert size distribution | Consistent with library prep [13] | Detects library preparation issues |
| GC content distribution | Matches reference profile [13] | Identifies amplification biases | |
| Strand balance | ~50% in normal samples [91] | Flags alignment artifacts | |
| Sample Integrity | Cross-sample contamination | <3% [8] | Critical for accurate somatic calling |
| Sex chromosome consistency | Matches reported sex [14] | Validates sample identity |
The precision of somatic variant calling exhibits a strong dependence on both sequencing depth and variant allele frequency (VAF). Research has demonstrated that for mutations with VAF ≥20%, a sequencing depth of ≥200X is sufficient to call approximately 95% of mutations, while for lower frequency variants (≤10%), simply increasing sequencing depth provides diminishing returns without addressing fundamental quality issues [4]. This relationship underscores the importance of optimizing both library preparation and sequencing parameters rather than relying exclusively on depth to recover performance with suboptimal BAM files.
BAM Quality Assessment Methodologies
Alignment Statistics and Mapping Quality
The initial assessment of BAM quality focuses on alignment characteristics using tools such as samtools flagstat and samtools stats [91]. Key metrics include the percentage of uniquely mapped reads, which should typically exceed 75% for human samples, with significantly lower values indicating potential issues with library complexity, reference genome mismatch, or excessive repetitive content [91]. The distribution of mapping qualities (MAPQ) provides additional insight, with high-quality alignments typically exhibiting MAPQ scores ≥30 [91]. The command samtools view -q 30 -c can be used to count high-confidence reads, establishing a quantitative basis for filtering prior to variant calling [91].
Duplicate reads, arising from PCR amplification during library preparation, should be identified and marked using tools such as Picard Tools or Sambamba [14]. In exome sequencing, duplicate rates typically range from 5-15%, with values exceeding this threshold indicating potential issues with limited input DNA or over-amplification [14]. For somatic variant calling, particularly in cancer samples with possible aneuploidy, careful interpretation of duplicate metrics is essential, as elevated duplication in regions of copy number loss may reflect genuine biological signals rather than technical artifacts.
Coverage Analysis and Uniformity
Coverage depth and uniformity represent critical determinants of somatic variant detection sensitivity, particularly for low-frequency subclonal mutations. The samtools depth command provides base-level coverage information, while more sophisticated tools like QualiMap and Mosdepth generate comprehensive coverage distributions across target regions [14] [13]. For somatic studies, the relationship between sequencing depth and variant detection sensitivity must be carefully considered; research has demonstrated that while increasing depth from 100X to 200X improves recall rates by 0.6-44%, further depth increases provide diminishing returns and may slightly reduce precision [4].
For targeted sequencing panels, the fraction of target bases achieving minimum coverage thresholds (e.g., 95% of targets at ≥30X) provides a key quality indicator [14]. Coverage uniformity can be assessed by calculating the fraction of target bases with coverage below 0.2× the mean, with values exceeding 30% indicating potential issues with capture efficiency or library complexity. QualiMap's "Genome Fraction Coverage" plot provides a visual representation of this metric, showing the percentage of the reference genome covered at or above a specified depth threshold [13].
Contamination and Sample Quality Assessment
Cross-sample contamination poses a significant challenge for somatic variant calling, particularly in detecting low-frequency variants. The GATK pipeline includes specific tools for contamination assessment, with GetPileupSummaries and CalculateContamination providing estimates of contamination fractions for each tumor sample [12] [8]. Unlike other contamination tools, CalculateContamination is designed to work effectively without a matched normal sample and performs robustly even in samples with significant copy number variation [8].
Additional sample quality checks include verification of expected sample relationships using tools such as the KING algorithm [14] and confirmation of sex chromosome consistency between reported sample sex and observed read distribution across sex chromosomes. For paired tumor-normal analyses, these checks are particularly important to prevent sample mix-ups that could severely compromise somatic variant identification.
Quality Control Visualization Workflows
Effective quality assessment requires both quantitative metrics and visual representations to identify potential issues. The following workflow diagrams illustrate key quality assessment processes and their relationship to the GATK somatic variant calling pipeline.
BAM Quality Assessment Workflow
Diagram 1: Comprehensive BAM quality assessment workflow showing sequential evaluation steps and decision points prior to variant calling.
GATK Somatic Variant Calling with Quality Integration
Diagram 2: GATK somatic variant calling workflow showing integration points for quality assessment metrics and filters.
The Scientist's Toolkit: Essential Research Reagents and Tools
Implementation of robust BAM quality assessment requires a comprehensive toolkit of software utilities and reference resources. Table 2 summarizes key tools and their specific applications within the quality assessment workflow.
Table 2: Essential Research Tools for BAM Quality Assessment
| Tool Name | Primary Function | Application in BAM QC | Key Output Metrics |
|---|---|---|---|
| Samtools [91] [14] | BAM file manipulation and statistics | Basic alignment statistics, read filtering, format conversion | Mapping percentages, read counts, coverage depth |
| Picard Tools [14] | NGS data processing utilities | Duplicate marking, insert size metrics, quality score distribution | Duplication rate, insert size histograms, GC bias |
| Qualimap [13] | Comprehensive quality control | Coverage analysis, GC content distribution, mapping quality | Coverage uniformity, genomic origin of reads, bias detection |
| GATK [92] [8] | Variant discovery toolkit | Contamination estimation, base quality recalibration | Contamination fractions, base error models |
| BEDTools [14] | Genome arithmetic utilities | Coverage analysis in target regions, intersection operations | Target coverage statistics, off-target reads |
| VerifyBamID [14] | Sample authentication | Contamination detection, sample mix-up prevention | Contamination estimates, sample comparisons |
In addition to these software tools, reference resources such as the Genome in a Bottle (GIAB) consortium data provide benchmark variant calls for established reference samples, enabling validation of variant calling performance using quality-tested BAM files [14]. For somatic variant calling, the integration of these tools into a structured workflow ensures comprehensive quality assessment at multiple levels, from basic alignment characteristics to sample-specific contamination checks.
Experimental Protocols for Key Quality Assessments
Protocol: Comprehensive BAM QC with Qualimap
Qualimap provides a multi-faceted approach to BAM quality assessment, particularly valuable for targeted sequencing data such as exomes and panels [13]. The following protocol outlines a standard Qualimap analysis:
Input Preparation: Ensure BAM files are coordinate-sorted and indexed. Prepare a BED file of target regions if analyzing targeted sequencing data.
Command Execution:
The
-gffparameter specifies target regions for enrichment-based sequencing, while-sdgenerates statistics for each chromosome and-cenables collection of GC-content metrics [13].Output Interpretation: Review the generated HTML report, paying particular attention to:
- Globals Section: Total reads, mapping percentages, and duplication rate
- Coverage Histograms: Distribution of coverage depth across the genome
- GC Content Distribution: Shift relative to reference genome may indicate biases
- Mapping Quality Distribution: Prevalence of low-quality alignments
- Insert Size Metrics: Consistency with library preparation specifications [13]
For RNA-seq data, Qualimap provides a specialized RNA-seq QC mode that evaluates reads genomic origin, junction analysis, and 5'-3' bias, which are critical for transcriptome analyses [13].
Protocol: Contamination Assessment with GATK
The GATK toolkit provides specialized tools for estimating contamination in tumor samples, a critical quality metric for somatic variant calling [12] [8]:
GetPileupSummaries: Generate summary counts of read support against known variant sites:
This step tabulates pileup metrics for inferring contamination, using a set of known common variants as reference [12].
CalculateContamination: Estimate contamination fraction:
This tool calculates the fraction of reads coming from cross-sample contamination, with specific design to work effectively even in samples with significant copy number variation [8].
Interpretation: Contamination estimates below 3% are generally acceptable, while values exceeding this threshold may require additional sample purification or bioinformatic filtering [8].
Protocol: Strand Bias and Orientation Artifact Assessment
For samples susceptible to substitution errors on a single strand before sequencing (such as FFPE tumors or samples sequenced on Illumina Novaseq instruments), assessment of read orientation artifacts is essential [12]:
During Mutect2 Execution: Collect F1R2 counts for orientation bias modeling:
LearnReadOrientationModel: Process the F1R2 counts to generate artifact priors:
Integration with Filtering: Pass the learned model to FilterMutectCalls using the
--ob-priorsargument [12]. This step is particularly important for FFPE-derived samples where orientation artifacts can significantly impact variant calling accuracy.
Establishing comprehensive quality metrics for input BAM assessment represents a foundational element in robust somatic variant discovery for drug development research. The framework presented in this guide integrates multiple dimensions of BAM quality evaluation, from basic alignment statistics to sample-specific contamination checks, all contextualized within the GATK somatic variant calling workflow. Implementation of these standardized assessments enables researchers to identify potential quality issues before variant calling, allocate computational resources efficiently, and interpret variant calling results with appropriate confidence.
For research organizations pursuing clinical applications, the establishment of laboratory-specific quality thresholds based on historical performance data provides an evidence-based approach to quality management. Continuous monitoring of quality metrics across sequencing batches further enhances quality control, enabling rapid detection of deviations from established baselines. Through rigorous application of these BAM quality assessment protocols, researchers and drug development professionals can ensure the reliability of their somatic variant discoveries, ultimately supporting the identification of valid therapeutic targets in oncology and other disease areas.
Best Practices for Clinical Sequencing and Variant Calling Benchmarking
In clinical genomics, the accuracy and reliability of variant calling are paramount, as the results directly influence patient diagnosis and treatment strategies. Next-generation sequencing (NGS), particularly whole-genome sequencing (WGS), is now firmly established in clinical diagnostics, with robust benchmarking practices serving as the foundation for ensuring data quality [93]. The transition towards WGS as a first-tier diagnostic test offers a more comprehensive view of genetic variation compared to targeted approaches, but simultaneously demands more rigorous and standardized bioinformatics practices to ensure clinical consensus, accuracy, reproducibility, and comparability across different laboratories and platforms [93] [94]. This guide outlines established best practices and emerging standards for benchmarking variant calling pipelines, with a specific focus on the context of somatic mutation detection using GATK tools.
Foundational Principles for Clinical Bioinformatics
Clinical bioinformatics operations must adhere to quality standards comparable to other clinical laboratory disciplines. Key recommendations for production environments include the adoption of the hg38 (GRCh38) genome build as the reference standard to ensure consistency and compatibility with modern annotation resources [93] [95]. Bioinformatics systems should utilize secure, off-grid, clinical-grade high-performance computing infrastructure to protect patient data, with strict version control for all software and pipelines [93].
A core principle for ensuring reproducibility is the use of containerized software environments (e.g., Docker, Singularity), which encapsulate all dependencies and guarantee consistent analysis results across different computing environments [93]. Furthermore, data integrity must be verified using file hashing, and sample identity must be confirmed through methods such as genetic fingerprinting and genetically inferred markers like sex and relatedness checks [93].
Core Analysis Types for Comprehensive Sequencing
A best-practice clinical NGS pipeline should be designed to detect a broad spectrum of genetic variation. The recommended core set of analyses includes [93] [94]:
- Single Nucleotide Variants (SNVs) and small insertions/deletions (indels)
- Copy Number Variants (CNVs) including deletions and duplications
- Structural Variants (SVs) including insertions, inversions, translocations, and complex rearrangements
- Short Tandem Repeats (STRs) and repeat expansions
- Mitochondrial SNVs and indels
For somatic variant calling in cancer, optional but highly informative analyses include Tumor Mutational Burden (TMB), Microsatellite Instability (MSI), and Homologous Recombination Deficiency (HRD) [93].
Benchmarking Methodologies and Performance Metrics
Rigorous benchmarking is essential for validating the performance of any variant calling pipeline before its deployment in a clinical or research setting.
Establishing Ground Truth with Reference Materials
The use of well-characterized reference samples with established "ground truth" variant sets is a cornerstone of reliable benchmarking. The Genome in a Bottle (GIAB) consortium has developed gold standard datasets for several human genomes (e.g., HG001, HG002, HG003) that provide high-confidence variant calls within defined high-confidence regions [95]. These are widely used for benchmarking germline small variant calls. For somatic variant calling, the SEQC2 consortium provides relevant reference materials [93].
Emerging benchmarks are also addressing more complex genomic regions and variant types. The Platinum Pedigree benchmark, for instance, uses Mendelian inheritance patterns in a large pedigree to create a comprehensive truth set that includes over 4.7 million single-nucleotide variants, 767,795 indels, 537,486 tandem repeats, and 24,315 structural variants, adding approximately 200 Mb of high-confidence regions to previous benchmarks [96].
Quantitative Performance Metrics
When comparing variant calls to a truth set, performance is quantitatively assessed using the following key metrics, calculated separately for SNVs and indels [95]:
- Precision (Positive Predictive Value) = TP / (TP + FP)
- Recall (Sensitivity) = TP / (TP + FN)
- F1 Score = 2 × (Precision × Recall) / (Precision + Recall)
Table 1: Key Performance Metrics for Variant Caller Benchmarking
| Metric | Calculation | Interpretation |
|---|---|---|
| True Positives (TP) | Variants correctly identified | Measure of accurate detection |
| False Positives (FP) | Variants incorrectly identified | Measure of noise/spurious calls |
| False Negatives (FN) | True variants missed by the caller | Measure of omissions |
| Precision | TP / (TP + FP) | Fraction of correct calls among all calls made |
| Recall | TP / (TP + FN) | Fraction of true variants successfully detected |
| F1 Score | 2 × (Precision × Recall) / (Precision + Recall) | Harmonic mean balancing precision and recall |
These metrics should be assessed genome-wide and within stratified genomic contexts (e.g., by functional region, GC-content, or segmental duplication status) to identify potential biases [95].
Benchmarking Experimental Protocol
A typical benchmarking workflow for a somatic variant caller involves the following steps [95]:
- Data Acquisition: Obtain publicly available sequencing data (e.g., from NCBI Sequence Read Archive) for a reference sample with a established truth set.
- Data Pre-processing: Align the raw sequencing reads (FASTQ) to the reference genome (GRCh38) to produce BAM files. This includes all standard pre-processing steps like duplicate marking and base quality score recalibration.
- Variant Calling: Run the variant calling software (e.g., Mutect2) on the processed BAM file(s) to generate a VCF file.
- Performance Assessment: Compare the output VCF file against the gold standard truth set using a specialized benchmarking tool like the Variant Calling Assessment Tool (VCAT) or hap.py [95]. This tool generates counts of TP, FP, and FN, and calculates precision, recall, and F1 scores.
- Analysis and Interpretation: Analyze the results to determine if the pipeline meets the required performance thresholds for clinical or research use.
Diagram 1: Variant Calling Benchmarking Workflow
Somatic Variant Calling with GATK Mutect2: Workflow and Input Requirements
The GATK Mutect2 tool is a widely adopted standard for calling somatic short variants (SNVs and Indels) from tumor sequencing data. Its workflow is designed to handle the specific challenges of somatic variant detection, such as tumor heterogeneity, contamination, and sequencing artifacts.
The Mutect2 Somatic Calling Workflow
The recommended best-practice workflow for Mutect2 involves multiple steps beyond the initial variant calling [8] [1] [12]:
- Call Candidate Variants: Mutect2 performs local assembly of haplotypes in active regions to call SNVs and indels simultaneously. It uses a Bayesian somatic likelihoods model to calculate the log odds of a variant being a true somatic event versus a sequencing error [8] [1].
- Calculate Contamination: Use
GetPileupSummariesandCalculateContaminationto estimate the fraction of reads in the tumor sample arising from cross-sample contamination. This step is robust to samples with significant copy number variation [8]. - Learn Orientation Bias Artifacts: For samples prone to artifacts like FFPE-derived DNA, run
Mutect2with the--f1r2-tar-gzargument to generate data forLearnReadOrientationModel. This produces a prior probabilities table for strand-specific errors [12]. - Filter Variants:
FilterMutectCallsapplies multiple filters to account for correlated errors, alignment artifacts, strand bias, germline variants, and contamination. It uses a learned model of the tumor's mutation spectrum and automatically optimizes a filtering threshold to balance sensitivity and precision (F-score) [8] [12]. - Annotate Variants: Tools like
Funcotatoradd functional context to the filtered variants, including gene effect, protein change, and population frequency from sources like GENCODE, dbSNP, gnomAD, and COSMIC [8].
Diagram 2: GATK Mutect2 Somatic Variant Calling Workflow
Critical Input BAM Requirements for Robust Somatic Analysis
The accuracy of Mutect2 is highly dependent on the quality of the input BAM files. Adherence to the following requirements is critical for research and clinical validity:
- Pre-processing: Input BAMs must be generated through a rigorous pre-processing pipeline as described in the GATK Best Practices. This includes proper alignment, duplicate marking, base quality score recalibration (BQSR), and quality control [8].
- Reference Genome: Alignment and variant calling must use the same reference genome build, preferably GRCh38 [93].
- Sample Information: For tumor-normal paired analysis, the normal sample must be correctly specified using the
-normalargument. Mutect2 uses this to pre-filter sites that are clearly germline, saving computational resources [1]. - Auxiliary Data Resources: The use of a germline resource (e.g., gnomAD) is recommended. This resource provides population allele frequencies, allowing Mutect2 to distinguish common germline polymorphisms from true somatic events. The
--af-of-alleles-not-in-resourceparameter is dynamically set based on the calling mode (tumor-only vs. tumor-normal) to handle variants absent from the resource [1] [12]. A Panel of Normals (PoN), created by calling variants across a set of normal samples usingCreateSomaticPanelOfNormals, is also highly recommended to filter out common technical artifacts present in the sequencing platform and methodology [12].
Table 2: Key Research Reagent Solutions for Sequencing and Variant Calling
| Item | Function / Application | Example / Source |
|---|---|---|
| Reference Genomes | Standardized sequence for read alignment and variant reporting. | GRCh38 (hg38) [93] |
| Gold Standard Genomes | Benchmarking and validation of variant calls. | GIAB samples (HG001, HG002, etc.) [95] |
| Germline Resource | Provides population allele frequencies to filter common germline variants. | gnomAD VCF [1] [12] |
| Panel of Normals (PoN) | A VCF of artifacts common to a specific lab's process/platform, used to filter non-somatic calls. | Created from in-house normal samples [12] |
| Truth Sets | High-confidence variant calls for comprehensive benchmarking. | GIAB, SEQC2, Platinum Pedigree [93] [96] [95] |
| Benchmarking Tools | Software for comparing VCFs to a truth set and calculating metrics. | VCAT, hap.py [95] |
| Containerized Software | Ensures pipeline reproducibility and consistency across environments. | Docker, Singularity [93] |
The implementation of standardized, benchmarked variant calling practices is non-negotiable for generating clinically actionable and research-grade genomic data. This involves a multi-faceted approach: adopting the GRCh38 reference genome, utilizing comprehensive benchmarking protocols with gold-standard samples like those from GIAB, and following best-practice workflows such as the one implemented in GATK Mutect2 for somatic variant discovery. The field continues to evolve with the development of more comprehensive benchmarks that include complex variant types and difficult-to-sequence genomic regions [96]. By adhering to these principles and continuously validating pipeline performance against community standards, researchers and clinical laboratories can ensure the accuracy, reproducibility, and reliability of their genomic analyses, thereby fully harnessing the diagnostic and therapeutic potential of clinical whole-genome sequencing.
Strategies for Balancing Sensitivity, Precision, and Computational Efficiency
The identification of somatic variants from high-throughput sequencing data is a cornerstone of cancer genomics, driving both research and clinical decision-making in personalized oncology. The analysis of tumor samples, provided as Binary Alignment Map (BAM) files, presents a fundamental bioinformatic challenge: maximizing the detection of true positive variants (sensitivity) while minimizing false positives (precision), all within feasible computational constraints. This balance is particularly critical in clinical contexts, where undetected therapeutic targets or misinterpreted variants can directly impact patient treatment strategies. The Genome Analysis Toolkit's Mutect2 has emerged as a widely adopted solution for somatic short variant discovery, yet its effective implementation requires careful consideration of multiple interdependent parameters and auxiliary data resources. This technical guide examines evidence-based strategies for optimizing somatic variant calling performance within the GATK framework, with particular emphasis on BAM file requirements, parameter configuration, and computational efficiency trade-offs relevant to researchers and drug development professionals.
Core Performance Trade-offs in Somatic Variant Calling
Sequencing Depth and Mutation Frequency
The relationship between sequencing depth, mutation frequency (variant allele fraction), and variant calling performance represents a fundamental consideration in experimental design. Evidence indicates that these factors interact in complex ways that directly impact the sensitivity-precision balance.
Table 1: Performance Metrics Across Sequencing Depths and Mutation Frequencies
| Mutation Frequency | Sequencing Depth | Recall Rate | Precision Rate | F-score | Recommended Use Case |
|---|---|---|---|---|---|
| ≥20% | 200X | 92-97% | >95% | 0.94-0.96 | Standard tumor profiling |
| 10% | 200X | 50-96% | 95.5-95.9% | 0.65-0.95 | Heterogeneous tumors |
| 5-10% | 500-800X | 48-96% | 95.5-96.5% | 0.63-0.95 | Subclonal detection |
| 1% | 500-800X | 32-50% | 68.9-100% | 0.32-0.51 | Liquid biopsy/ctDNA |
| 1% | 100-300X | 2.7-34.5% | 68.9-100% | 0.05-0.19 | Limited material |
Empirical data demonstrates that for mutations with higher allele fractions (≥20%), a sequencing depth of 200X is generally sufficient to detect over 95% of variants with high precision [4]. However, as mutation frequency decreases to 5-10%, significantly higher sequencing depths (500-800X) become necessary to maintain reasonable sensitivity. At very low mutation frequencies (1%), even ultra-deep sequencing (800X) achieves only modest recall rates (32-50%), suggesting fundamental limitations in variant detection that cannot be overcome solely by increasing sequencing depth [4]. For contexts with extremely low tumor content, such as circulating tumor DNA (ctDNA) analysis, specialized approaches like unique molecular identifiers (UMIs) or modified bioinformatic parameters are recommended rather than simply increasing sequencing depth [72].
Computational Efficiency Comparisons
The choice of variant calling tools significantly impacts computational requirements and processing time, an important consideration for large-scale studies or clinical applications with rapid turnaround needs.
Table 2: Computational Performance of Somatic Variant Callers
| Variant Caller | Processing Speed | Sensitivity Profile | Precision Profile | Optimal Use Context |
|---|---|---|---|---|
| Mutect2 | Baseline (1X) | Better at <10% VAF | >95% across depths | Standard somatic calling |
| Strelka2 | 17-22X faster | Better at ≥20% VAF | >95% across depths | High-throughput pipelines |
| DRAGEN | Not specified | High for SNVs/INDELs | High for SNVs/INDELs | Clinical-grade calling |
| BWA-Mutect2 | Moderate | High for SNVs | High for SNVs | Open-source solution |
Benchmarking analyses reveal that Strelka2 performs 17-22 times faster than Mutect2 on average while maintaining comparable accuracy profiles [4]. However, their sensitivity profiles differ across the mutation frequency spectrum: Strelka2 shows slightly better performance at higher mutation frequencies (≥20%), while Mutect2 demonstrates advantages at lower frequencies (<10%) [4]. Recent evaluations of multiple aligner-caller combinations indicate that DRAGEN exhibits superior overall performance, while among open-source solutions, the combination of BWA with Mutect2 achieves the highest mean F1 scores for single-nucleotide variant (SNV) detection (0.949) [97].
Optimization Strategies for Mutect2 Implementation
Parameter Tuning for Specific Applications
Mutect2 incorporates several operational modes specifically designed for different experimental contexts, each with optimized default parameters that balance sensitivity and precision according to the biological scenario.
Tumor-Normal Mode: This is the standard approach when matched normal tissue is available. Mutect2 employs logic to skip emitting variants that are clearly present in the germline based on the matched normal evidence, conserving computational resources [1]. The tool dynamically sets --af-of-alleles-not-in-resource to 1e-6 in this mode, which represents the prior probability that a novel allele (not in the germline resource) is a somatic variant [1].
Tumor-Only Mode: When no matched normal is available, Mutect2 adjusts its default for --af-of-alleles-not-in-resource to 5e-8, reflecting a more conservative prior that helps compensate for the absence of a matched control [1]. This mode requires additional filtering resources including a panel of normals (PoN) and germline resource to identify technical artifacts and common germline variants.
Mitochondrial Mode: Activated with the --mitochondria flag, this mode optimizes parameters for calling variants in mitochondrial DNA, including setting --af-of-alleles-not-in-resource to 4e-3 and adjusting lod thresholds to 0 to accommodate the unique characteristics of mitochondrial genomics [1].
Force-Calling Mode: This mode enables researchers to specifically evaluate predefined alleles of interest while still discovering novel variants, useful for monitoring known resistance mutations or validated cancer hotspots [1].
Advanced Filtering Configuration
The FilterMutectCalls tool implements a sophisticated Bayesian approach that automatically optimizes the trade-off between sensitivity and precision. Key advancements include:
Unified Filtering Strategy: FilterMutectCalls now utilizes a single probabilistic threshold based on the probability that a variant is a true somatic mutation, replacing the previous multiple-threshold approach (which included separate parameters for -normal-artifact-lod, -max-germline-posterior, and others) [12]. The tool automatically determines the optimal threshold to maximize the F-score (the harmonic mean of sensitivity and precision), though users can adjust the balance using the -f-score-beta parameter [12].
Somatic Clustering Model: This innovative approach models the spectrum of subclonal allele fractions using a Dirichlet process binomial mixture model to help distinguish true somatic variants from artifacts. By learning the characteristic allele fraction distribution of each tumor, the filter becomes more sensitive to variants that fit the expected pattern and more skeptical of outliers that might represent errors [12].
Orientation Bias Artifact Detection: For samples susceptible to substitution errors occurring on a single strand before sequencing (such as FFPE tumors or NovaSeq data), Mutect2 implements a specialized three-step workflow [12]:
- Generate F1R2 counts during Mutect2 execution using
--f1r2-tar-gz - Learn the orientation bias model with LearnReadOrientationModel
- Apply the model during filtering with FilterMutectCalls using
-ob-priors
Experimental Design and Benchmarking Protocols
Benchmarking Data Generation Methodologies
Robust assessment of variant calling performance requires carefully designed benchmarking datasets with known ground truth variants. Recent approaches have developed innovative methodologies for creating reference materials:
Mosaic Genome Construction: This approach creates specialized benchmarking samples by extracting reads from validated somatic variants in original tumor-normal samples and inserting them into a simulated WGS genome generated from the GRCh37 reference [98]. The process preserves the intrinsic noise and properties from sample preparation and sequencing while enabling controlled assessment. The methodology involves:
- Selecting validated variants from consensus callsets (e.g., 32,267 variants from PCAWG and HMF datasets)
- Extracting all reads mapped within 2kb windows surrounding each variant
- Inserting these reads into a simulated WGS background while removing artificial reads overlapping the window spans
- Generating multiple mosaic genomes grouped by consistent insert sizes [98]
Tumorized Samples: For precision assessment, hybrid samples are created by combining reads from Genome in a Bottle (GIAB) reference samples (NA12878 and HG002) with synthetically reproduced true somatic variants from consensus callsets [98]. This approach modifies only 0.2% of reads to represent variant sequences while preserving 99.8% of original reads, creating realistic precision benchmarks that can be openly shared.
Artificial Dataset Generation: For low-frequency variant benchmarking, specialized workflows simulate ultra-deep targeted sequencing data without relying on pre-existing data templates [72]:
- Generate artificial normal reads using NEAT with sequencing error models
- Create random somatic variants at specified VAFs using BAMSurgeon
- Spike variants into normal BAM files, verifying successful incorporation
- Validate using multiple variant callers with tuned parameters
Table 3: Key Research Reagent Solutions for Somatic Variant Calling
| Resource Type | Specific Examples | Function | Impact on Performance |
|---|---|---|---|
| Germline Resource | gnomAD (af-only-gnomad.vcf.gz) | Provides population allele frequencies to distinguish rare germline variants from somatics | Critical for reducing false positives in tumor-only mode |
| Panel of Normals | CreateSomaticPanelOfNormals output | Identifies technical artifacts systematic across normal samples | Significantly reduces false positives from sequencing artifacts |
| Contamination Resource | chr17smallexaccommon3_grch38.vcf.gz | Known variant sites for calculating cross-sample contamination | Prevents false positives from sample contamination |
| Benchmarking Datasets | Mosaic genomes, tumorized samples | Validated variant sets for pipeline optimization | Enables objective performance assessment and harmonization |
| Read Orientation Model | LearnReadOrientationModel output | Quantifies strand-specific sequencing artifacts | Essential for FFPE and NovaSeq data to reduce false positives |
Implementation Framework and Best Practices
Integrated Variant Discovery Workflow
A comprehensive somatic variant calling pipeline extends beyond initial variant calling to include multiple validation and annotation steps that collectively ensure result reliability.
Harmonization Across Analysis Centers
As multi-center collaborations become increasingly important in cancer genomics, pipeline harmonization emerges as a critical challenge. Studies have demonstrated significant heterogeneity in performance across different research centers, particularly for indels and structural variants [98]. Solutions like the ONCOLINER platform provide actionable recommendations for improving and aligning somatic variant calling pipelines across facilities [98]. Key harmonization strategies include:
- Implementing common benchmarking using validated reference datasets
- Establishing consensus filtering thresholds based on shared performance metrics
- Adopting standardized annotation and reporting formats
- Regular cross-center proficiency testing and parameter calibration
Optimizing somatic variant calling performance requires a multifaceted approach that strategically balances sensitivity, precision, and computational efficiency. The most effective implementations combine appropriate experimental design (sequencing depth selection informed by expected mutation frequencies), thoughtful pipeline configuration (leveraging Mutect2's specialized operational modes and auxiliary resources), and rigorous validation using biologically-informed benchmarking materials. As the field advances toward increasingly standardized analysis frameworks, these strategies will ensure that somatic variant detection pipelines generate reliable, reproducible results that effectively support both cancer research and clinical decision-making in drug development.
Conclusion
Successful somatic variant calling with GATK Mutect2 fundamentally depends on proper input BAM file preparation and understanding of analysis mode-specific requirements. From foundational processing to advanced optimization for challenging scenarios like low allele fractions or mitochondrial DNA, each step in the workflow builds upon quality-controlled sequencing data. By implementing the troubleshooting strategies and validation frameworks outlined, researchers can achieve reliable mutation detection critical for both cancer genomics and non-cancer somatic mosaicism studies. Future directions will continue to emphasize integration of long-read sequencing, improved artifact correction, and standardized benchmarking to enhance clinical applicability across diverse research contexts.
References
- 1. Mutect2 - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2]
- 2. Mutect2 - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360036485152-Mutect2]
- 3. FAQ for Mutect2 – GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360050722212-FAQ-for-Mutect2]
- 4. Systematic comparison of somatic variant calling ... [https://www.nature.com/articles/s41598-020-60559-5]
- 5. Best practices for variant calling in clinical sequencing [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-020-00791-w]
- 6. FilterByOrientationBias (EXPERIMENTAL) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360037422851-FilterByOrientationBias-EXPERIMENTAL]
- 7. SAM or BAM or CRAM - Mapped sequence data formats - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035890791-SAM-or-BAM-or-CRAM-Mapped-sequence-data-formats]
- 8. Somatic short variant discovery (SNVs + Indels) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035894731-Somatic-short-variant-discovery-SNVs-Indels]
- 9. Data pre-processing for variant discovery - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035535912-Data-pre-processing-for-variant-discovery]
- 10. Understanding and manipulating SAM/BAM alignment files [http://homer.ucsd.edu/homer/basicTutorial/samfiles.html]
- 11. QUALITY CONTROL AND BAM PROCESSING [https://docs.nvidia.com/clara/parabricks/v3.5/text/qc_metrics.html]
- 12. (How to) Call somatic mutations using GATK4 Mutect2 – GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035531132--How-to-Call-somatic-mutations-using-GATK4-Mutect2]
- 13. Analysis types — Qualimap 2.3 documentation [http://qualimap.conesalab.org/doc_html/analysis.html]
- 14. Best practices for variant calling in clinical sequencing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7586657/]
- 15. sarek: Output [https://nf-co.re/sarek/2.5.2/docs/output/]
- 16. How should I pre-process data from multiplexed sequencing ... [https://gatk.broadinstitute.org/hc/en-us/articles/360035889471-How-should-I-pre-process-data-from-multiplexed-sequencing-and-multi-library-designs]
- 17. Read groups - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360035890671-Read-groups]
- 18. About the GATK Best Practices [https://gatk.broadinstitute.org/hc/en-us/articles/360035894711-About-the-GATK-Best-Practices]
- 19. Alfred: interactive multi-sample BAM alignment statistics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6612896/]
- 20. Sensitive detection of somatic point mutations in impure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3833702/]
- 21. wf-somatic-variation [https://nanoporetech.com/document/epi2me-workflows/wf-somatic-variation]
- 22. De novo detection of somatic mutations in high-throughput ... [https://www.nature.com/articles/s41587-023-01863-z]
- 23. What input files does the GATK accept or require? [https://gatk.broadinstitute.org/hc/en-us/articles/360035889451-What-input-files-does-the-GATK-accept-or-require]
- 24. Workflow overview: tumour-normal sequencing [https://nanoporetech.com/resource-centre/workflow-tumour-normal-sequencing]
- 25. Conpair: concordance and contamination estimator for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5048070/]
- 26. Tumor Sample Page [https://support.illumina.com/help/BaseSpace_App_TumorNormal_help/Content/Vault/Informatics/Sequencing_Analysis/Apps/swSEQ_mAPP_Tune_TumorSamplePage.htm]
- 27. ClairS-TO: a deep-learning method for long-read tumor ... [https://www.nature.com/articles/s41467-025-64547-z]
- 28. gatk-workflows/gatk4-data-processing [https://github.com/gatk-workflows/gatk4-data-processing]
- 29. Fast, accurate, and racially unbiased pan-cancer tumor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9825621/]
- 30. Somatic mutation and selection at population scale [https://www.nature.com/articles/s41586-025-09584-w]
- 31. Mitochondrial somatic mutation and selection throughout ... [https://www.nature.com/articles/s41559-024-02338-3]
- 32. Resolution of Optimal Mitochondrial and Nuclear DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11393944/]
- 33. Leveraging new methods for comprehensive characterization ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-024-01319-2]
- 34. Mutect2 – GATK [https://gatk.broadinstitute.org/hc/en-us/articles/30332058799003-Mutect2]
- 35. mutect2 multi-sample - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/community/posts/360062528691-mutect2-multi-sample]
- 36. Panel of Normals (PON) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360035890631-Panel-of-Normals-PON]
- 37. (How to) Call somatic mutations using GATK4 Mutect2 ... [https://gatk.broadinstitute.org/hc/en-us/articles/360035889791--How-to-Call-somatic-mutations-using-GATK4-Mutect2-Deprecated]
- 38. CreateSomaticPanelOfNormals (BETA) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/13832769396635-CreateSomaticPanelOfNormals-BETA]
- 39. BAM File Format [https://support.illumina.com/help/BS_App_ENR_OLH_15050961/Content/Source/Informatics/BAM-Format.htm]
- 40. What are SAM & BAM Files? [https://www.zymoresearch.com/blogs/blog/what-are-sam-and-bam-files?srsltid=AfmBOop3rJkhtiVCsbtuEQbdHCWh6GQH0noYTYN4D7Iaksv4eIlW3vuI]
- 41. BAM Track Format [https://genome.ucsc.edu/goldenpath/help/bam.html]
- 42. Input data | GATK-SV - Broad Institute [https://broadinstitute.github.io/gatk-sv/docs/gs/inputs/]
- 43. (How to) Map and clean up short read sequence data efficiently [https://gatk.broadinstitute.org/hc/en-us/articles/360039568932--How-to-Map-and-clean-up-short-read-sequence-data-efficiently]
- 44. GATK4 command-line syntax – GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360035531892-GATK4-command-line-syntax]
- 45. Toward better understanding of artifacts in variant calling from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4271055/]
- 46. Standard operating procedure for somatic variant refinement ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6450397/]
- 47. FilterAlignmentArtifacts (EXPERIMENTAL) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/13832707791771-FilterAlignmentArtifacts-EXPERIMENTAL]
- 48. A simple consensus approach improves somatic mutation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3978449/]
- 49. Accelerated somatic mutation calling for whole-genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11146589/]
- 50. NeuSomatic: Deep convolutional neural networks for ... [https://github.com/bioinform/neusomatic]
- 51. Multi-platform discovery of haplotype-resolved structural ... [https://www.nature.com/articles/s41467-018-08148-z]
- 52. Short-Read and Long-Read Whole Genome Sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12031342/]
- 53. Benchmarking nanopore sequencing and rapid genomics ... [https://www.nature.com/articles/s41525-024-00445-5]
- 54. BAMscale: quantification of next-generation sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7175505/]
- 55. (How to) Fix a badly formatted BAM - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360037872491--How-to-Fix-a-badly-formatted-BAM]
- 56. Chapter 19 Alignment of sequence data to a reference ... [https://eriqande.github.io/eca-bioinf-handbook/alignment-of-sequence-data-to-a-reference-genome-and-associated-steps.html]
- 57. bamCoverage — deepTools 3.5.6 documentation [https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html]
- 58. A comprehensive performance evaluation, comparison ... [https://www.sciencedirect.com/science/article/pii/S1532046424000431]
- 59. CalculateContamination - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360037225192-CalculateContamination]
- 60. GATK VARIANT CALLING BEST PRACTICE WORKFLOW [https://snakemake-wrappers.readthedocs.io/en/v6.1.0/meta-wrappers/bio/gatk_mutect2_calling.html]
- 61. CalculateContamination - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360036888972-CalculateContamination]
- 62. GetPileupSummaries (BETA) - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/360036456532-GetPileupSummaries-BETA]
- 63. Introducing GATK "Biggest Practices" for Joint Calling ... [https://gatk.broadinstitute.org/hc/en-us/articles/16957867036315-Introducing-GATK-Biggest-Practices-for-Joint-Calling-Supersized-Cohorts]
- 64. GATK4: CalculateContamination — Janis documentation [https://janis.readthedocs.io/en/stable/tools/bioinformatics/gatk4/gatk4calculatecontamination.html]
- 65. CHARR efficiently estimates contamination from DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10716339/]
- 66. Comparative analysis of somatic variant calling on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7336445/]
- 67. LearnReadOrientationModel - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/4405451237147-LearnReadOrientationModel]
- 68. Filtering Mutect output from FFPE based targeted panel ... [https://gatk.broadinstitute.org/hc/en-us/community/posts/7610560664219-Filtering-Mutect-output-from-FFPE-based-targeted-panel-sequencing-data]
- 69. Proteogenomic characterization reveals tumorigenesis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11897189/]
- 70. Multi-omics analyses reveal biological and clinical insights ... [https://www.nature.com/articles/s41467-024-55068-2]
- 71. Spatiotemporal genomic profiling of intestinal metaplasia ... [https://www.sciencedirect.com/science/article/pii/S1535610823003604]
- 72. Integrated approach to generate artificial samples with low ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11077792/]
- 73. Detecting very low allele fraction variants using targeted DNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-016-3425-4]
- 74. The use of technical replication for detection of low-level ... [https://www.nature.com/articles/s41467-019-09026-y]
- 75. Errors about input files having missing or incompatible contigs [https://gatk.broadinstitute.org/hc/en-us/articles/360035891131-Errors-about-input-files-having-missing-or-incompatible-contigs]
- 76. Errors about contigs in BAM or VCF files not being properly ... [https://sites.google.com/a/broadinstitute.org/legacy-gatk-documentation/solutions-to-problems/1328-Errors-about-contigs-in-BAM-or-VCF-files-not-being-properly-ordered-or-sorted]
- 77. Question about bam format errors and handling large contigs [https://gatk.broadinstitute.org/hc/en-us/community/posts/16175388734747-Question-about-bam-format-errors-and-handling-large-contigs]
- 78. Genome in a Bottle | NIST [https://www.nist.gov/programs-projects/genome-bottle]
- 79. The GIAB genomic stratifications resource for human ... [https://www.nature.com/articles/s41467-024-53260-y]
- 80. usnistgov/giab-HG002-mosaic-benchmark [https://github.com/usnistgov/giab-HG002-mosaic-benchmark]
- 81. A guide to reviewing BAM files [https://epi2me.nanoporetech.com/reviewing-bam/]
- 82. A benchmarking study of individual somatic variant callers ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11790059/]
- 83. Towards an accurate and robust analysis pipeline for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9705725/]
- 84. Assessment of MuTect2 and VarScan2 for somatic ... [https://research-archive.org/index.php/rars/preprint/view/1207]
- 85. What variant calling method should I use? - GATK [https://gatk.broadinstitute.org/hc/en-us/community/posts/21209287099931-What-variant-calling-method-should-I-use]
- 86. Integrated approach to generate artificial samples with low ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05793-8]
- 87. Empirical evaluation of variant calling accuracy using ultra ... [https://www.nature.com/articles/s41598-018-38346-0]
- 88. Optimal sequencing depth design for whole genome re ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3164-z]
- 89. Variant calling: Considerations, practices, and developments [https://pmc.ncbi.nlm.nih.gov/articles/PMC9545713/]
- 90. bamCompare — deepTools 3.5.6 documentation [https://deeptools.readthedocs.io/en/develop/content/tools/bamCompare.html]
- 91. Introduction to RNA-Seq using high-performance computing [https://hbctraining.github.io/Intro-to-rnaseq-hpc-O2/lessons/04_alignment_quality.html]
- 92. Tool Documentation Index - GATK [https://gatk.broadinstitute.org/hc/en-us/articles/4413079147803--Tool-Documentation-Index]
- 93. Recommendations for bioinformatics in clinical practice [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-025-01543-4]
- 94. Best practices for the interpretation and reporting of clinical ... [https://www.nature.com/articles/s41525-022-00295-z]
- 95. Benchmarking of variant calling software for whole-exome ... [https://www.nature.com/articles/s41598-025-97047-7]
- 96. a long-read benchmark for genetic variants - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/40759746/]
- 97. Evaluating Discordant Somatic Calls Across Mutation ... [https://www.sciencedirect.com/science/article/pii/S1525157825001357?dgcid=rss_sd_all]
- 98. ONCOLINER: A new solution for monitoring, improving, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11480849/]