How PCA Reduces Gene Expression Data Dimensionality: A Guide for Biomedical Researchers
This article provides a comprehensive exploration of Principal Component Analysis (PCA) and its pivotal role in simplifying high-dimensional gene expression data.
How PCA Reduces Gene Expression Data Dimensionality: A Guide for Biomedical Researchers
Abstract
This article provides a comprehensive exploration of Principal Component Analysis (PCA) and its pivotal role in simplifying high-dimensional gene expression data. Tailored for researchers, scientists, and drug development professionals, it covers foundational concepts from its statistical origins to its function as a 'data summary' technique. The piece details methodological applications across transcriptomics and spatial transcriptomics, addresses critical troubleshooting aspects like overfitting and noise sensitivity, and validates PCA's utility through performance comparisons with alternative methods. By synthesizing theory and practical application, this guide empowers professionals to leverage PCA for efficient, interpretable analysis in complex biomedical datasets.
The Core of PCA: From Statistical Theory to Genomic Data Summary
Defining the High-Dimensionality Problem in Gene Expression Data
Gene expression data, particularly from high-throughput technologies like RNA sequencing (RNA-seq) and single-cell RNA sequencing (scRNA-seq), is inherently high-dimensional. A typical experiment generates expression measurements for tens of thousands of genes (features) across a much smaller number of biological samples (observations). This "high-dimension, low-sample size" (HDLSS) scenario presents significant challenges for statistical analysis, visualization, and biological interpretation [1] [2]. The core problem lies in the curse of dimensionality, where the vast feature space leads to data sparsity, increased computational complexity, and elevated risks of overfitting and identifying spurious correlations. Within this context, dimensionality reduction techniques—particularly Principal Component Analysis (PCA)—have become indispensable tools for making high-dimensional gene expression data computationally manageable and biologically interpretable. This technical guide examines the high-dimensionality problem and elucidates how PCA and related methods provide solutions within gene expression research, equipping scientists with practical frameworks for analyzing complex transcriptomic datasets.
The Nature of High-Dimensionality in Gene Expression Data
Fundamental Characteristics and Statistical Challenges
In bioinformatics, high-dimensionality refers to datasets where the number of features (p) vastly exceeds the number of observations (n). A standard RNA-seq experiment might profile 20,000-30,000 genes across only a dozen or so samples, creating a 20,000×12 data matrix [1]. This structure violates the fundamental assumptions of many classical statistical tests and machine learning algorithms designed for contexts where n >> p. The resulting data sparsity means that as dimensionality increases, data points become increasingly distant from each other, making it difficult to detect true clusters or patterns. Furthermore, the high feature-to-sample ratio dramatically increases the multiple testing burden in differential expression analysis, requiring stringent correction methods that can obscure genuinely significant findings [3].
Consequences for Analysis and Interpretation
The high-dimensional nature of gene expression data introduces several specific analytical challenges that impact biological interpretation. Table 1 summarizes the core problems and their implications for transcriptomic research.
Table 1: Key Challenges in High-Dimensional Gene Expression Data Analysis
| Challenge | Description | Impact on Analysis |
|---|---|---|
| Data Sparsity | As dimensions increase, data points become increasingly distant in feature space, occupying only a tiny fraction of the possible space. | Reduces effectiveness of clustering algorithms; makes it difficult to detect true patterns and relationships. |
| Computational Burden | Processing and storing large matrices (e.g., 30,000 genes × 1,000 samples) demands significant memory and processing power. | Slows down analysis; requires high-performance computing resources for large-scale studies. |
| Risk of Overfitting | Models with excessive parameters relative to observations may fit noise rather than true biological signal. | Produces models that fail to generalize to new datasets; leads to false discoveries. |
| Multiple Testing Problem | Evaluating thousands of genes simultaneously dramatically increases the chance of false positives. | Requires stringent statistical corrections (e.g., FDR) that may obscure true biological signals. |
| Visualization Difficulty | Humans cannot directly perceive relationships in spaces with thousands of dimensions. | Hinders exploratory data analysis and intuitive understanding of dataset structure. |
Principal Component Analysis (PCA): A Mathematical Foundation for Dimensionality Reduction
Core Conceptual Framework
Principal Component Analysis (PCA) is a linear dimensionality reduction technique that identifies the most informative directions of variance in high-dimensional data [2]. Mathematically, PCA performs an orthogonal transformation of the potentially correlated original variables (gene expression levels) into a new set of linearly uncorrelated variables called principal components (PCs). These components are ordered such that the first PC (PC1) captures the largest possible variance in the data, the second PC (PC2) captures the second largest variance while being orthogonal to the first, and so on. The resulting low-dimensional representation preserves the most significant patterns in the data while filtering out noise.
Mathematical Formulation
Given a centered gene expression matrix X with n samples and p genes, PCA is typically solved via eigen decomposition of the covariance matrix C = (1/(n-1))XᵀX. The principal components are the eigenvectors of C, and the amount of variance explained by each component is proportional to its corresponding eigenvalue. Alternatively, PCA can be computed via Singular Value Decomposition (SVD) of the centered data matrix: X = UΣVᵀ, where the columns of V are the principal components (loadings), and the product UΣ represents the principal component scores [2]. The loadings indicate the contribution of each original gene to each PC, while the scores represent the projection of each sample onto the new components, enabling low-dimensional visualization of sample relationships.
PCA in Practice: An Experimental Protocol for Gene Expression Analysis
Standardized Workflow for RNA-seq Data
Implementing PCA effectively requires a structured approach to data preprocessing, computation, and interpretation. The following protocol outlines key steps for applying PCA to RNA-seq count data, integrating best practices from established bioinformatics pipelines [1] [4].
Table 2: Key Research Reagent Solutions for PCA-Based Gene Expression Analysis
| Tool/Resource | Function | Implementation |
|---|---|---|
| DESeq2 | Normalizes raw count data to account for library size and RNA composition biases. | R/Bioconductor package |
| pcaExplorer | Interactive exploration of PCA results; generates publication-ready graphs and reports. | R/Bioconductor package |
| SingleCellExperiment | S4 class for storing and manipulating single-cell expression data. | R/Bioconductor package |
| Scanpy | Preprocessing, PCA, and visualization for large-scale single-cell data. | Python package |
| Factoextra | Visualizes PCA results, including scree plots and sample biplots. | R package |
| ideal | Interactive differential expression analysis integrating PCA for quality assessment. | R/Bioconductor package |
Step 1: Data Input and Normalization Begin with a raw count matrix (genes as rows, samples as columns) and associated sample metadata. Normalize the counts to correct for technical variations like library size and RNA composition. While PCA can be applied to raw counts, variance-stabilizing transformations (e.g., via DESeq2) or log-transformation of normalized counts (e.g., Counts Per Million) are often recommended to prevent highly expressed genes from dominating the variance structure [1] [5].
Step 2: Feature Selection and Data Scaling Select the most variable genes (e.g., top 500-5000) before performing PCA to reduce noise and computational load. Center the data (ensure each gene has a mean of zero) as PCA is sensitive to the mean. Scaling (setting variance to unity for each gene) is debated; it prevents high-expression genes from dominating but may inflate the influence of low-expression noise genes. The decision should align with the biological question [2].
Step 3: PCA Computation and Dimension Selection
Perform PCA using optimized functions (prcomp in R, scanpy.tl.pca in Python). The output includes sample scores (coordinates in PC space) and gene loadings (contributions to each PC). Determine the number of meaningful components to retain using a scree plot (variance vs. component number) or the elbow method, selecting components that capture significant biological variance [1] [3].
Step 4: Visualization and Interpretation Visualize samples in 2D or 3D plots using the first few PCs (e.g., PC1 vs. PC2) to assess sample clustering, batch effects, or outliers. Color points by experimental conditions from the metadata. Examine the gene loadings to identify which genes drive the separation observed along each component, linking patterns back to biology [1] [4].
Step 5: Functional and Reproducibility Analysis
Interpret the biological meaning of components by performing gene set enrichment analysis on genes with high absolute loadings for each significant PC. Ensure reproducibility by generating automated reports with tools like pcaExplorer, which captures the analysis state and parameters [1] [6].
The following diagram illustrates the logical relationships and workflow between these key steps.
Beyond Linearity: Advanced and Complementary Dimensionality Reduction Techniques
Expanding the Methodological Arsenal
While PCA is powerful for capturing global linear structures, gene expression data often contains complex nonlinear relationships. Several advanced techniques address this limitation, offering complementary approaches to dimensionality reduction [2] [7].
t-Distributed Stochastic Neighbor Embedding (t-SNE) focuses on preserving local neighborhood structures, often creating visually compelling cluster separations. However, t-SNE is computationally intensive and its global structure preservation can be sensitive to parameter tuning [3]. Uniform Manifold Approximation and Projection (UMAP) often provides a superior balance, preserving more of the global data structure than t-SNE while maintaining strong local clustering and offering faster computation [4] [2]. For the most complex patterns, Autoencoders (AEs) and Variational Autoencoders (VAEs) use deep learning to learn nonlinear latent representations, providing exceptional flexibility for capturing intricate hierarchical structures in large-scale datasets like scRNA-seq [8] [7].
Table 3: Comparison of Key Dimensionality Reduction Methods for Gene Expression Data
| Method | Type | Key Strength | Key Limitation | Typical Use Case |
|---|---|---|---|---|
| PCA [1] [2] | Linear | Computationally efficient; preserves global variance; interpretable. | Cannot capture nonlinear relationships. | Initial exploratory analysis; quality control; detecting batch effects. |
| t-SNE [3] | Nonlinear | Excellent at visualizing local clusters and complex groupings. | Computationally heavy; loses global structure. | Visualizing cell types or subtypes in scRNA-seq. |
| UMAP [4] [2] | Nonlinear | Better preservation of global structure than t-SNE; faster. | Can be sensitive to parameters; less interpretable than PCA. | Large-scale single-cell data visualization; trajectory inference. |
| Autoencoders [8] [7] | Nonlinear (DL) | Highly flexible; can model complex, hierarchical patterns. | "Black-box" nature; requires large datasets and expertise. | Analyzing very large, complex datasets (e.g., million-cell datasets). |
Method Selection and Hybrid Approaches
Choosing the appropriate method depends on the data scale and biological question. PCA remains the optimal starting point for most analyses due to its speed and interpretability. For detailed visualization of cellular heterogeneity in single-cell studies, UMAP or t-SNE are preferred. A powerful strategy involves combining methods: using PCA for initial noise reduction before applying UMAP or t-SNE, or employing PCA on a nonlinear latent space discovered by an autoencoder [2] [3]. Recent innovations like the Boosting Autoencoder (BAE) further enhance interpretability by identifying small, biologically relevant gene sets that explain latent dimensions, directly linking patterns back to specific genes and pathways [7].
The relationships between these methods, from linear to nonlinear and from traditional to deep learning-based, are visualized below.
The high-dimensionality of gene expression data presents fundamental challenges that are effectively addressed through dimensionality reduction. PCA stands as a cornerstone technique, providing a computationally efficient, interpretable framework for exploratory analysis, quality control, and initial data compression. Its linear nature offers a transparent foundation for understanding major sources of variation. However, the complexity of biological systems often necessitates complementary nonlinear methods like UMAP and deep learning approaches such as autoencoders for uncovering intricate patterns. The ongoing innovation in this space, including hybrid models and interpretable deep learning, continues to enhance our capacity to extract meaningful biological insights from increasingly complex genomic datasets. By strategically implementing these techniques within a structured analytical workflow, researchers can transform the high-dimensionality problem from an analytical obstacle into a source of discovery.
Principal Component Analysis (PCA) stands as a cornerstone multivariate technique in modern data science, with profound implications for dimensionality reduction in gene expression research. This whitepaper traces the methodological foundations of PCA to Karl Pearson's seminal 1901 work, elucidates its core statistical mechanics, and provides detailed experimental protocols for its application in transcriptomic data analysis. Within the context of gene expression research, PCA enables researchers to transform high-dimensional genomic data into lower-dimensional spaces, preserving essential biological signals while mitigating computational challenges. We demonstrate how proper application of PCA facilitates exploratory data analysis, outlier detection, and biological interpretation in pharmaceutical development and basic research.
Historical Foundations: The Pearsonian Revolution
Karl Pearson's Geometrical Innovation
The genesis of Principal Component Analysis dates to Karl Pearson's seminal 1901 paper "On Lines and Planes of Closest Fit to Systems of Points in Space," which introduced a fundamental shift in statistical reasoning [9]. Pearson addressed a critical limitation of traditional least-squares regression, which treated independent variables (x) as error-free and dependent variables (y) as subject to deviation. He recognized that in biological and physical systems, both variables often contain inherent uncertainty, necessitating a symmetrical approach to error minimization [9].
Pearson's novel conceptualization defined the core objective of PCA: to identify the "best-fitting" linear subspaces that represent multidimensional data. His approach minimized perpendicular distances from data points to the model subspace (Figure 1), in contrast to conventional regression that minimized vertical distances assuming certainty only in the y-variable. This geometrical innovation established the foundation for extracting principal components as the directions of maximum variance in correlated data [9].
From Pearson to Modern Multivariate Analysis
Pearson's initial framework has evolved through various methodological traditions under different names—including Factor Analysis, Singular Value Decomposition, Karhunen-Loève transformation, and Essential Dynamics—while retaining the core principles he established [9]. This cross-disciplinary dissemination underscores PCA's versatility as a "hypothesis-generating tool" that creates a statistical mechanics framework for biological systems modeling without requiring strong a priori theoretical assumptions [9]. In the era of high-throughput genomics, Pearson's approach provides precisely the methodological foundation needed to explore complex biological datasets, particularly in gene expression analysis where numerous correlated variables (gene expression levels) must be reduced to their essential features.
Statistical and Geometrical Basis of PCA
The Mathematics of Dimensionality Reduction
PCA operates on a fundamental principle: transforming correlated variables into a set of uncorrelated principal components that are linear combinations of the original variables. These components are derived in sequence, with each subsequent component capturing the maximum remaining variance while being orthogonal to all previous components [10].
Mathematically, principal components are constructed according to the formula: PC = aX₁ + bX₂ + cX₃ + ... + kXₙ where X₁–Xₙ represent the original variables (e.g., gene expression values) and the coefficients a, b, c,..., k are determined through eigen decomposition to maximize variance capture [9]. The resulting components provide the "best summary" of information in the original data in a least-squares sense while eliminating multicollinearity [9].
The PCA Workflow: A Step-by-Step Statistical Procedure
The implementation of PCA follows a systematic workflow that operationalizes Pearson's geometrical insight:
Standardization and Centering - Variables are transformed to comparable scales by subtracting means and dividing by standard deviations, ensuring that variables with larger ranges do not dominate the analysis [10].
Covariance Matrix Computation - The covariance matrix captures inter-variable relationships, with diagonal elements representing variances and off-diagonal elements representing covariances between variable pairs [10].
Eigen Decomposition - Eigenvectors and eigenvalues of the covariance matrix are computed, where eigenvectors define the directions of principal components (new axes), and eigenvalues quantify the variance captured by each component [10].
Component Selection - Components are ranked by decreasing eigenvalues, and a subset is selected based on variance contribution, typically using scree plots or variance retention thresholds (e.g., 95% cumulative variance) [11].
Data Projection - The original data is projected onto the selected component axes, creating a transformed dataset with reduced dimensionality [10].
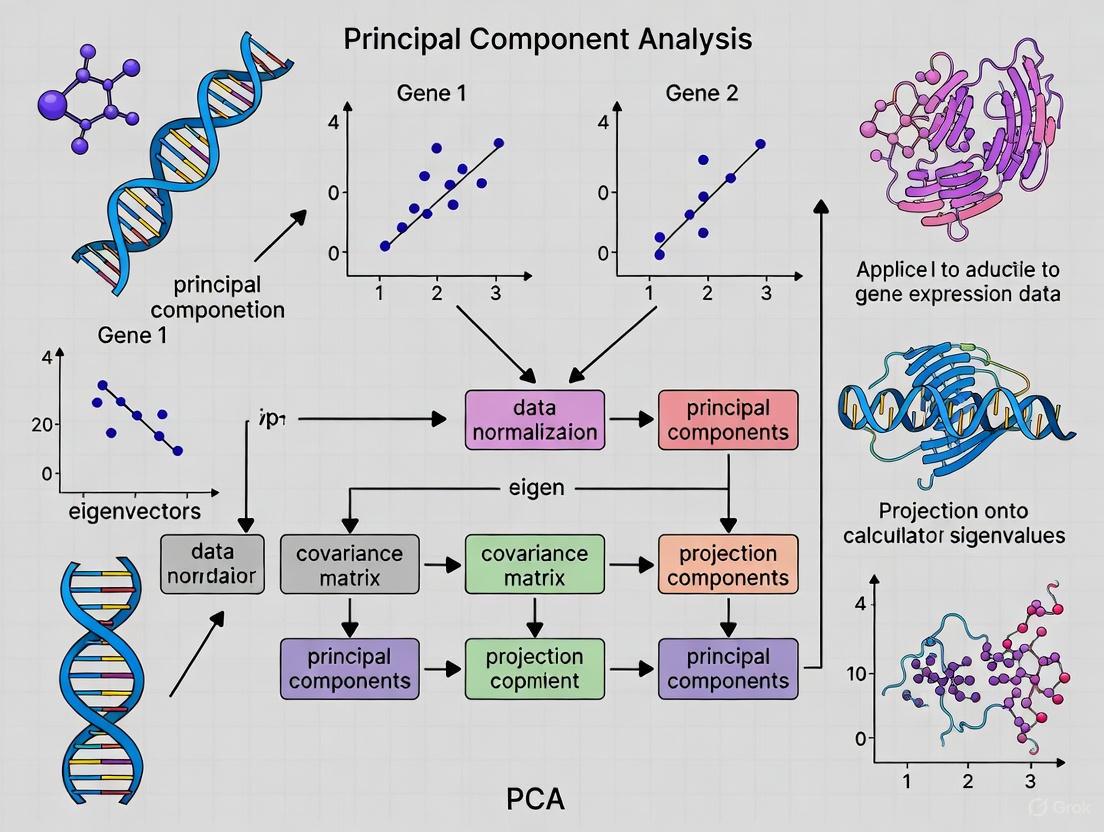
The following diagram illustrates this workflow in the context of gene expression analysis:
Figure 1: PCA Workflow for Gene Expression Data
Variance Interpretation and Component Selection
The information content of principal components is quantified through eigenvalues, with larger eigenvalues indicating components that capture more variance. The proportion of total variance explained by each component is calculated as the ratio of its eigenvalue to the sum of all eigenvalues [10]. In gene expression studies, the cumulative variance approach typically retains components that collectively explain 70-95% of total variance, though biological interpretability often supersedes strict percentage thresholds [11].
PCA in Gene Expression Data Analysis
The Dimensionality Challenge in Transcriptomics
Modern transcriptomic datasets present substantial analytical challenges with tens of thousands of genes measured across multiple samples, creating high-dimensional spaces where traditional analysis methods struggle [11]. The "curse of dimensionality" manifests through data sparsity, increased risk of overfitting, and computational inefficiency [11]. PCA addresses these issues by identifying the dominant patterns of variation, effectively reducing thousands of gene expression measurements to a manageable number of composite variables while preserving essential biological information [12].
Normalization Critical Considerations
The application of PCA to RNA-sequencing data requires careful normalization prior to analysis. Different normalization methods significantly impact PCA results and biological interpretation [13]. A comprehensive evaluation of twelve normalization methods for RNA-seq data demonstrated that while PCA score plots may appear similar across normalization techniques, the biological interpretation of components can vary substantially [13]. Without proper normalization, technical artifacts such as sequencing depth can dominate the first principal component, obscuring biological signals [12].
Table 1: Variance Explained by Principal Components in Gene Expression Studies
| Principal Component | Percentage of Variance Explained | Typical Biological Interpretation |
|---|---|---|
| PC1 | 15-30% | Sequencing depth, major tissue type, or dominant biological factor |
| PC2 | 8-15% | Secondary biological factor, batch effects |
| PC3 | 5-10% | Tertiary biological factor, additional batch effects |
| PC4+ | <5% each | Subtle biological signals, noise |
| Cumulative (PC1-PC10) | 40-70% | Total variance captured for downstream analysis |
Experimental Protocol: PCA for Transcriptomic Data
The following protocol provides a standardized approach for applying PCA to RNA-sequencing data, incorporating critical considerations for normalization and interpretation:
Data Preprocessing
- Data Acquisition: Obtain raw count data from RNA-sequencing experiments. Publicly available sources include TCGA, GTEx, and ICGC [14].
- Filtering: Remove genes with zero counts across all samples and apply expression thresholding (e.g., counts per million > 1 in at least 20% of samples) [14].
- Normalization: Apply appropriate normalization method to address sequencing depth and compositional effects. Options include:
- Quantile normalization [14]
- TMM (Trimmed Mean of M-values)
- DESeq2's median of ratios
- Transformation: Convert normalized counts to log2 scale to stabilize variance [14].
- Standardization: Center each gene to zero mean and unit variance [10].
PCA Implementation
- Covariance Matrix Computation: Calculate the gene-by-gene covariance matrix on the preprocessed data [10].
- Eigen Decomposition: Perform singular value decomposition on the covariance matrix to obtain eigenvectors (principal components) and eigenvalues (variances) [11].
- Component Selection:
- Generate scree plot of eigenvalues
- Calculate cumulative variance explained
- Select components that collectively explain >70% variance or until eigenvalue <1 (Kaiser criterion)
- Result Interpretation:
- Examine component loadings to identify genes contributing most to each PC
- Project samples into PC space for visualization
- Correlate PC scores with sample metadata (e.g., tissue type, treatment)
Validation and Downstream Analysis
- Stability Assessment: Perform cross-validation to evaluate component stability [15].
- Biological Interpretation: Conduct gene set enrichment analysis on high-loading genes for each component [13].
- Visualization: Generate biplots to simultaneously visualize samples and variables in reduced space [15].
Information Content in Higher Components
While early transcriptomic studies suggested limited utility beyond the first 2-3 principal components, recent evidence indicates important biological signals reside in higher components. Research on large heterogeneous gene expression datasets (7,100 samples) revealed that while the first three components explained approximately 36% of variance, significant tissue-specific information remained in higher components [16]. When analyzing specific tissue subsets (e.g., brain regions), components beyond the first three captured biologically relevant differentiation [16].
The following diagram illustrates how PCA reveals hierarchical biological structure in gene expression data:
Figure 2: Hierarchical Biological Structure in PCA of Gene Expression Data
Applications in Drug Discovery and Development
Quantitative Structure-Activity Relationships (QSAR)
PCA has become an indispensable tool in QSAR studies, where it analyzes matrices of molecular descriptors to identify dominant structural properties influencing biological activity [17]. In a case study on CCR5 antagonists, PCA enabled researchers to navigate complex descriptor spaces, identify outliers, and select appropriate modeling approaches for further analysis [17]. By reducing hundreds of molecular descriptors to a few principal components, PCA facilitates the understanding of structure-activity relationships and guides rational drug design.
Network Pharmacology and Systems Biology
The emergence of network pharmacology requires methods that can capture system-level responses to therapeutic interventions. PCA provides a framework for analyzing complex correlation structures in biological networks, identifying latent factors that underlie observed phenotypic responses [9]. This approach aligns with Pearson's original vision of capturing system behavior through correlation structure analysis, enabling researchers to move beyond reductionist models to more comprehensive representations of biological complexity [9].
Table 2: PCA Applications in Drug Discovery Pipeline
| Application Area | PCA Function | Impact on Drug Development |
|---|---|---|
| Target Identification | Analysis of gene expression patterns across tissues and conditions | Identifies disease-relevant pathways and potential therapeutic targets |
| Lead Optimization | Reduction of molecular descriptor space in QSAR | Prioritizes compound series with desirable properties, reduces synthetic effort |
| Toxicology Assessment | Pattern recognition in metabolomic and transcriptomic data | Identifies safety liabilities early in development process |
| Biomarker Discovery | Identification of dominant variation patterns in patient populations | Stratifies patient populations, identifies predictive biomarkers |
| Clinical Trial Analysis | Multidimensional assessment of efficacy endpoints | Provides comprehensive view of drug effects, supports regulatory decisions |
Methodological Considerations and Best Practices
Data Preprocessing Critical Impact
The choice of data preprocessing methods significantly influences PCA outcomes in transcriptomic studies. Research comparing RNA-Seq data preprocessing pipelines demonstrated that normalization, batch effect correction, and scaling decisions affect downstream classification performance [14]. Interestingly, while batch effect correction improved performance when applied to certain datasets (TCGA to GTEx), it worsened performance with other independent test sets (ICGC/GEO) [14]. This underscores the context-dependent nature of preprocessing decisions and the need for careful pipeline validation.
Alternative Dimensionality Reduction Techniques
While PCA remains the most widely used linear dimensionality reduction technique, several alternatives offer complementary approaches:
Table 3: Comparison of Dimensionality Reduction Methods for Biological Data
| Method | Input Data | Distance Measure | Best Application Context |
|---|---|---|---|
| PCA | Original feature matrix | Covariance/Euclidean | Linear data, feature extraction, exploratory analysis |
| PCoA | Distance matrix | Any distance metric (Bray-Curtis, Jaccard) | Ecological distances, beta-diversity analysis |
| NMDS | Distance matrix | Rank-order preservation | Complex datasets, non-linear relationships |
| t-SNE | Similarity matrix | Probability distributions | Visualization of high-dimensional data |
| UMAP | Nearest neighbor graph | Topological preservation | Visualization, clustering preservation |
PCA is particularly effective for linear data structures and serves as an excellent first step in exploratory analysis, while PCoA and NMDS offer advantages when analyzing ecological distances or non-linear relationships [18]. The selection of method should align with data characteristics and research objectives.
Table 4: Essential Computational Tools for PCA in Gene Expression Analysis
| Tool/Resource | Function | Application Context |
|---|---|---|
| R Statistical Environment | Programming platform for statistical computing | Primary analysis environment with specialized packages |
| Python (scikit-learn) | Machine learning library with PCA implementation | Integrative analysis pipelines, machine learning workflows |
| TCGAbiolinks | Bioconductor package for TCGA data access | Standardized retrieval of cancer transcriptomic data |
| DESeq2/edgeR | Differential expression analysis | Normalization and preprocessing of RNA-seq count data |
| ggplot2/plotly | Visualization libraries | Generation of publication-quality PCA score plots |
| FactoMineR | Multivariate analysis package | Comprehensive PCA implementation with enhanced diagnostics |
| Enrichr/clusterProfiler | Gene set enrichment analysis | Biological interpretation of principal components |
Karl Pearson's century-old geometrical insight continues to fuel modern scientific discovery, particularly in the high-dimensional landscape of gene expression research. PCA provides an indispensable framework for reducing dimensionality while preserving essential biological information, enabling researchers to navigate complex transcriptomic datasets and extract meaningful patterns. As genomic technologies generate increasingly large and complex datasets, Pearson's legacy endures through the ongoing evolution and application of principal component analysis in biological research and drug development. The method's unique combination of mathematical elegance and practical utility ensures its continued relevance in an era of data-intensive biology.
Principal Component Analysis (PCA) stands as a foundational dimensionality reduction technique in computational biology, transforming high-dimensional gene expression data into a lower-dimensional space of synthetic variables known as principal components (PCs). Within gene expression analysis, these PCs function conceptually as "metagenes"—linear combinations of original genes that capture coordinated expression patterns across biological samples. This whitepaper provides an in-depth technical examination of PCA's mathematical framework, its implementation as a feature extraction method in transcriptomics, and its critical role in elucidating biological structure through metagenes. We further present standardized protocols for applying PCA to single-cell RNA sequencing (scRNA-Seq) data, benchmark performance against alternative methods, and discuss both interpretative advantages and limitations relevant to drug discovery and biomedical research.
Modern genomic technologies, particularly single-cell RNA sequencing (scRNA-Seq), generate data of extreme dimensionality, where each of thousands or millions of cells is characterized by expression measurements for 20,000-30,000 genes [19]. This high-dimensional space presents significant challenges for computational analysis, visualization, and biological interpretation—a phenomenon known as the "curse of dimensionality" [20].
In this context, dimensionality reduction techniques like PCA become essential preprocessing steps that address multiple analytical challenges:
- Computational efficiency: Reducing dataset size enables faster downstream analysis [21]
- Noise reduction: Filtering out technical variance and highlighting biologically meaningful signals [19]
- Visualization: Enabling exploration of data structure in 2D or 3D plots [10] [22]
- Overfitting prevention: Mitigating the risk of models memorizing noise rather than learning biological patterns [21]
The concept of "metagenes" emerges naturally from PCA's mathematical foundation, representing latent variables that capture coordinated biological programs across cell populations or experimental conditions [19].
Mathematical Foundation of PCA
Core Principles
PCA is an orthogonal linear transformation that converts correlated variables into a set of uncorrelated principal components, ordered by the amount of variance they explain from the original data [22]. The transformation is designed such that:
- The first principal component (PC1) captures the maximum possible variance in the data
- Each subsequent component captures the maximum remaining variance while being orthogonal to previous components [10] [22]
Mathematically, given a data matrix ( X ) with dimensions ( n × p ) (where ( n ) is the number of samples and ( p ) is the number of genes), PCA seeks the set of eigenvectors ( w ) that satisfy: [ w{(1)} = \arg \max{\|w\|=1} \left{ \sumi (x{(i)} · w)^2 \right} ] where ( w_{(1)} ) is the first principal component [22].
The Five Steps of PCA Implementation
The practical implementation of PCA follows a standardized five-step process [10]:
Standardization: Centering and scaling each variable to mean=0 and variance=1, ensuring equal contribution from all genes regardless of their original expression ranges [10]
Covariance Matrix Computation: Constructing a ( p × p ) symmetric matrix that captures how all pairs of genes vary together, with entries indicating:
- Positive covariance: Genes increase or decrease together
- Negative covariance: One gene increases as the other decreases
- Zero covariance: No linear relationship between genes [10]
Eigen Decomposition: Calculating eigenvectors and eigenvalues of the covariance matrix, where:
- Eigenvectors represent the principal components (directions of maximum variance)
- Eigenvalues indicate the amount of variance carried by each component [10]
Feature Selection: Sorting eigenvectors by decreasing eigenvalues and selecting the top ( k ) components that capture sufficient cumulative variance [10]
Data Projection: Transforming the original data into the new subspace via matrix multiplication ( T = XW ), where ( W ) contains the selected eigenvectors [10]
Metagenes as Biological Constructs
In the context of gene expression data, each principal component constitutes a metagene—a weighted linear combination of original genes where loadings (eigenvector entries) indicate each gene's contribution to the component [19]. These metagenes represent latent expression programs that may correspond to:
- Coregulated gene modules under common transcriptional control
- Biological pathways activated in specific cell states
- Technical artifacts or batch effects requiring statistical correction
Table 1: Interpretation of PCA Outputs in Gene Expression Studies
| PCA Output | Mathematical Meaning | Biological Interpretation |
|---|---|---|
| Eigenvectors | Direction of maximum variance | "Metagenes" - coordinated expression programs |
| Eigenvalues | Variance along each component | Importance of each expression program |
| Projections (Scores) | Sample coordinates in new space | Sample expression profile based on metagenes |
| Loadings | Gene contributions to components | Individual gene weights in each metagene |
PCA in Single-Cell RNA Sequencing Workflows
Standardized Protocol for scRNA-Seq Data
The following experimental protocol details PCA implementation specifically for single-cell transcriptomics:
Input Requirements:
- Normalized count matrix (cells × genes) with batch effects mitigated
- Minimum quality control: Removal of low-quality cells, dead cells, and empty droplets
- Recommended: Log-transformation and normalization for sequencing depth
Procedure:
- Feature Selection: Identify highly variable genes (HVGs) across the cell population
- Data Scaling: Center and scale HVG expression values to mean=0, variance=1
- PCA Execution: Perform eigendecomposition on scaled expression matrix
- Component Selection: Determine significant PCs using elbow method or statistical tests
- Downstream Application: Use PC embeddings for clustering, visualization, or trajectory inference
Component Selection Criteria:
- Elbow method: Visual identification of variance explained "elbow" in scree plot [19]
- Statistical approaches: Tracy-Widom test for significant deviation from random noise [23]
- Arbitrary thresholds: Retain components explaining >90% cumulative variance [19]
The following workflow diagram illustrates the standard PCA pipeline in scRNA-Seq analysis:
Research Reagents and Computational Tools
Table 2: Essential Research Reagents and Software for PCA in Genomic Studies
| Resource | Type | Function/Purpose |
|---|---|---|
| Cell Ranger [19] | Software Pipeline | Preprocessing of 10x Genomics scRNA-Seq data |
| EIGENSOFT/SmartPCA [23] | Specialized Software | Population genetics-focused PCA implementation |
| Seurat / Scanpy [19] | Analysis Toolkit | Comprehensive scRNA-Seq analysis including PCA |
| Scikit-learn [10] | Python Library | General-purpose PCA implementation |
| UMI Filtering [19] | Molecular Technology | Unique Molecular Identifiers for accurate gene counting |
Benchmarking PCA Against Alternative Dimensionality Reduction Methods
Performance Comparison in Biological Contexts
Recent benchmarking studies have evaluated PCA alongside numerous linear and nonlinear dimensionality reduction techniques specifically for transcriptomic data [24]. The table below summarizes key findings:
Table 3: Benchmarking of Dimensionality Reduction Methods for Transcriptomic Data
| Method | Type | Key Strengths | Limitations in Genomic Context |
|---|---|---|---|
| PCA [24] | Linear | Computational efficiency, interpretability, preservation of global structure | Limited capture of nonlinear relationships |
| t-SNE [24] | Nonlinear | Excellent local structure preservation, effective clustering | Computational intensity, loss of global structure |
| UMAP [24] | Nonlinear | Balance of local/global structure, runtime efficiency | Parameter sensitivity, less interpretable |
| PaCMAP [24] | Nonlinear | Strong performance without parameter tuning | Less established in biological domains |
| PHATE [24] | Nonlinear | Superior capture of branching trajectories | Specialized primarily for trajectory inference |
In drug discovery contexts, PCA has demonstrated particular utility for analyzing drug-induced transcriptomic changes in resources like the Connectivity Map (CMap), where it effectively groups compounds with similar mechanisms of action while separating those with distinct targets [24].
Theoretical Foundations: The Genealogical Interpretation
A fundamental theoretical framework connects PCA to population genetics concepts, demonstrating that for single nucleotide polymorphism (SNP) data, sample projections onto principal components directly reflect average coalescence times between pairs of haploid genomes [25]. This genealogical interpretation provides a population-genetic foundation for understanding PCA outputs:
[ \textbf{E}[X{ij}] = \textbf{E}[\delta{is} - \mus] = \textbf{Cov}(\delta{is}, \delta{js}) - \textbf{Cov}(\delta{is}, \mus) - \textbf{Cov}(\delta{js}, \mus) + \textbf{Var}(\mus) ]
where ( \delta{is} ) represents allele frequency differences and ( \mus ) represents population means [25].
This framework enables interpretation of PCA results in terms of underlying demographic processes, including:
- Population migration and admixture events
- Geographical isolation and genetic drift
- Historical bottlenecks and founder effects
The following diagram illustrates the mathematical relationships underlying the genealogical interpretation of PCA:
Critical Considerations and Limitations
Methodological Artifacts and Biases
Despite its widespread application, PCA presents significant limitations that researchers must consider:
Sensitivity to Data Composition: PCA outcomes are strongly influenced by sample composition, where inclusion or exclusion of specific populations can dramatically alter component orientations [23]. In one comprehensive evaluation, PCA demonstrated vulnerability to generating "artifacts of the data" rather than true biological structure [23].
Variance Interpretation Challenges: In population genetics, the proportion of variance explained by principal components may not reliably reflect biological importance, as major axes of variation can represent technical artifacts or evolutionarily neutral structure [23].
Linearity Assumption: PCA's fundamental limitation as a linear method constrains its ability to capture complex nonlinear relationships common in biological systems, potentially necessitating complementary nonlinear approaches [24].
Validation and Interpretation Guidelines
To ensure robust biological interpretation of PCA results:
Technical Validation:
- Assess batch effects across experimental conditions
- Perform sensitivity analysis with sample subsets
- Compare multiple normalization strategies
Biological Corroboration:
- Integrate with complementary methods (e.g., clustering, trajectory inference)
- Correlate metagene expression with known biological markers
- Validate findings through experimental perturbation
Statistical Rigor:
- Apply appropriate significance testing for component selection
- Avoid overinterpretation of low-variance components
- Report variance explained alongside visualization
Future Directions and Advanced Applications
Integrative Approaches
Emerging methodologies combine PCA with other analytical frameworks to enhance feature selection and interpretation:
PCA-MCDM Fusion: Integration of PCA with Multi-Criteria Decision-Making (MCDM) methods provides a robust framework for unsupervised feature selection in bioinformatics, evaluating genes against multiple criteria simultaneously [26].
Deep Learning Extensions: Variational autoencoders and other neural architectures extend PCA concepts to capture nonlinear relationships while maintaining interpretability [19].
Specialized Applications in Drug Development
PCA-derived metagenes enable several advanced applications in pharmaceutical research:
Mechanism of Action Prediction: Metagenes representing conserved transcriptional programs facilitate classification of novel compounds by mechanism of action through pattern matching against reference databases [24].
Dose-Response Modeling: Subtle transcriptomic changes across drug concentrations can be captured through specialized application of PCA and related methods [24].
Biomarker Discovery: Metagenes representing coordinated pathway activity serve as robust biomarkers for patient stratification and treatment response prediction.
Principal Component Analysis serves as an essential transformation in the genomic data analysis pipeline, converting high-dimensional gene expression measurements into interpretable metagenes that capture coordinated biological programs. While limitations regarding sensitivity to data composition and linearity assumptions necessitate complementary approaches, PCA's computational efficiency, mathematical elegance, and interpretability ensure its continued relevance. For drug development professionals and biomedical researchers, understanding both the theoretical foundations and practical implementation of PCA remains crucial for extracting meaningful biological insights from complex transcriptomic datasets. Future methodological developments will likely focus on hybrid approaches that maintain PCA's interpretability while capturing the nonlinear relationships inherent in biological systems.
In the analysis of gene expression data, researchers are frequently confronted with the "large d, small n" problem, where the number of genes (d) vastly exceeds the number of samples (n) [27]. Principal Component Analysis (PCA) addresses this challenge by performing a linear dimensionality reduction that transforms the high-dimensional gene expressions into a lower-dimensional set of orthogonal principal components (PCs), often referred to as "metagenes" or "super genes" in bioinformatics literature [27]. This transformation allows researchers to project samples with tens of thousands of gene expressions onto just two or three dimensions for visualization, identify sample clusters, and detect batch effects [28]. The technique is computationally simple and has become fundamental to bioinformatics data analysis, enabling researchers to overcome the curse of dimensionality while preserving essential patterns in the data.
Mathematical Foundations of PCA
Core Mathematical Framework
PCA is fundamentally an orthogonal linear transformation that maps data to a new coordinate system [22]. Consider a gene expression dataset represented as an ( n \times p ) matrix ( \mathbf{X} ), where ( n ) is the number of samples and ( p ) is the number of genes. Each element ( x_{ij} ) represents the expression level of gene ( j ) in sample ( i ). PCA seeks a set of new variables (principal components) that are linear combinations of the original genes:
[ PCk = w{1k}x1 + w{2k}x2 + \cdots + w{pk}x_p ]
where ( PCk ) is the ( k )-th principal component, and ( \mathbf{w}{(k)} = (w{1k}, w{2k}, \dots, w{pk}) ) is the weight vector for the ( k )-th component [22]. The first weight vector ( \mathbf{w}{(1)} ) must satisfy:
[ \mathbf{w}{(1)} = \arg\max{\|\mathbf{w}\|=1} \left{ \sumi (t1){(i)}^2 \right} = \arg\max{\|\mathbf{w}\|=1} \left{ \sumi (\mathbf{x}{(i)} \cdot \mathbf{w})^2 \right} ]
where ( t_1 ) represents the scores of the first principal component [22]. This optimization problem can be solved via eigendecomposition of the covariance matrix ( \mathbf{X}^T\mathbf{X} ) or singular value decomposition (SVD) of the data matrix ( \mathbf{X} ) itself [27] [22].
Geometric Interpretation and Variance Maximization
Geometrically, PCA can be understood as fitting a p-dimensional ellipsoid to the data, where each axis of the ellipsoid represents a principal component [22]. The principal components are the directions along which the data exhibits maximum variance. The process begins with mean-centering each variable in the dataset, followed by computation of the covariance matrix. The eigenvectors of this covariance matrix form the principal components, while the corresponding eigenvalues represent the amount of variance explained by each component [22]. The proportion of total variance explained by the ( k )-th principal component is calculated as:
[ \text{Proportion}k = \frac{\lambdak}{\sum{j=1}^p \lambdaj} ]
where ( \lambda_k ) is the eigenvalue corresponding to the ( k )-th principal component [29]. In practical terms, PCA rotates the original set of ( n ) axes (corresponding to the ( n ) measured variables) to find a new axis (the first principal component) that explains as much of the total variance as possible [30]. Subsequent components are then found sequentially, each explaining the maximum possible residual variance while being orthogonal to all previous components.
Figure 1: Geometric Interpretation of PCA as Axis Rotation
Core Mathematical Concepts: Loadings, Scores, and Variance
Loadings: The Building Blocks of Components
Loadings (also called eigenvectors or weights) define the orientation of the principal components relative to the original variables [30] [29]. Mathematically, the loadings are the coefficients ( w{1k}, w{2k}, \dots, w_{pk} ) in the linear combination that defines each principal component. These coefficients indicate the relative contribution of each original variable to that specific principal component.
For a dataset with two original variables, the loadings for the first principal component are defined by the cosine of its angle of rotation relative to each of the original axes [30]. If PC1 has a rotation angle of θ relative to variable 1, then its loading for variable 1 is cos(θ), and for variable 2 is cos(90° - θ) (or sin(θ)) [30]. Since individual loadings are defined by a cosine function, they are limited to the range -1 to +1 [30]. The sign of the loading indicates how a variable contributes to the principal component: a positive loading suggests the variable's presence contributes to the component, while a negative loading indicates its absence contributes [30].
Scores: The Projected Coordinates
Scores represent the coordinates of the original data points in the new principal component space [30]. After determining the principal component axes (loadings), the score for a given sample on the k-th principal component is calculated as the linear combination of its original variable values weighted by the loadings:
[ tk(i) = x{(i)} \cdot w{(k)} = w{1k}x{1(i)} + w{2k}x{2(i)} + \cdots + w{pk}x_{p(i)} ]
where ( tk(i) ) is the score of sample i on the k-th principal component, ( x{j(i)} ) is the value of variable j for sample i, and ( w_{jk} ) is the loading of variable j on component k [22]. In gene expression analysis, these scores allow researchers to visualize high-dimensional data in a reduced space (typically 2D or 3D), enabling the identification of sample clusters, outliers, and patterns [27] [30].
Variance Explained: Measuring Information Content
The variance explained by each principal component quantifies its importance in capturing the variability within the dataset [29]. The total variance in the data is the sum of the variances of all original variables (when variables are standardized). The eigenvalue ( \lambda_k ) corresponding to the k-th principal component represents the variance captured by that component [29]. The proportion of total variance explained by the k-th component is:
[ \text{Proportion}k = \frac{\lambdak}{\sum{j=1}^p \lambdaj} ]
The cumulative variance explained by the first m components is the sum of their individual proportions [29]. In practice, researchers often retain only the first few principal components that explain a sufficient amount of variance (e.g., 70-90%), effectively reducing dimensionality while preserving most meaningful information [27] [29].
Table 1: Interpretation of PCA Outputs in Gene Expression Studies
| Component | Loadings Interpretation | Scores Interpretation | Variance Explained |
|---|---|---|---|
| PC1 | Genes with highest absolute loadings contribute most; reveals primary expression pattern | Positions samples along dominant expression pattern; reveals major sample groupings | Typically explains largest variance percentage; may represent major biological factor |
| PC2 | Genes orthogonal to PC1 pattern; reveals secondary expression signature | Positions samples along secondary axis; may represent subtler biological effects | Usually less than PC1; combined with PC1 often explains substantial total variance |
| Subsequent PCs | Progressively capture residual patterns; may represent noise or subtle biological signals | Finer sample discrimination; may correlate with minor biological factors or technical artifacts | Diminishing returns; later components often discarded as noise |
PCA Workflow for Gene Expression Data
Standard Analytical Procedure
The application of PCA to gene expression data follows a systematic workflow designed to transform raw expression measurements into meaningful biological insights. A comprehensive protocol includes the following steps:
Data Preprocessing: Gene expression data should be properly normalized before PCA. Typically, expression values are centered to mean zero, and sometimes scaled to unit variance to make genes more comparable [27]. This step is crucial as PCA is sensitive to variable scales.
Covariance Matrix Computation: Calculate the sample variance-covariance matrix based on the normalized expression values across all samples [27]. For p genes, this produces a p × p covariance matrix that captures the relationships between all gene pairs.
Eigendecomposition: Perform eigendecomposition of the covariance matrix to extract eigenvalues and eigenvectors [27] [22]. This can be achieved using standard singular value decomposition (SVD) techniques available in most statistical software packages [27].
Component Selection: Sort the eigenvectors by decreasing magnitude of their corresponding eigenvalues [29]. The eigenvector with the largest eigenvalue becomes the first principal component, and so on. Determine how many components to retain based on eigenvalues (>1 according to Kaiser criterion), scree plot analysis, or cumulative variance explained (typically 70-90%) [29].
Score Calculation: Compute principal component scores for each sample by projecting the original data onto the selected components [30] [29]. These scores serve as new coordinates for samples in the reduced-dimensional space.
Interpretation and Visualization: Create biplots showing both sample scores and variable loadings, scree plots displaying variance explained by each component, and score plots colored by experimental conditions to identify patterns and clusters [27] [22].
Figure 2: PCA Workflow for Gene Expression Data Analysis
Research Reagents and Computational Tools
Table 2: Essential Computational Tools for PCA in Bioinformatics
| Tool/Software | Specific Function/Package | Application Context |
|---|---|---|
| R Statistical Environment | prcomp(), princomp(), FactoMineR | Comprehensive PCA implementation with extensive visualization capabilities |
| Python Scientific Stack | scikit-learn (decomposition.PCA), SciPy, NumPy | Custom analysis pipelines and integration with machine learning workflows |
| SAS | Procedures PRINCOMP and FACTOR | Enterprise-level statistical analysis with robust validation |
| SPSS | Factor function (Data Reduction menu) | User-friendly interface for basic PCA applications |
| MATLAB | princomp(), pca() functions | High-performance numerical computation for large-scale datasets |
| Specialized Bioinformatics Tools | NIA Array Analysis Tool | Domain-specific implementations for genomic data |
Advanced PCA Applications in Drug Discovery
Network and Pathway-Based PCA
Traditional PCA applied to entire gene expression datasets has evolved into more sophisticated approaches that incorporate biological domain knowledge. Recent methodologies conduct PCA on genes within predefined biological pathways or network modules rather than across all genes simultaneously [27]. In this framework, PCA is performed separately on genes within each pathway, and the resulting principal components are used as covariates in downstream analyses [27]. These components effectively represent the coordinated activity of entire pathways, providing a more biologically interpretable reduction of dimensionality.
This pathway-based PCA approach offers significant advantages for drug discovery applications. First, it aligns dimensionality reduction with biologically meaningful units (pathways) known to be important in disease mechanisms. Second, it naturally accommodates interaction effects between pathways. For instance, when studying two pathways containing different gene sets, researchers can conduct PCA on each pathway separately and include interaction terms between the resulting components in predictive models [27]. This enables the detection of non-additive effects between biological pathways that might be missed by conventional analyses.
Supervised and Sparse PCA Variations
Standard PCA is an unsupervised technique that doesn't incorporate sample labels or outcomes. Supervised PCA extends the method by incorporating response variable information to guide the dimension reduction, often resulting in components with better predictive performance for specific biological outcomes [27]. This approach is particularly valuable in drug discovery when trying to relate gene expression patterns to drug response phenotypes.
Sparse PCA addresses another limitation of traditional PCA: the fact that each principal component typically includes nonzero loadings for all genes, making biological interpretation challenging [27]. Sparse PCA introduces regularization constraints that force many loadings to exactly zero, resulting in components defined by smaller, more interpretable gene sets. This enhances the ability to identify specific genes driving the observed patterns, which is crucial for identifying potential drug targets.
Table 3: Comparison of PCA Variants in Pharmaceutical Research
| Method | Key Features | Advantages | Limitations |
|---|---|---|---|
| Standard PCA | Unsupervised; Maximizes variance; All genes contribute to all components | Computational simplicity; Widely implemented; Preserves global data structure | Difficult biological interpretation; No incorporation of outcome variables |
| Supervised PCA | Incorporates response variables; Outcome-guided dimension reduction | Improved predictive performance; Enhanced relevance to specific phenotypes | Risk of overfitting; More complex implementation |
| Sparse PCA | Regularization forces zero loadings; Sparse component structure | More interpretable results; Identifies key driver genes | Additional tuning parameters; May miss subtle coordinated effects |
| Functional PCA | Designed for time-course data; Models temporal patterns | Captures dynamic biological processes; Suitable for longitudinal studies | Increased mathematical complexity; Requires careful experimental design |
Biomarker Identification and Drug Response Prediction
PCA-based approaches have demonstrated significant utility in identifying biomarkers and predicting drug response in pharmaceutical applications. In transcriptomic studies of drug response, PCA and other dimensionality reduction methods have been benchmarked for their ability to capture drug-induced gene expression changes [31]. These methods help in separating distinct drug responses and grouping drugs with similar molecular mechanisms of action, facilitating drug repositioning and mechanism elucidation [31].
Beyond transcriptomic data, PCA finds applications in structural biology and molecular dynamics simulations relevant to drug discovery. When analyzing molecular dynamics trajectories of protein-ligand complexes, PCA can reduce the high-dimensional atomic coordinate data to reveal essential collective motions [32]. This helps in understanding how different ligands influence protein conformational sampling, which has direct implications for rational drug design and optimizing compound selectivity.
Principal Component Analysis provides a powerful mathematical framework for addressing the high-dimensionality challenges inherent in gene expression research and drug discovery. Through the decomposition of data into orthogonal components defined by loadings, scores, and explained variance, PCA enables researchers to distill complex genomic information into interpretable patterns. The continuous evolution of PCA methodologies—including supervised, sparse, and pathway-aware variants—ensures its ongoing relevance in extracting biologically meaningful insights from increasingly complex datasets. As pharmaceutical research continues to embrace systems-level approaches, PCA's ability to reveal latent structures in high-dimensional data will remain indispensable for connecting molecular profiles to therapeutic outcomes.
Within high-dimensional biological research, particularly gene expression analysis, the challenge of modeling relationships amidst numerous variables is paramount. This article delineates a fundamental paradigm shift between Principal Component Analysis (PCA), a dimensionality reduction technique, and Classical Regression. While classical regression models an outcome variable based on predictor variables, explicitly modeling and minimizing prediction error, PCA adopts a fundamentally different approach. It is an unsupervised method designed to reduce data dimensionality by creating new, uncorrelated variables that successively capture maximum variance from the entire dataset without reference to a specific outcome [33]. Framed within the context of single-cell RNA sequencing (scRNA-seq) data analysis, this whitepaper explores how PCA's variance-centric model, in contrast to the error-minimization model of regression, provides a powerful foundation for interpreting complex genomic data, enhancing computational efficiency, and facilitating downstream discovery in drug and therapeutic development.
The advent of technologies like single-cell RNA sequencing (scRNA-seq) has revolutionized genomics by enabling the measurement of gene expression at the resolution of individual cells. However, this power comes with a significant challenge: the "curse of dimensionality." scRNA-seq data is inherently high-dimensional and sparse, where each of the thousands of genes represents a dimension, and each cell is a data point in this vast space [34] [35]. This high-dimensionality, compounded by technical artifacts like "dropout events" (an abundance of zero counts for truly expressed genes), poses major obstacles for visualization, analysis, and interpretation [35].
In this context, dimensionality reduction is not merely a preprocessing step but a critical necessity to transform data into a lower-dimensional space that retains essential biological information [35]. It mitigates computational load, reduces noise, and enables the identification of latent structures, such as novel cell types or states, which are crucial for understanding disease mechanisms and identifying drug targets [34] [36]. Principal Component Analysis (PCA) stands as one of the most widely used techniques for this purpose, but its core philosophy represents a significant departure from the classical statistical modeling approach of regression.
Theoretical Foundations: Error Modeling vs. Variance Capture
Classical Regression: Predicting Outcomes and Minimizing Error
Classical linear regression is a supervised learning technique. Its primary goal is to model the relationship between a set of independent variables (predictors) and a dependent variable (outcome) to predict future outcomes. The model takes the form:
Y = Xβ + ε
Here, Y is the dependent variable, X is the matrix of independent variables, β represents the unknown regression coefficients, and ε represents the error term [37]. The model is fundamentally concerned with this error term; parameter estimation, typically via ordinary least squares (OLS), aims to find the coefficients β that minimize the sum of squared errors between the observed and predicted values of Y [37]. The entire framework is built around accurately predicting a specific target and quantifying the error in that prediction.
Principal Component Analysis: Capturing Data Structure through Variance
PCA, in contrast, is an unsupervised technique. It requires no outcome variable and is instead concerned with the internal structure of the entire dataset X. Its objective is not prediction but exploratory data analysis and dimensionality reduction [33]. PCA seeks a set of new, uncorrelated variables—the principal components (PCs)—which are linear combinations of the original variables. These components are ordered such that the first PC (PC1) captures the direction of maximum variance in the data, the second PC (PC2) captures the next greatest variance while being orthogonal to the first, and so on [11] [33].
Mathematically, this is achieved by solving an eigenvalue-eigenvector problem on the covariance matrix S of the data:
Sa = λa
The eigenvectors ak are the PC loadings, defining the directions of the new feature space, and the eigenvalues λk represent the amount of variance captured by each component [33]. The projected data points in this new space are the PC scores. Thus, the fundamental "model" of PCA is one of variance decomposition, not error minimization.
The Paradigm Shift Summarized
The table below summarizes the core differences between these two approaches, highlighting the fundamental shift in their objectives and methodologies.
Table 1: Core Philosophical Differences Between Classical Regression and PCA
| Feature | Classical Regression | Principal Component Analysis (PCA) |
|---|---|---|
| Primary Goal | Predict a specific outcome variable [37] | Reduce dimensionality; explore data structure [33] |
| Model Core | Minimizes prediction error (ε) [37] | Maximizes captured variance (λ) [33] |
| Variable Role | Distinction between dependent (Y) and independent (X) variables | No distinction; analyzes all variables collectively |
| Learning Type | Supervised | Unsupervised |
| Output | A predictive equation for Y | A set of principal components (loadings and scores) |
| Key Metric | R-squared, p-values | Explained variance ratio [11] |
This shift in objective from explaining an outcome to explaining the structure of the predictor set itself is what makes PCA uniquely suited for the initial exploration of high-dimensional genomic data, where a single, pre-specified outcome of interest may not exist.
PCA in Practice: Dimensionality Reduction for Gene Expression Data
The Standard PCA Workflow in scRNA-seq Analysis
In scRNA-seq analysis, where the data matrix X consists of cells (observations) and genes (variables), PCA is applied to reduce the thousands of gene dimensions into a smaller set of latent variables [35]. The standard workflow, as implemented in pipelines such as those in Seurat or Scanpy [36], involves several key steps. The following diagram illustrates this sequential process.
Figure 1: The Standard PCA Workflow for scRNA-seq Data.
- Data Preprocessing: The raw gene count matrix is normalized and scaled to account for differences in sequencing depth and other technical variations between cells [35].
- Centering: The data matrix is column-centered, meaning the mean expression of each gene is subtracted. This is crucial as PCA is sensitive to the mean of the variables [33].
- Covariance Matrix: The covariance matrix S of the centered data is computed. This matrix encodes how the expression levels of all genes vary with one another.
- Eigendecomposition: The covariance matrix is decomposed into its eigenvalues and eigenvectors. The eigenvectors are the principal components (the new axes), and the eigenvalues indicate the variance each PC explains.
- Component Selection: The top k eigenvectors (PCs) are selected based on their eigenvalues, often by looking for an "elbow" in a scree plot (a plot of eigenvalues in descending order) or by retaining enough components to capture a pre-defined percentage (e.g., 95%) of the total variance [35].
- Projection: The original data is projected onto the selected PCs to obtain the PC scores matrix T, which is the low-dimensional representation of the data used for downstream tasks like clustering and visualization [34].
Key Advantages for Genomic Data
- Noise Reduction: By discarding lower-order principal components, which often capture technical noise or irrelevant biological variation, PCA effectively denoises the data [11].
- Computational Efficiency: Reducing dimensionality drastically lowers the computational cost of subsequent analyses, such as clustering, which is essential for large-scale scRNA-seq datasets containing tens of thousands of cells [34] [38].
- Preservation of Variance: The method ensures that the most biologically meaningful signals, which often contribute the most to variance in the data, are retained in the lower-dimensional space.
Experimental Benchmarking and Protocol
Benchmarking PCA in Genomic Studies
The performance of PCA is often benchmarked against other dimensionality reduction techniques in terms of computational efficiency and the quality of downstream analysis, such as clustering. A recent large-scale benchmarking study on scRNA-seq data provides quantitative insights into PCA's performance [34].
Table 2: Performance Benchmark of Dimensionality Reduction Methods on scRNA-seq Data [34]
| Method | Type | Key Strength | Computational Speed | Clustering Quality (Example Metrics) |
|---|---|---|---|---|
| PCA (Full SVD) | Linear | Maximizes variance capture | Slower on very large datasets | High, serves as a common baseline |
| Randomized SVD | Linear | Approximation of full PCA | Faster than full SVD | Comparable to full PCA |
| Gaussian Random Projection (GRP) | Random | Distance preservation | Very Fast | Rivals/Potentially exceeds PCA in some cases |
| Sparse Random Projection (SRP) | Random | Memory efficiency | Very Fast | Competes well with PCA |
| t-SNE | Non-linear | Preserves local structure | Slow | High for visualization, separates clusters |
| UMAP | Non-linear | Preserves local/global structure | Moderate-Fast | High for visualization and clustering |
This benchmarking demonstrates that while PCA remains a robust and interpretable standard, random projection methods have emerged as strong competitors, sometimes surpassing PCA in speed and rivaling it in clustering effectiveness [34]. This is particularly relevant for ultra-large-scale datasets in drug discovery.
Detailed Experimental Protocol: Applying PCA to an scRNA-seq Dataset
The following is a generalized protocol for applying PCA to a typical scRNA-seq dataset, as described in the literature [34] [36] [35].
Objective: To reduce the dimensionality of a scRNA-seq gene expression matrix for downstream cell clustering and population identification.
Materials and Reagents (Computational): Table 3: Essential Research Reagent Solutions for scRNA-seq PCA Analysis
| Reagent / Tool | Function / Explanation | Example |
|---|---|---|
| scRNA-seq Data Matrix | The input data; rows = cells, columns = genes. | UMI count matrix from 10x Genomics [35]. |
| High-Performance Computing (HPC) Cluster | Provides the computational power needed for large matrix operations. | Local server or cloud computing environment. |
| Normalization Software | Corrects for technical variation (e.g., sequencing depth). | scran package in R or scanpy.pp.normalize_total in Python. |
| PCA Implementation | The algorithm that performs the principal component analysis. | prcomp() in R or sklearn.decomposition.PCA in Python [36]. |
| Visualization Package | For generating scree plots and 2D PCA plots. | ggplot2 in R or matplotlib in Python [11]. |
Methodology:
Data Input and Quality Control (QC):
- Load the gene-cell count matrix. A common starting point is a matrix with dimensions [3,000 cells x 20,000 genes].
- Perform QC to filter out low-quality cells (e.g., based on high mitochondrial gene percentage) and poorly detected genes.
Normalization and Transformation:
- Normalize the counts to correct for library size differences between cells (e.g., counts per million (CPM) or log-normalization).
- Identify Highly Variable Genes (HVGs): To reduce noise and focus on biologically informative genes, select a subset of genes (e.g., 1,000-5,000) that exhibit the highest cell-to-cell variation. This step is critical for enhancing the signal in PCA.
Scaling and Centering:
- Center the data: subtract the mean expression for each gene so that each gene has a mean of zero.
- Scale the data (optional but often recommended): divide each gene's expression by its standard deviation. This puts all genes on the same scale, preventing highly expressed genes from dominating the PCs simply due to their magnitude.
Perform PCA:
- Apply the PCA algorithm to the preprocessed (normalized, HVG-selected, and scaled) data matrix.
- The output includes:
- Loadings (
A_k): The eigenvectors, showing the contribution (weight) of each original gene to each PC. - Scores (
T): The projected coordinates of each cell in the new PC space. - Explained Variance Ratio (
λ_k / Σλ): The proportion of total variance explained by each PC.
- Loadings (
Component Selection:
- Plot the explained variance ratio for each PC (Scree Plot).
- Identify the "elbow" point where the marginal gain in explained variance drops significantly. Alternatively, retain enough PCs to capture ~80-90% of the total cumulative variance. For a typical scRNA-seq dataset, this might be the top 20-50 PCs.
Downstream Analysis:
- Use the PC scores matrix
T(containing only the top k PCs) as input for graph-based clustering algorithms (e.g., Louvain, Leiden) to identify cell populations. - Visualize the first two PCs in a scatter plot to observe broad separation of cell types or states.
- Use the PC scores matrix
Discussion: Implications for Research and Drug Development
The variance-capture model of PCA has profound implications for biomedical research. For scientists and drug development professionals, PCA is not an endpoint but a critical enabling technology. By transforming high-dimensional gene expression data into a lower-dimensional latent space, PCA facilitates the discovery of novel cell subtypes associated with disease, the identification of biomarker genes (by examining PC loadings), and the understanding of developmental trajectories [34] [35].
The relationship between PCA and regression is not purely antagonistic; they can be synergistic. Principal Component Regression (PCR) is a technique that uses the principal components as new regressors for a outcome variable, elegantly combining the two philosophies [37]. This approach helps overcome multicollinearity in high-dimensional datasets, as the PCs are, by definition, uncorrelated.
Looking forward, the field continues to evolve. While PCA remains a cornerstone, benchmarks show that random projection methods offer compelling advantages in speed for very large datasets [34]. Furthermore, non-linear methods like UMAP are superior for final visualization, as they can capture complex manifold structures that linear PCA cannot [36] [39]. However, PCA's mathematical transparency, computational efficiency, and strength as a variance-preserving first step ensure it will remain an indispensable tool in the computational biologist's and drug developer's toolkit for the foreseeable future.
This whitepaper has articulated the fundamental conceptual shift from the error-minimizing framework of classical regression to the variance-maximizing model of Principal Component Analysis. Within the domain of gene expression research, PCA's unsupervised, variance-centric approach is uniquely suited to tackle the curse of dimensionality inherent in modern genomic technologies like scRNA-seq. It provides a robust, mathematically sound foundation for data compression, noise reduction, and exploratory analysis. By enabling the visualization and clustering of cells in a reduced space that captures the most significant biological signals, PCA directly empowers researchers to uncover novel biology, characterize disease states, and ultimately accelerate the pipeline of drug discovery and development.
Implementing PCA in Practice: From scRNA-seq to Spatial Transcriptomics
Principal Component Analysis (PCA) stands as a cornerstone dimensionality reduction technique in computational biology, enabling researchers to distill high-dimensional gene expression data into its core components of variation. This whitepaper details the standardized PCA workflow—encompassing critical data normalization, eigenvector-based transformation, and biological interpretation of components—within the broader thesis of how PCA effectively reduces data dimensionality to uncover latent biological patterns. By providing a rigorous methodological framework and benchmarking against emerging techniques, this guide equips researchers and drug development professionals with the foundational knowledge to implement PCA in genomic studies, thereby facilitating the identification of novel biomarkers and therapeutic targets.
The advent of high-throughput technologies has rendered genomic data, particularly from single-cell RNA sequencing (scRNA-seq), both high-dimensional and sparse. A typical dataset may measure the expression levels of over 20,000 genes (variables, P) across only hundreds of samples or cells (observations, N), creating a scenario where P ≫ N [20]. This "curse of dimensionality" presents significant challenges for analysis, including computational intractability, difficulty in visualization, and an increased risk that machine learning models will capture noise rather than biological signal, leading to overfitting [40] [41].
Dimensionality reduction techniques mitigate these issues by transforming the data into a lower-dimensional space while preserving essential biological information. PCA achieves this by identifying new, uncorrelated variables—the principal components—that are linear combinations of the original genes and that sequentially capture the maximum possible variance in the data [41] [42]. This process simplifies the data structure, making it amenable to visualization, clustering, and further downstream analysis, which is crucial for tasks like identifying distinct cell populations or characterizing gene expression dynamics in disease research [34].
Mathematical Foundations of PCA
At its core, PCA is a statistical procedure that leverages linear algebra to redefine a dataset in terms of its directions of maximum variance.
Key Concepts and Linear Algebra
The mathematical engine of PCA is the eigenvalue decomposition of the data's covariance matrix. Given a data matrix ( X ) with ( n ) observations (cells) and ( p ) features (genes), and assuming the data has been centered (each column has mean zero), the covariance matrix ( \Sigma ) is calculated as: [ \Sigma = \frac{1}{n-1} X^T X ] Eigenvalue decomposition solves the equation: [ \Sigma v = \lambda v ] where:
- ( v ) is an eigenvector, representing a direction of maximum variance in the data (a principal component direction).
- ( \lambda ) is an eigenvalue, a scalar that quantifies the amount of variance captured by its associated eigenvector [40] [41].
A dataset will have ( min(n-1, p) ) such eigenvector-eigenvalue pairs. The principal components themselves are the projections of the original data onto these eigenvectors. The first principal component (PC1) is the projection onto the eigenvector with the largest eigenvalue, the second principal component (PC2) onto the eigenvector with the next largest eigenvalue, and so on [41].
The Critical Role of Normalization
Before performing eigenvalue decomposition, a crucial preprocessing step is the normalization—more accurately, standardization—of the data. This involves subtracting the mean and dividing by the standard deviation for each gene, so that all features have a mean of 0 and a standard deviation of 1 [42] [43].
This step is essential in the context of gene expression data. Genes naturally have different baseline expression levels and variances. A highly expressed gene (e.g., one with values in the range of 0-1000) would inherently have a much larger variance than a lowly expressed gene (e.g., with values from 0-1). If PCA were performed on this unnormalized data, the algorithm would be biased towards the highly variable genes, and the first principal components would predominantly reflect these technical differences rather than the most meaningful biological variation [42] [43]. Standardization ensures that each gene contributes equally to the analysis, allowing biologically relevant, coordinated shifts in expression to drive the principal components.
Table 1: Key Linear Algebra Concepts in PCA
| Concept | Mathematical Representation | Interpretation in PCA |
|---|---|---|
| Covariance Matrix | ( \Sigma = \frac{1}{n-1} X^T X ) | Summarizes how all pairs of genes vary together; a square, symmetric matrix. |
| Eigenvector | ( v ) | Represents a principal component direction—an axis of maximum variance in the data. |
| Eigenvalue | ( \lambda ) | Quantifies the amount of variance explained by its corresponding eigenvector. |
| Principal Component | ( Z = X v ) | The actual projected coordinates of the data along the eigenvector direction. |
The Standard PCA Workflow: A Step-by-Step Guide
This section outlines the definitive workflow for applying PCA to a gene expression dataset, from raw data to interpretation.
Data Preprocessing and Normalization
- Data Cleaning: Handle missing values, typically by imputation or removal of genes/cells with excessive missing data.
- Centering: For each gene, subtract the mean expression across all cells. This centers the data around the origin, a requirement for the covariance matrix calculation.
- Scaling (Standardization): For each gene, divide the centered expression values by the gene's standard deviation across all cells. This ensures all genes are on a comparable scale [41] [42]. The resulting standardized data matrix is used for all subsequent steps.
Computation of Principal Components
- Covariance Matrix Calculation: Compute the covariance matrix of the standardized data matrix. This ( p \times p ) matrix captures the pairwise covariances between all genes.
- Eigenvalue Decomposition: Perform eigenvalue decomposition on the covariance matrix to obtain the eigenvectors and their corresponding eigenvalues.
- Sort Components: Sort the eigenvector-eigenvalue pairs in descending order of the eigenvalues. The eigenvector with the highest eigenvalue is the first principal component (PC1), and so on [40] [41].
Selection of Principal Components
The sorted eigenvalues indicate the importance of each component. A common approach is to choose the top ( k ) components that collectively explain a sufficient proportion of the total variance (e.g., 95%). This is visualized using a Scree Plot (a bar plot of individual explained variances) or a plot of Cumulative Explained Variance [40] [41].
Table 2: Metrics for Component Selection
| Metric | Calculation | Interpretation and Use |
|---|---|---|
| Explained Variance per PC | ( \lambda_i ) | The variance captured by a single PC. Used in scree plots to visually identify an "elbow" point. |
| Explained Variance Ratio | ( \lambda_i / \sum(\lambda) ) | The proportion of total variance explained by a single PC. |
| Cumulative Explained Variance | ( \sum{i=1}^k \lambdai / \sum(\lambda) ) | The total proportion of variance explained by the first ( k ) PCs. Used to select the final number of components. |
Projection and Interpretation
- Projection: Transform the original standardized data into the new principal component space. This is done by multiplying the data matrix by the matrix of the top ( k ) eigenvectors: ( X{\text{pca}} = X{\text{std}} \cdot Vk ), where ( Vk ) contains the first ( k ) eigenvectors as columns. The result is a new dataset with ( n ) observations and ( k ) features (the principal components) [40].
- Interpretation: Analyze the "loadings" of the principal components, which are the weights (coefficients) in the eigenvectors. Genes with large absolute loadings (positive or negative) in a given PC have a strong influence on that component. Biologically, these genes often represent co-regulated gene sets or pathways that drive the major axes of variation, such as differences between cell types or response to a treatment [41] [7].
Figure 1: The Standard PCA Workflow. This diagram outlines the sequential steps from raw data to biological interpretation.
PCA in Gene Expression Research: Protocols and Reagents
The following section provides a detailed methodology for a typical gene expression analysis pipeline incorporating PCA.
Experimental Protocol: scRNA-seq Cluster Analysis
Objective: To identify distinct cell populations and their defining marker genes from a single-cell RNA sequencing dataset.
- Data Input: Load a gene expression count matrix (cells x genes). Publicly available datasets, such as the Sorted PBMC Dataset (2,882 cells, 7,174 genes) [34], are suitable for benchmarking.
- Quality Control & Normalization: Filter out low-quality cells and genes. Normalize counts (e.g., by library size) and log-transform the data. Standardize the data post-transformation.
- Dimensionality Reduction with PCA: Apply the standard workflow from Section 3. Select the number of components,
k, such that the cumulative explained variance is >80-95%. - Clustering & Visualization: Use the top
kprincipal components as input to a clustering algorithm (e.g., Hierarchical Clustering, K-Means). Visualize the cells in 2D or 3D using the first 2 or 3 principal components as axes. - Marker Gene Identification: For each cluster, perform differential expression analysis. Cross-reference the results with the PCA loadings; genes with high loadings in the PCs that separate the clusters are strong candidate marker genes [3] [34].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Tools for PCA-Based Gene Expression Studies
| Item / Reagent | Function / Application |
|---|---|
| scRNA-seq Kit (e.g., 10x Genomics) | Generation of the primary high-dimensional gene expression count matrix from single cells. |
| Computational Environment (e.g., R/Python) | Platform for performing data normalization, PCA computation, and downstream statistical analysis. |
| PCA Software Library (e.g., scikit-learn, Scanpy) | Implements the efficient computational algorithms for performing PCA on large datasets. |
| Clustering Algorithm (e.g., Spherical K-Means) | Used on the PCA-reduced data to group cells into putative populations based on expression similarity [34]. |
| Differential Expression Tool (e.g., MAST, Wilcoxon test) | Statistically validates the identity of cell clusters identified via PCA and clustering by finding marker genes. |
Advanced Considerations and Benchmarking
While PCA is a powerful and widely used linear technique, the complex, non-linear relationships often present in biological systems have spurred the development of alternative and advanced methods.
Limitations of Standard PCA
- Linearity Assumption: PCA identifies linear combinations of genes. It may fail to capture complex, non-linear relationships [34].
- Sensitivity to Outliers: Extreme values can disproportionately influence the principal components, skewing results [44].
- Dense Output: All genes contribute to all principal components (non-zero loadings), which can complicate biological interpretation by making it difficult to isolate small, defining gene sets [7].
Emerging Techniques and Extensions
To address these limitations, several advanced variants have been developed:
- Kernel PCA: Maps data into a higher-dimensional space where non-linear patterns can be captured linearly, useful for complex gene regulatory dynamics [45].
- Sparse PCA (SPCA): Incorporates L1 regularization to force the loadings of many genes to zero, resulting in components defined by small, easier-to-interpret gene sets [7].
- Boosting Autoencoder (BAE): A recent deep learning approach that combines autoencoders with boosting to produce highly sparse and interpretable latent dimensions, each characterized by a small, distinct set of genes [7].
- Random Projections (RP): An extremely computationally efficient alternative to PCA that projects data onto a random lower-dimensional subspace, often rivaling PCA in clustering performance for very large datasets [34].
Figure 2: A Taxonomy of Dimensionality Reduction Techniques. PCA exists within a broader ecosystem of methods, each with distinct strengths.
Benchmarking Performance
A 2025 preprint benchmarking study provides a comparative look at PCA's performance against Random Projections (RP) [34]. The study evaluated multiple methods on scRNA-seq datasets using metrics like clustering accuracy and computational speed.
Table 4: Benchmarking PCA Against Random Projections (RP) on scRNA-seq Data
| Method | Computational Speed | Preservation of Variability | Clustering Quality | Key Characteristic |
|---|---|---|---|---|
| Standard PCA (SVD) | Baseline (Slowest) | High | High | The gold-standard, linear method. |
| Randomized SVD | Faster than Standard PCA | High (Approximate) | High | A faster approximation of full PCA. |
| Gaussian RP (GRP) | Fastest | Moderate | Rivals PCA | Uses a dense random matrix. |
| Sparse RP (SRP) | Very Fast | Moderate | Rivals/Surpasses PCA | Uses a sparse random matrix; memory efficient. |
The study concluded that while PCA remains a robust and reliable method, RP techniques not only offer significant computational advantages but also rival and sometimes surpass PCA in the quality of downstream analyses like clustering, making them attractive for rapidly exploring very large datasets [34].
The standard PCA workflow—comprising rigorous normalization, linear transformation via eigenvalue decomposition, and careful interpretation of components—is an indispensable tool for making high-dimensional gene expression data tractable. By reducing dimensionality while preserving maximal biological variance, PCA provides a powerful, unsupervised foundation for exploratory data analysis, visualization, and hypothesis generation. Despite the emergence of powerful non-linear and sparse alternatives, PCA's mathematical rigor, computational efficiency, and interpretability ensure its continued relevance. Its proper application will remain fundamental for researchers and drug developers seeking to decipher the complexity of genomic data and translate it into meaningful biological insights and therapeutic innovations.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the examination of gene expression at the level of individual cells, thereby revealing hidden cellular diversity within tissues [19]. This technology investigates the transcriptome of single cells, in contrast to bulk RNA-Seq which provides only an average expression for each gene across all cells in a sample [19]. The standard scRNA-seq workflow begins with single-cell isolation from tissue, followed by cell lysis and RNA isolation, reverse transcription of mRNA with amplification via PCR, and finally library preparation and sequencing [19]. The raw output is typically a FASTQ file, which undergoes computational preprocessing to produce a gene expression matrix, usually in the form of gene read counts or unique molecular identifiers (UMIs) for droplet-based platforms [19].
The resulting scRNA-seq data presents significant analytical challenges due to its inherent high-dimensionality and sparsity [19]. High-dimensionality stems from the simultaneous analysis of thousands of cells and tens of thousands of genes, while sparsity arises from an abundance of zero counts for genes that are genuinely expressed in other cells of the same type—a phenomenon known as "dropout events" [19]. These zeros can be attributed to the low mRNA quantities extracted from individual cells, the stochastic nature of gene expression, and cell-specific expression patterns [19]. To extract meaningful biological signals from this complex data landscape, robust computational methods for dimensionality reduction are essential. Principal Component Analysis (PCA) serves as a foundational technique in this domain, transforming high-dimensional gene count data into lower-dimensional spaces while retaining critical biological information [19].
The Role of Principal Component Analysis in scRNA-seq
Fundamental Concepts of PCA for scRNA-seq
In the context of scRNA-seq data, each cell can be represented as a data point in a high-dimensional Euclidean space where the number of dimensions corresponds to the number of genes in the dataset, and the coordinates represent each gene's expression level in that cell [19]. Dimensionality reduction refers to the transformation of this high-dimensional data into a lower-dimensional representation while preserving most of the original information content [19]. This process reduces computational resource requirements, decreases memory usage, and shortens algorithm execution times—critical considerations when analyzing large-scale scRNA-seq datasets [46].
PCA is a statistical method that performs an orthogonal linear transformation of the original data points to create new variables called principal components (PCs) [19]. These components are uncorrelated with each other and capture decreasing proportions of the total variance present in the original dataset [19]. When applied to scRNA-seq data with cells as data points, PCs represent linear combinations of genes, often referred to as "latent genes" [19]. As an unsupervised method that requires no prior information about cell identity, PCA captures linear associations within scRNA-seq gene expressions, producing a low-dimensional dataset with the same number of cells but far fewer latent genes than the original dataset [19].
Computational Considerations for Large-Scale scRNA-seq Data
The application of PCA to large-scale scRNA-seq datasets requires careful consideration of computational efficiency. Traditional PCA algorithms that load entire data matrices into memory become impractical for datasets comprising millions of cells due to excessive memory demands [46]. Benchmarking studies have identified that PCA algorithms based on Krylov subspace methods and randomized singular value decomposition (SVD) offer optimal combinations of speed, memory efficiency, and accuracy for large-scale scRNA-seq applications [46]. These efficient algorithms are particularly valuable in research environments where analytical workflows undergo repeated refinement through trial-and-error cycles with parameter adjustments and data modifications [46].
Table 1: PCA Algorithm Comparison for scRNA-seq Data
| Algorithm Type | Computational Efficiency | Memory Usage | Accuracy | Best Use Cases |
|---|---|---|---|---|
| Krylov Subspace Methods | High | Low | High | Large datasets (>10,000 cells) |
| Randomized SVD | High | Low | High | Very large datasets (>100,000 cells) |
| Similarity Transformation | Medium | Medium | High | Medium datasets (1,000-10,000 cells) |
| Downsampling-based | High | Low | Low | Exploratory analysis only |
| Gradient Descent-based | Variable | Low | Medium | Streaming data applications |
Preprocessing and Normalization: Critical Precursors to PCA
Normalization Methods and Their Impact on PCA
Normalization of gene expression count data represents an essential preprocessing step in scRNA-seq analysis, significantly influencing subsequent PCA results and their biological interpretation [13]. A comprehensive evaluation of twelve normalization methods revealed that while PCA score plots may appear similar across different normalization techniques, the biological interpretation of these models depends heavily on the specific method applied [13]. Normalization affects correlation patterns within the data, which subsequently influences PCA model complexity, sample clustering quality in low-dimensional space, and gene ranking within the model [13]. These differences ultimately translate to variations in biological insights derived from gene enrichment and pathway analyses [13].
The sensitivity of PCA to normalization methods stems from its reliance on variance structure within the data. Different normalization approaches alter gene expression distributions and relationships between variables, thereby changing which dimensions PCA identifies as most significant [13]. Consequently, researchers must carefully select normalization techniques aligned with their biological questions and explicitly report the methods used to ensure reproducible and interpretable results [13].
Experimental Protocol: Standard scRNA-seq Preprocessing for PCA
A robust preprocessing protocol ensures that PCA captures biologically meaningful signals rather than technical artifacts:
Quality Control and Filtering: Remove low-quality cells based on metrics including total counts, number of detected genes, and mitochondrial gene percentage [19]. Exclude genes expressed in only a small fraction of cells.
Normalization: Apply a normalization method appropriate for the experimental design. Common approaches include:
- Library size normalization with log transformation
- Regularized negative binomial regression (e.g., SCTransform)
- Depth-adjusted negative binomial models (e.g., DEseq2)
Feature Selection: Identify highly variable genes (HVGs) that exhibit cell-to-cell variation beyond technical noise. Typically, 1,000-5,000 HVGs are selected for PCA input [19].
Scaling: Standardize expression values for each gene to have zero mean and unit variance, ensuring that genes with high expression levels don't disproportionately influence PCs.
Table 2: Key Research Reagents and Computational Tools for scRNA-seq PCA
| Resource Name | Type | Function in Analysis |
|---|---|---|
| Cell Ranger | Software Pipeline | Processing 10x Genomics scRNA-seq data from raw sequences to gene count matrices [19] |
| Seurat | R Toolkit | Comprehensive scRNA-seq analysis including normalization, PCA, and downstream applications [47] |
| Scanpy | Python Toolkit | Scalable Python-based analysis of single-cell gene expression data including PCA implementations [47] |
| 10X Genomics Chromium | Hardware/Reagent System | Single-cell partitioning and barcoding platform for high-throughput scRNA-seq [46] |
| Unique Molecular Identifiers (UMIs) | Molecular Barcodes | Sequence tags that label individual mRNA molecules to correct for PCR amplification bias [19] |
Figure 1: scRNA-seq Preprocessing Workflow for PCA. This diagram outlines the essential steps required to prepare single-cell RNA sequencing data for principal component analysis, from raw sequencing files to normalized and scaled data suitable for dimensionality reduction.
Advanced PCA Applications in scRNA-seq Analysis
Spatial Transcriptomics Extensions
Spatial transcriptomics (ST) technologies have emerged that measure gene expression while preserving spatial location information within tissues, creating new opportunities and challenges for dimensionality reduction [48] [47]. Unlike conventional scRNA-seq where spatial context is lost during cell dissociation, ST data contains both gene expression profiles and spatial coordinates, enabling the development of spatially-aware PCA extensions [47]. Methods like GraphPCA incorporate spatial neighborhood structures as graph constraints within the PCA framework, enforcing that adjacent spots in the original tissue are positioned near each other in the low-dimensional embedding [47]. This approach has demonstrated superior performance in spatial domain detection compared to standard PCA and other competing methods [47].
Randomized Spatial PCA (RASP) represents another innovation specifically designed for large-scale ST datasets, employing randomized linear algebra techniques for computational efficiency while incorporating spatial smoothing through k-nearest neighbor thresholding [48]. Benchmarking experiments on diverse ST technologies including 10x Visium, Stereo-Seq, MERFISH, and 10x Xenium demonstrated that RASP achieves tissue domain detection comparable to or superior than existing methods while being orders-of-magnitude faster [48]. This computational efficiency becomes particularly valuable as ST technologies advance toward subcellular resolution and larger sample sizes [48].
Interpretable PCA through Structural Constraints
A significant limitation of conventional PCA in scRNA-seq applications is the difficulty in biologically interpreting principal components, as they typically represent linear combinations of hundreds or thousands of genes [7]. The Boosting Autoencoder (BAE) approach addresses this challenge by combining the dimensionality reduction capabilities of autoencoders with the variable selection properties of componentwise boosting [7]. This integration produces a sparse mapping to the latent space where each dimension is characterized by a small set of explanatory genes, facilitating biological interpretation [7].
The BAE framework can incorporate structural assumptions about the data through customizable constraints in the variable selection process [7]. For instance, a disentanglement constraint ensures that genes selected for each dimension explain complementary information to that encoded in other dimensions, effectively capturing distinct sets of genes that characterize different cell subgroups [7]. In time-series scRNA-seq data, constraints can encode knowledge about gradually evolving gene expression dynamics, helping identify gene programs that change across developmental trajectories [7]. This approach has proven particularly valuable for characterizing small cell populations that might be overlooked in global clustering analyses [7].
Figure 2: Advanced PCA Extensions for Specialized Applications. This diagram illustrates how standard PCA has been extended to address specific challenges in single-cell and spatial transcriptomics, including spatial pattern preservation and biological interpretability.
Experimental Framework for PCA in scRNA-seq Studies
Protocol: Implementing PCA for Cell Type Identification
The following protocol details a standard approach for utilizing PCA in scRNA-seq analysis to identify cell types and states:
Data Preparation: Begin with a normalized, scaled, and highly-variable-gene-selected count matrix as described in Section 3.2.
PCA Computation:
- For datasets with <10,000 cells: Use full SVD implementation (e.g., prcomp in R)
- For datasets with 10,000-100,000 cells: Implement randomized SVD for memory efficiency
- For datasets with >100,000 cells: Utilize Krylov subspace methods or out-of-core PCA algorithms [46]
Determining Significant PCs:
- Apply the elbow method to a scree plot of explained variance
- Use JackStraw permutation testing to identify statistically significant components
- Consider biological context when selecting the number of PCs for downstream analysis
Downstream Applications:
- Visualize cells in 2D or 3D space using the first 2-3 PCs for initial data exploration
- Use top PCs (typically 10-50) as input for clustering algorithms (e.g., Louvain, Leiden)
- Employ PCs for trajectory inference in developmental studies
Biological Interpretation:
- Examine gene loadings for each PC to identify driving genes
- Correlate PC scores with known marker genes or experimental conditions
- Perform gene set enrichment analysis on high-loading genes for significant PCs
Methodological Considerations and Troubleshooting
Successful application of PCA to scRNA-seq data requires attention to several methodological considerations. First, the choice of number of PCs retained significantly impacts downstream analyses. While the elbow method provides a visual heuristic, it can be subjective, particularly when no clear elbow is present [19]. Alternative approaches include retaining PCs that explain a predetermined percentage of total variance (e.g., 80-90%) or using more formal statistical criteria like parallel analysis [19].
PCA's sensitivity to outliers and technical artifacts necessitates robust quality control procedures. Extreme values can disproportionately influence principal component directions, potentially obscuring biologically relevant patterns [49]. Additionally, the rotational ambiguity of PCA solutions, while mathematically equivalent in terms of variance explanation, can affect interpretability [49]. Small rotations of PC axes sometimes enhance biological interpretability without substantially altering the overall variance structure [49].
Table 3: Troubleshooting Common PCA Challenges in scRNA-seq Analysis
| Problem | Potential Causes | Solutions |
|---|---|---|
| Poor separation of known cell types | Insufficient sequencing depth, inappropriate normalization, excessive technical variation | Re-evaluate QC thresholds, try alternative normalization methods, include covariance drivers as covariates |
| First PCs dominated by technical effects | Batch effects, sequencing depth variation, cell cycle confounding | Implement batch correction, include technical factors as covariates, regress out cell cycle scores |
| Instability in PC directions | Outliers, small population of highly variable cells | Implement robust PCA variants, remove outliers, increase cell numbers for rare populations |
| Discrepancy between biological knowledge and PC loadings | Biological complexity, non-linear relationships | Apply non-linear dimensionality reduction (t-SNE, UMAP) after PCA, use interpretable PCA variants [7] |
Principal Component Analysis remains a cornerstone method for dimensionality reduction in single-cell RNA sequencing data analysis, providing a robust framework for navigating high-dimensional gene expression spaces. While the mathematical foundations of PCA are well-established, their effective application to scRNA-seq data requires careful consideration of preprocessing strategies, computational implementation, and interpretation within biological context. Recent methodological advances have extended PCA in two significant directions: incorporation of spatial information for transcriptomics data that preserves tissue architecture, and enhancement of interpretability through integration with variable selection techniques. These developments ensure that PCA continues to evolve alongside sequencing technologies, maintaining its relevance as an essential tool for extracting biological insights from increasingly complex and large-scale single-cell genomics datasets. As the field progresses toward multi-omic single-cell measurements and integration of complementary data modalities, the principles of dimensionality reduction embodied by PCA will remain fundamental to exploratory biological data analysis.
Principal Component Analysis (PCA) serves as a foundational dimension reduction technique in bioinformatics, transforming high-dimensional genomic data into a lower-dimensional set of uncorrelated principal components (PCs) that capture maximum variance [27]. In gene expression analysis, where datasets typically contain thousands of genes (dimensions) measured across far fewer samples, PCA addresses the "curse of dimensionality" by creating linear combinations of original variables called principal components [27] [20]. These components, often referred to as "metagenes" or "latent genes," provide an orthogonal basis that simplifies complex biological data while preserving essential patterns [27]. The mathematical foundation of PCA lies in singular value decomposition (SVD) of the data covariance matrix, where eigenvectors define new directions in feature space and eigenvalues quantify variance captured by each component [27] [11]. This transformation enables researchers to project high-dimensional gene expression profiles onto a reduced space where biological patterns become more apparent, forming the basis for advanced applications in pathway, network, and interaction analysis.
Core Principles: How PCA Reduces Gene Expression Dimensionality
Mathematical and Computational Foundations
PCA operates on the fundamental principle of identifying directions of maximum variance in high-dimensional data through eigen decomposition of the covariance matrix [11]. Given a gene expression matrix ( X ) with ( m ) samples (rows) and ( n ) genes (columns), where each element ( x{ij} ) represents the expression level of gene ( j ) in sample ( i ), PCA begins by centering the data to have zero mean for each variable. The covariance matrix ( C ) is computed as ( C = \frac{1}{m-1}X^TX ), capturing relationships between all gene pairs [27]. Eigen decomposition of ( C ) yields eigenvectors (principal components) and eigenvalues (variance explained), with PCs sorted in descending order of explained variance [11]. The first PC (( PC1 )) represents the direction of maximum variance, the second PC (( PC2 )) captures the next highest variance orthogonal to ( PC1 ), and so on [11]. This process generates a new coordinate system where the axes are uncorrelated and ranked by their importance in describing data variability.
Table 1: Key Properties of Principal Components in Gene Expression Analysis
| Property | Mathematical Representation | Biological Interpretation |
|---|---|---|
| Orthogonality | ( PCi \cdot PCj = 0 ) for ( i \neq j ) | Independent biological factors |
| Variance Explanation | ( \lambda1 \geq \lambda2 \geq \cdots \geq \lambda_n ) | Relative importance of each biological effect |
| Dimensionality Reduction | ( X{m \times n} \rightarrow T{m \times k} ) with ( k \ll n ) | Focus on major sources of variation |
| Linear Combinations | ( PCj = a1x1 + a2x2 + \cdots + anx_n ) | Combined gene effects representing biological processes |
Practical Implementation for Gene Expression Data
In practice, PCA implementation for gene expression data requires careful preprocessing and parameter decisions. The standard workflow begins with data normalization to make features comparable, often using z-score standardization (mean-centering and division by standard deviation) to prevent highly expressed genes from dominating the analysis [50] [13]. The prcomp() function in R or similar implementations in Python (e.g., sklearn.decomposition.PCA) then performs the core PCA computation [27] [51]. A critical step involves determining the number of components to retain, typically achieved through scree plots of eigenvalues or by selecting components that collectively explain a predetermined percentage of total variance (e.g., 80-95%) [11]. For gene expression data, the resulting PCs can be interpreted biologically by examining their "loadings" (coefficients assigned to each gene), where genes with large absolute loadings strongly influence that component's direction [51]. Sample "scores" indicate positions along PC directions, enabling visualization of sample relationships in reduced dimensions [51].
Figure 1: PCA workflow for gene expression data analysis
Advanced Pathway-Centric PCA Applications
Pathway-Level Representation and Interpretation
Traditional PCA applications analyze genome-wide expression data, but pathway-centric approaches offer enhanced biological interpretability by incorporating prior knowledge of gene groupings [27]. In this framework, PCA is applied separately to genes within predefined pathways (e.g., from KEGG, Reactome, or GO databases), generating pathway-level scores that represent the coordinated activity of functionally related genes [27]. Each pathway becomes represented by its principal components, which capture the major patterns of variation within that biological process [27]. For example, in a study of congenital adrenal hyperplasia, researchers applied PCA to steroidogenesis pathway genes, creating endocrine profiles that successfully distinguished patients according to treatment efficacy and disease form (classical vs. non-classical) with high accuracy (AUC=92% for classical CAH) [52]. This pathway-PCA approach transforms the analysis from examining thousands of individual genes to investigating hundreds of pathway activities, maintaining statistical power while dramatically improving biological interpretability.
Table 2: Comparison of Standard PCA vs. Pathway-Centric PCA
| Aspect | Standard PCA | Pathway-Centric PCA |
|---|---|---|
| Analysis Unit | Individual genes (20,000+) | Biological pathways (200-500) |
| Dimensionality | Very high (gene space) | Reduced (pathway space) |
| Biological Interpretation | Challenging, often requires post-hoc gene set enrichment | Direct, built into analysis framework |
| Statistical Power | Limited by high multiple testing burden | Improved due to reduced dimensionality |
| Handling of Coordinated Expression | Captured but not explicitly modeled | Explicitly models co-regulated gene sets |
| Implementation | Single PCA on full expression matrix | Multiple PCAs on pathway-specific gene subsets |
Methodological Protocol for Pathway PCA
Implementing pathway-centric PCA requires systematic methodology to ensure robust and reproducible results. Begin by acquiring pathway definitions from standard databases (KEGG, Reactome, BioCarta, or GO terms) and mapping genes in the expression dataset to these pathways. For each pathway, extract expression values for member genes and apply PCA, retaining components that explain a predetermined percentage of variance (typically 70-80%) or those with eigenvalues >1 [52] [27]. The resulting pathway-PCs serve as features in downstream analyses, such as regression models predicting clinical outcomes or clustering samples based on pathway activities. Critically, pathway-PCs must be computed independently in training and validation sets to avoid overfitting, with pathway definitions potentially refined through bootstrap stability analysis [27]. This approach effectively addresses the "large p, small n" problem common in genomic studies by reducing thousands of gene expressions to hundreds of pathway activities while respecting biological organization.
Network and Interaction Analysis Through PCA
Network Module Identification and Characterization
PCA provides powerful approaches for analyzing gene regulatory networks by identifying functionally coherent modules within larger interaction networks. In this application, the network is first decomposed into modules of tightly connected genes using community detection algorithms, then PCA is applied to each module to capture its dominant expression patterns [27]. The resulting module PCs represent the collective behavior of gene groups with strong regulatory relationships, potentially reflecting activities of shared transcription factors or coordinated responses to perturbations [27]. For example, in single-cell RNA sequencing analyses, PCA-based network modules have revealed cell-type-specific regulatory programs and transition states during differentiation [34]. This approach effectively reduces complexity while preserving the network topology information, enabling researchers to move from analyzing individual gene-gene interactions to studying coordinated activity of functional modules. The module PCs can subsequently be used to characterize sample relationships, identify regulatory drivers through loading analysis, or track temporal changes in network states across experimental conditions.
Modeling Gene Interactions Using PCA
Beyond identifying network modules, PCA facilitates explicit modeling of gene-gene interactions through innovative applications of the technique. Rather than analyzing pathways independently, interaction-aware PCA incorporates cross-pathway effects by creating composite variables that capture between-pathway relationships [27]. One advanced implementation involves generating all pairwise products between genes from different pathways, then applying PCA to these interaction terms to identify dominant interaction patterns [27]. Formally, for two pathway gene sets ( A = {a1, a2, ..., am} ) and ( B = {b1, b2, ..., bn} ), the interaction set ( I = {aibj | i=1..m, j=1..n} ) is created, followed by PCA on ( I ) to reduce dimensionality while preserving major interaction effects [27]. This approach makes the otherwise intractable problem of testing all gene-gene interactions computationally feasible by focusing on the dominant interaction patterns captured by the first few PCs. Applications in pharmacogenomics have revealed novel pathway crosstalk mechanisms affecting drug response, demonstrating how interaction PCA can uncover biological relationships invisible to standard single-pathway analyses.
Figure 2: PCA workflow for network module analysis
Experimental Design and Methodological Protocols
Comprehensive Protocol for Pathway Interaction PCA
Implementing PCA for pathway and interaction analysis requires meticulous experimental design and execution. The following step-by-step protocol ensures robust analysis:
Data Preprocessing: Begin with quality control of gene expression data, followed by normalization (e.g., min-max, z-score, or log transformation) to address technical variability [50] [13]. For RNA-seq data, carefully select normalization methods as they significantly impact PCA results and biological interpretation [13].
Pathway Definition: Curate pathway definitions from standard databases (KEGG, Reactome, MSigDB), ensuring proper gene identifier mapping. Filter pathways to include those with 10-200 genes to balance specificity and robustness.
Pathway PCA Implementation: For each pathway, extract expression matrix of member genes. Apply PCA using
prcomp()in R or equivalent, retaining components with eigenvalue >1 or those needed to explain ~80% of variance [52]. Record component scores (sample coordinates) and loadings (gene weights).Interaction Term Construction: For pathway pairs of interest, create interaction terms by multiplying standardized expressions of genes between pathways, generating all possible pairwise products.
Interaction PCA: Apply PCA to the interaction term matrix, retaining significant components. The resulting interaction-PCs represent dominant patterns of pathway crosstalk.
Validation Framework: Use cross-validation or bootstrap resampling to assess stability of pathway and interaction PCs. Compute variance explained metrics and component reproducibility across resampling iterations.
This protocol systematically reduces dimensionality while preserving biological structure, enabling detection of pathway activities and their interactions in a statistically robust framework.
Validation and Interpretation Strategies
Robust validation is essential for credible pathway and interaction PCA results. Apply the following validation strategies:
Component Stability: Assess component reliability through bootstrap resampling (100+ iterations), calculating the mean and confidence intervals for eigenvalues and loadings [27]. Stable components show narrow confidence intervals and consistent loading patterns.
Biological Replication: Validate identified pathway activities in independent datasets when available, testing reproducibility of component patterns across studies.
Permutation Testing: Establish statistical significance by comparing observed variance explained to null distributions generated by permuting sample labels 1000+ times.
Out-of-Sample Projection: Validate models by projecting new data into established PCA spaces, assessing whether expected biological separations remain apparent.
For interpretation, focus on genes with extreme loadings (top and bottom 5%) on each PC, as these drive the component's biological meaning. Conduct gene set enrichment analysis on high-loading genes to verify alignment with expected pathway functions. For interaction PCs, examine whether high-loading interaction terms involve biologically plausible gene pairs with established physical or functional relationships.
Research Reagents and Computational Tools
Table 3: Essential Research Reagents and Computational Tools for PCA-Based Analysis
| Tool/Resource | Type | Function | Implementation Notes |
|---|---|---|---|
| PCA Algorithms | Software | Core dimension reduction | R: prcomp(), factoextra; Python: sklearn.decomposition.PCA |
| Pathway Databases | Data | Biological context | KEGG, Reactome, MSigDB, Gene Ontology - provide gene sets for pathway PCA |
| Normalization Methods | Algorithm | Data preprocessing | Min-max, z-score, log transformation - critical for RNA-seq data [13] |
| Network Analysis Tools | Software | Module identification | Cytoscape, WGCNA, igraph - identify gene modules for network PCA |
| Visualization Packages | Software | Results interpretation | R: ggplot2, ggbiplot; Python: matplotlib, seaborn - create PCA plots |
| Benchmarking Frameworks | Methodology | Method validation | Custom scripts for comparing PCA vs. alternatives (Random Projection) [34] |
Comparative Analysis with Alternative Methods
While PCA remains widely used for dimension reduction in gene expression analysis, several alternative methods offer complementary strengths. Random Projection (RP) methods have emerged as computationally efficient alternatives, particularly valuable for large-scale single-cell RNA-seq datasets [34]. Unlike PCA, which identifies directions of maximum variance through computationally intensive eigen decomposition, RP projects data onto a random lower-dimensional subspace while approximately preserving pairwise distances [34]. Benchmarking studies demonstrate that RP methods (Sparse RP and Gaussian RP) can surpass PCA in computational speed while rivaling or sometimes exceeding PCA in preserving data structure and clustering quality [34]. However, PCA generally provides more interpretable components linked to biological processes, as PCs are linear combinations of genes with large loadings often belonging to coordinated pathways or networks.
Non-linear dimension reduction techniques like t-SNE and UMAP excel at preserving local data structure and creating visually appealing cluster separations, making them popular for exploratory analysis [34]. However, these methods typically lack the mathematical framework for projecting new samples into existing embeddings, limiting their utility for predictive modeling. PCA remains preferred when the analytical goal involves understanding major axes of variation, creating features for downstream statistical models, or maintaining a linear transformation for new sample projection. The choice between PCA and alternatives thus depends on specific research objectives, with PCA particularly well-suited for pathway and network analyses where component interpretability is paramount.
PCA continues to evolve beyond its traditional role as a dimension reduction tool into a sophisticated framework for pathway, network, and interaction analysis in genomic research. By incorporating biological structure into the analytical process through pathway-centric and network-aware implementations, PCA bridges statistical methodology and biological interpretation in ways that address fundamental challenges in high-dimensional genomics. The applications discussed—from pathway activity profiling to interaction modeling and network module analysis—demonstrate how PCA's linear algebraic foundation can be adapted to extract meaningful biological signals from complex gene expression data.
Future developments will likely focus on integrating PCA with other dimension reduction techniques, developing sparse implementations for enhanced interpretability, and creating specialized versions for emerging data types like single-cell multi-omics. As genomic datasets continue growing in size and complexity, PCA's ability to provide a "statistical mechanics" framework for biological systems—extracting collective parameters from microscopic details—will remain invaluable [53]. By maintaining a balance between computational efficiency, mathematical rigor, and biological interpretability, PCA-based approaches will continue enabling researchers to translate high-dimensional genomic measurements into meaningful biological insights and therapeutic discoveries.
Principal Component Analysis (PCA) stands as one of the oldest and most fundamental dimensionality reduction approaches in data science, invented by Karl Pearson in 1901 and later independently developed by Harold Hotelling in the 1930s [22]. In bioinformatics, PCA has become an indispensable tool for analyzing high-throughput genomic measurements that characteristically possess a "large d, small n" paradigm, where the number of features (genes) far exceeds the number of samples [27]. Traditional PCA operates by identifying orthogonal linear combinations of the original variables called principal components (PCs), which are sequenced to capture decreasing amounts of variance in the data [22]. This linear transformation allows researchers to project high-dimensional gene expression data into a lower-dimensional space that preserves essential biological signals while reducing computational complexity and noise [27] [54].
The advent of spatial transcriptomics (ST) technologies has revolutionized genomic analysis by enabling transcriptomic profiling while preserving crucial spatial localization information within tissues [55] [56]. These groundbreaking technologies range from sequencing-based methods like Slide-seq and 10x Visium to imaging-based approaches such as MERFISH and STARmap [56]. However, this spatial context introduces new computational challenges that conventional PCA cannot adequately address. Standard scRNA-seq dimension reduction methods assume independence among cells and fail to incorporate spatial dependency information, resulting in suboptimal performance for spatial transcriptomics where neighboring locations typically exhibit similar gene expression patterns due to shared microenvironmental influences and cellular interactions [57] [56]. This limitation has catalyzed the development of spatially-aware PCA methods that explicitly model spatial relationships while performing dimensionality reduction, thereby preserving critical spatial correlation structures in the resulting low-dimensional representations [57] [56].
The Computational Foundation of Spatial PCA Methods
Core Mathematical Principles
Spatially-aware PCA methods build upon the foundation of standard probabilistic PCA but incorporate crucial modifications to account for spatial dependencies. The fundamental innovation lies in explicitly modeling the spatial correlation structure across tissue locations through the use of spatial kernel matrices or Gaussian processes [56]. In SpatialPCA, for instance, the method uses a kernel matrix to model spatial correlation, allowing the low-dimensional components to preserve neighboring similarity patterns present in the original spatial data [56]. Similarly, SMOPCA assumes each latent factor follows a multivariate normal prior distribution with a covariance matrix calculated based on spatial location information to explicitly capture spatial dependencies in the latent space [57].
The mathematical formulation of these methods typically extends the standard PCA objective function to incorporate spatial regularization terms. Whereas standard PCA aims to find weight vectors that maximize the variance captured (w(1) = argmax∥w∥=1 {wᵀXᵀXw}), spatial PCA methods introduce additional constraints that enforce similarity between neighboring locations in the latent space [22] [56]. This spatial regularization ensures that the resulting principal components not only explain global variance in the gene expression data but also respect the local spatial structure of the tissue being analyzed.
Comparative Methodological Framework
Table 1: Key Spatially-Aware PCA Methods and Their Technical Approaches
| Method | Spatial Modeling Approach | Multi-omics Capability | Computational Scalability | Primary Applications |
|---|---|---|---|---|
| SpatialPCA [56] | Kernel matrix-based spatial correlation | Single modality | Moderate (suited for standard resolution ST) | Spatial domain detection, trajectory inference, high-resolution map construction |
| SMOPCA [57] | Multivariate normal prior with spatial covariance | Multi-omics integration | High (efficient for diverse dataset sizes) | Spatial domain detection, multi-omics integration, downstream analysis enhancement |
| RASP [58] | Configurable spatial smoothing with randomized algorithms | Supports non-transcriptomic covariates | Very high (scales to 100,000+ locations) | High-resolution ST analysis, rapid spatial domain detection |
| KSRV [59] | Kernel PCA with domain adaptation | RNA velocity integration | Moderate | Spatial RNA velocity inference, trajectory reconstruction |
Implementation and Workflow of Spatial PCA
Core Computational Pipeline
The implementation of spatially-aware PCA follows a structured workflow that integrates both expression data and spatial coordinates. The following diagram illustrates the generalized computational pipeline shared across multiple spatial PCA methods:
Diagram 1: Generalized workflow for spatially-aware PCA methods
The workflow begins with two primary inputs: the gene expression matrix (typically preprocessed and normalized) and the spatial coordinates of cells or spots within the tissue [57] [56]. The spatial kernel construction phase computes similarity metrics between locations based on their physical proximity, which can be implemented using various approaches including Gaussian kernels, graph-based adjacency matrices, or covariance functions derived from spatial coordinates [56]. This spatial kernel is then incorporated into the dimension reduction process through spatial regularization, constraining the solution to favor similar latent representations for spatially proximate locations [57]. The output is a low-dimensional representation that encapsulates both transcriptional variation and spatial context, which can subsequently fuel various downstream analyses including spatial domain detection, trajectory inference, and high-resolution spatial mapping [56].
Experimental Protocol for Spatial Domain Detection
For researchers implementing spatial PCA methods for tissue domain detection, the following step-by-step protocol provides a standardized approach:
Data Preprocessing: Begin with raw spatial transcriptomics data (from 10X Visium, Slide-seq, MERFISH, or similar platforms). Perform standard quality control, normalization, and log-transformation of expression counts. Select highly variable genes (typically 2000-3000) to reduce noise and computational burden [54].
Spatial Coordinate Alignment: Ensure spatial coordinates are properly aligned with tissue geometry. For array-based technologies like 10X Visium, verify spot alignment; for imaging-based approaches, confirm coordinate registration with tissue morphology.
Method Configuration: Initialize the chosen spatial PCA method (SpatialPCA, SMOPCA, etc.) with appropriate parameters. For SpatialPCA, this includes specifying the spatial kernel bandwidth; for SMOPCA, define the covariance structure for the multivariate normal prior [57] [56].
Model Fitting: Execute the spatial PCA algorithm to compute spatial principal components. This typically involves iterative optimization to maximize the likelihood function incorporating both expression variance and spatial constraints.
Component Selection: Determine the number of spatial PCs to retain for downstream analysis. This can be based on variance explained thresholds (e.g., PCs explaining >1% variance) or more automated approaches like elbow plots or permutation testing [54].
Spatial Clustering: Apply clustering algorithms (K-means, Louvain, etc.) to the spatial PCs to identify spatially coherent domains. Spatial constraints may be explicitly incorporated during this step or implicitly through the spatially-informed PCs.
Validation and Interpretation: Compare identified domains with known tissue histology, marker gene expression, or orthogonal clustering methods. Perform differential expression analysis between domains to identify domain-specific markers.
This protocol has been validated across multiple spatial transcriptomics technologies and tissue types, demonstrating robust performance in identifying anatomically and functionally distinct tissue regions [57] [56].
Performance Benchmarking and Comparative Analysis
Quantitative Performance Metrics
Table 2: Performance Comparison of Spatial PCA Methods Across Multiple Metrics
| Method | Spatial Domain Detection Accuracy (ARI) | Computational Speed | Scalability to Large Datasets | Multi-omics Integration | Resolution Enhancement |
|---|---|---|---|---|---|
| SpatialPCA [56] | 0.942 (simulated single-cell data) | Moderate | Good for standard resolutions | Limited | Yes, through kriging |
| SMOPCA [57] | Superior to single-modal methods | High | Excellent across dataset sizes | Native support for multiple modalities | Built-in capability |
| RASP [58] | Comparable or superior to benchmarks | Orders-of-magnitude faster | Designed for 100,000+ locations | Supports covariates | Configurable smoothing |
| KSRV [59] | Accurate trajectory inference | Moderate | Suitable for standard ST data | RNA velocity integration | Single-cell resolution |
Empirical evaluations demonstrate that spatially-aware PCA methods significantly outperform conventional dimension reduction approaches for spatial transcriptomics data. In comprehensive simulations using cortex tissue data with known ground truth spatial domains, SpatialPCA achieved a median adjusted Rand index (ARI) of 0.942, substantially exceeding the performance of non-spatial methods including standard PCA (ARI ~0.367) and other spatial domain detection methods like SpaGCN (ARI 0.625) and HMRF (ARI 0.773) in scenarios with complex cell-type compositions [56]. Similarly, SMOPCA has demonstrated superior performance in spatial domain detection across diverse technologies and tissue structures, particularly when analyzing multi-omics spatial data where it effectively integrates complementary information from different molecular modalities [57].
The computational efficiency of these methods varies considerably, with recent innovations like Randomized Spatial PCA (RASP) specifically designed to address scalability challenges in high-resolution spatial transcriptomics. RASP employs a randomized two-stage PCA framework with sparse matrix operations, achieving orders-of-magnitude speed improvements while maintaining comparable or superior accuracy in tissue domain detection [58]. This enhanced computational efficiency enables researchers to analyze datasets with over 100,000 spatial locations, which is increasingly important as spatial technologies advance toward subcellular resolution.
Integration Capabilities and Method Specialization
The landscape of spatial PCA methods has evolved to address specialized analytical needs beyond basic spatial domain detection. SMOPCA represents a significant advancement for multi-omics integration, simultaneously modeling different data modalities while preserving spatial dependencies [57]. Unlike earlier approaches that could only handle up to two modalities, SMOPCA can seamlessly integrate three or more omics layers without requiring architectural redesign, addressing a critical gap in spatial multi-omics analysis [57].
KSRV (Kernel PCA-Based Spatial RNA Velocity) demonstrates how spatial PCA principles can be extended to dynamic biological processes. By employing kernel PCA to integrate scRNA-seq with spatial transcriptomics data, KSRV enables inference of RNA velocity fields within spatial contexts, revealing differentiation trajectories directly in tissue architecture [59]. This approach addresses the fundamental limitation that most spatial transcriptomics technologies cannot simultaneously capture both spliced and unspliced transcripts necessary for conventional RNA velocity analysis.
The following diagram illustrates the architectural differences between these specialized spatial PCA approaches:
Diagram 2: Specialized spatial PCA methods for different analytical scenarios
Table 3: Essential Research Resources for Spatial PCA Implementation
| Resource Category | Specific Tools/Methods | Primary Function | Implementation Considerations |
|---|---|---|---|
| Spatial Technologies | 10X Visium, MERFISH, Slide-seq, STARmap | Generate spatial transcriptomics data | Technology choice balances resolution, gene coverage, and spatial capture area |
| Spatial PCA Algorithms | SpatialPCA, SMOPCA, RASP, KSRV | Perform spatially-aware dimension reduction | Selection depends on data type, scale, and analytical objectives |
| Computational Frameworks | R, Python, Bioconductor, Scanpy | Provide computational infrastructure | R environments often have specialized packages for spatial genomics |
| Downstream Analysis Tools | Clustering (K-means, Louvain), Trajectory Inference (Slingshot, PAGA) | Extract biological insights from spatial PCs | Method choice should align with biological questions |
| Validation Approaches | Histological alignment, marker gene expression, orthogonal clustering | Verify biological relevance of results | Essential for establishing methodological credibility |
Discussion and Future Perspectives
Spatially-aware PCA methods represent a significant evolution in dimensionality reduction technique that addresses the unique characteristics of spatial transcriptomics data. By explicitly incorporating spatial dependencies into the dimension reduction framework, these methods overcome fundamental limitations of conventional PCA when applied to spatially-resolved genomics data. The resulting low-dimensional representations not only capture transcriptional variation but also preserve critical spatial context, enabling more accurate spatial domain detection, trajectory inference, and tissue structure characterization [57] [56].
The development of these methods reflects a broader trend in bioinformatics toward designing specialized computational approaches that address the particularities of emerging genomic technologies. Just as single-cell RNA-seq required specialized analytical methods to account for technical artifacts and biological heterogeneity, spatial transcriptomics demands algorithms that respect spatial dependencies and tissue architecture [55] [56]. Spatially-aware PCA methods successfully bridge this gap by integrating principles from spatial statistics with established dimension reduction frameworks.
Looking forward, several emerging trends will likely shape the next generation of spatially-aware dimension reduction methods. As spatial multi-omics technologies mature, developing integrated analysis approaches that can simultaneously model transcriptomic, epigenomic, and proteomic data within spatial contexts will become increasingly important [57] [55]. Additionally, as spatial technologies achieve higher resolutions and larger capture areas, computational scalability will remain a persistent challenge, necessitating continued innovation in efficient algorithms and data structures [58]. Finally, tighter integration with experimental validation approaches will be essential for translating computational predictions into biological insights, particularly in clinical and drug development contexts where understanding spatial organization of cell types and states can illuminate disease mechanisms and therapeutic opportunities [55].
In conclusion, spatially-aware PCA methods have established themselves as essential tools in the spatial transcriptomics toolkit, enabling researchers to extract meaningful biological signals from complex spatial genomics data while respecting the spatial context that is fundamental to tissue function and organization.
High-dimensional data is a fundamental challenge in modern genomics, particularly in gene expression analysis where the number of measured genes (features) vastly exceeds the number of biological samples (observations). This "small sample size, high dimensionality" characteristic makes direct analysis computationally intensive and statistically problematic [60]. Principal Component Analysis (PCA) has served as a foundational technique for overcoming this challenge by transforming complex datasets into a lower-dimensional space while preserving essential patterns and structures [61]. In biotechnology research, PCA simplifies intricate multi-dimensional data from genomics, proteomics, and metabolomics, enabling researchers to visualize sample relationships, identify clusters, detect outliers, and uncover biological insights that would otherwise remain hidden in thousands of variables [62].
Despite its widespread adoption, standard PCA faces significant limitations when applied to gene expression data. Its dense loading vectors make biological interpretation difficult, as all genes contribute to each principal component. Furthermore, PCA assumes linear relationships between variables and struggles to capture the complex nonlinear structures prevalent in biological systems [63]. These constraints have motivated the development of specialized PCA variants that address the unique demands of genomic research. This technical guide examines three critical advancements: Sparse PCA for interpretable feature selection, Kernel PCA for nonlinear pattern recognition, and model-based alternatives specifically designed for count-based sequencing data, framing their development within the ongoing effort to extract meaningful biological signals from high-dimensional genomic data.
Methodological Foundations and Comparative Framework
Standard PCA: Mathematical Formulation and Workflow
Principal Component Analysis operates on the fundamental principle of identifying orthogonal directions of maximal variance in high-dimensional data. Given a standardized data matrix ( X ) with dimensions ( n \times p ) (where ( n ) represents samples and ( p ) represents genes), PCA performs an eigen decomposition of the covariance matrix ( \Sigma = \frac{1}{n-1}X^TX ) [62]. The resulting eigenvectors (principal components) form a new coordinate system where the axes are ordered by the amount of variance they explain, quantified by their corresponding eigenvalues.
The standardized workflow for PCA implementation in genomic studies involves several critical steps [61] [38]:
- Data Standardization: Normalizing each variable to have zero mean and unit variance ensures that all genes contribute equally to the analysis, preventing highly expressed genes from dominating the variance structure.
- Covariance Matrix Computation: Calculating the covariance matrix reveals how genes vary together across samples, capturing the core relationships in the expression data.
- Eigenvalue Decomposition: Extracting eigenvectors and eigenvalues from the covariance matrix identifies the principal components and their relative importance.
- Component Selection: Choosing the top ( k ) components based on eigenvalue magnitude or variance explained (e.g., retaining components that collectively explain >80% of total variance).
- Data Projection: Transforming the original data into the new subspace via linear combination ( Y = XW ), where ( W ) contains the selected eigenvectors [62].
Table 1: Standard PCA Variants in Genomic Applications
| Variant | Key Mechanism | Genomic Application Context | Computational Complexity |
|---|---|---|---|
| Standard PCA | Orthogonal projection to maximize variance | Exploratory analysis, data visualization, noise reduction | ( O(nd^2) ) [2] |
| Incremental PCA | Mini-batch processing for large datasets | Streaming genomic data, memory-efficient processing | ( O(ndk) ) [2] |
| Multilinear PCA | Mode-wise decomposition for tensor data | Multi-modal genomic integration, imaging genetics | ( O(n\prod{m=1}^M dm) ) [2] |
| Probabilistic PCA | Bayesian framework with Gaussian noise modeling | Uncertainty quantification in expression measurements | ( O(nd^2) ) [2] |
Limitations of Standard PCA in Genomic Contexts
While invaluable for initial exploration, standard PCA exhibits several limitations when applied to gene expression data. The principal components generated are linear combinations of all genes in the dataset, creating challenges for biological interpretation as researchers struggle to identify which specific genes drive the observed sample separations [60]. This deficiency becomes critical in biomarker discovery, where identifying specific gene signatures is paramount.
Additionally, the assumption of linear relationships fails to capture the complex nonlinear patterns inherent in gene regulatory networks and cellular responses [63]. Biological systems frequently exhibit threshold effects, synergistic interactions, and other nonlinear behaviors that linear PCA cannot adequately represent. These limitations have driven the development of specialized variants that extend PCA's utility while addressing its core constraints for genomic applications.
Sparse PCA for Interpretable Genomics
Methodological Framework of Sparse PCA
Sparse PCA (SPCA) introduces regularization to the standard PCA framework to produce loading vectors with many zero coefficients, effectively selecting a subset of meaningful genes for each component. The fundamental optimization problem for the first sparse principal component can be formulated as:
[\min{u1,v1} { \|X - u1v1^T\|F^2 + \text{pen}(v_1) }]
where (\text{pen}(v1)) represents a sparsity-inducing penalty term, most commonly the Lasso ((\ell1)-norm) penalty: (\text{pen}(v1) = \lambda\sum{j=1}^p |v_{1j}|) with (\lambda > 0) controlling the sparsity strength [60]. This formulation yields principal components defined by only a few non-zero loadings, dramatically enhancing biological interpretability.
The integrative Sparse PCA (iSPCA) extension further addresses the need to analyze multiple genomic datasets simultaneously. iSPCA leverages the homogeneity model, which assumes comparable studies share the same sparsity structure, and employs a group penalty to jointly identify relevant genes across datasets:
[\text{pen}(v1^{(1)}, \ldots, v1^{(M)}) = \lambda1 \sum{j=1}^p {(v{1j}^{(1)})^2 + \ldots + (v{1j}^{(M)})^2}^{1/2}]
This approach identifies stable gene signatures that consistently contribute to principal components across multiple independent studies, increasing the reliability of discovered biomarkers [60].
Experimental Implementation and Workflow
Implementing Sparse PCA in genomic studies requires careful attention to several methodological considerations. The following workflow outlines the key stages for applying SPCA to gene expression data:
- Data Preprocessing: Standardize each dataset to zero mean and unit variance. For multi-dataset integration, ensure consistent gene matching across platforms.
- Parameter Tuning: Determine the optimal sparsity parameter ( \lambda ) through cross-validation, evaluating reconstruction error versus sparsity strength. Use stability selection to identify robust genes.
- Model Fitting: Apply the SPCA algorithm to obtain sparse loading vectors. For multi-dataset integration, implement iSPCA with contrasted penalties.
- Component Interpretation: Analyze non-zero loadings to identify gene sets driving each component. Perform enrichment analysis on high-weight genes.
- Biological Validation: Relate sparse components to sample characteristics (e.g., disease status, treatment response). Validate identified gene signatures in independent cohorts.
Table 2: Sparse PCA Variants and Their Genomic Applications
| Variant | Sparsity Mechanism | Advantages in Genomics | Limitations |
|---|---|---|---|
| Standard SPCA | Lasso ((\ell_1)) penalty on loadings | Improved interpretability, feature selection | Potential instability, requires tuning |
| Integrative SPCA (iSPCA) | Group penalty across multiple datasets | Identifies robust biomarkers, enhances reproducibility | Assumes homogeneous sparsity across studies |
| Sparse PCA with Contrasted Penalties | Differential penalties based on prior knowledge | Incorporates biological knowledge, improves signal detection | Requires careful penalty specification |
Figure 1: Integrative Sparse PCA Workflow for Multi-Dataset Genomic Analysis
Kernel PCA for Nonlinear Genomic Relationships
Theoretical Foundation of Kernel PCA
Kernel PCA extends classical PCA to capture nonlinear patterns by leveraging the kernel trick, a method that implicitly maps data to a higher-dimensional feature space where linear separability becomes possible [63]. Given gene expression data ( xi ) in the original input space, Kernel PCA applies a nonlinear mapping ( \Phi(x) ) to transform the data into a higher-dimensional feature space ( F1 ). The key innovation circumvents the need to compute coordinates in the high-dimensional space; instead, operations are performed using the kernel function ( K(xi, xj) = \langle \Phi(xi), \Phi(xj) \rangle ) that represents inner products in the transformed space [64].
The radial basis function (RBF) kernel has proven particularly effective for genomic applications:
[K(xi, xj) = \exp\left(-\frac{\|xi - xj\|^2}{2h^2}\right)]
where ( h ) represents the kernel bandwidth parameter controlling the flexibility of the nonlinear mapping [64]. This kernel can model complex gene interaction networks and expression patterns that linear methods cannot capture, making it particularly valuable for identifying subtle biological signatures in high-dimensional genomic data.
Bandwidth Optimization and Implementation
The performance of Kernel PCA critically depends on appropriate bandwidth selection for the RBF kernel. A data-driven bandwidth selection criterion related to least squares cross-validation for kernel density estimation has been developed specifically for genomic applications [64]. This approach balances model flexibility and stability, ensuring the kernel captures meaningful biological signal without overfitting to noise.
The computational workflow for Kernel PCA in genomic analysis involves:
- Kernel Selection: Choose an appropriate kernel function (RBF recommended for initial applications).
- Bandwidth Optimization: Tune the bandwidth parameter ( h ) using cross-validation or data-driven criteria.
- Kernel Matrix Computation: Calculate the kernel matrix ( K ) where each entry ( K{ij} = K(xi, x_j) ).
- Kernel Matrix Centering: Center the kernel matrix to ensure feature space data has zero mean.
- Eigenvalue Decomposition: Perform eigendecomposition on the centered kernel matrix.
- Component Projection: Project data onto the dominant eigenvectors of the kernel matrix.
Figure 2: Kernel PCA Nonlinear Transformation Workflow
Comparative Analysis of PCA Variants in Genomic Studies
Table 3: Performance Comparison of PCA Variants on Gene Expression Data
| Method | Linearity Assumption | Interpretability | Genomic Data Suitability | Key Applications |
|---|---|---|---|---|
| Standard PCA | Linear | Moderate - dense loadings | Moderate - captures global structure | Initial exploration, data visualization |
| Sparse PCA | Linear | High - sparse loadings | High - identifies key genes | Biomarker discovery, feature selection |
| Kernel PCA | Nonlinear | Moderate - implicit mapping | High - captures complex patterns | Nonlinear pattern recognition, pathway analysis |
| Integrative SPCA | Linear | Very High - stable sparse loadings | Very High - multi-study integration | Cross-platform biomarker validation |
Model-Based Alternatives for Count-Based Genomic Data
Non-negative Matrix Factorization for Count Data
While not explicitly designed for count data, Non-negative Matrix Factorization (NMF) has emerged as a valuable dimensionality reduction technique for non-negative genomic data such as RNA-seq counts and chromatin accessibility measurements. NMF factorizes a non-negative data matrix ( V ) into two lower-dimensional non-negative matrices: ( V \approx WH ), where ( W ) represents basis components (metagenes) and ( H ) represents coefficient weights across samples [65].
The non-negativity constraint aligns with the natural characteristics of count-based genomic measurements, where negative values are biologically implausible. This often produces more interpretable components than PCA for certain applications, as NMF effectively performs parts-based decomposition that can identify co-expressed gene modules or combinatorial regulatory patterns [65]. Sequential NMF variants further enhance stability by sequentially constructing components, making them particularly suitable for analyzing the temporal dynamics of gene expression.
Generalized Linear Model-Based Alternatives
For count-based sequencing data with specific distributional characteristics, model-based alternatives built on generalized linear models (GLMs) often outperform standard dimensionality reduction techniques. The most effective approaches include:
- Poisson PCA: Models count data using Poisson distributions with log-link functions, directly addressing the mean-variance relationship in sequencing data.
- Negative Binomial Factor Models: Extends Poisson PCA to handle overdispersed count data common in RNA-seq experiments, incorporating sample-specific scaling parameters.
- Zero-Inflated Factor Models: Addresses the excess zeros frequently observed in single-cell RNA sequencing data through mixture modeling frameworks.
These model-based approaches explicitly account for the statistical characteristics of sequencing data, providing more biologically meaningful decompositions than standard PCA applied to transformed count data.
Comparative Evaluation and Research Applications
Performance Assessment in Transcriptomic Studies
Recent large-scale evaluations have quantified the relative performance of dimensionality reduction methods in transcriptomic data analysis. A comprehensive assessment of 71 bulk transcriptomic datasets compared PCA, multidimensional scaling (MDS), t-SNE, and UMAP, revealing that UMAP (a nonlinear manifold technique) generally outperformed PCA in revealing biologically meaningful sample clusters, particularly for capturing batch effects and identifying predefined biological groups [66].
In predictive modeling applications, regularized models without dimensionality reduction sometimes achieved the highest predictive performance for binary phenotype classification [67]. However, autoencoders and independent component analysis (ICA) with transfer learning demonstrated comparable performance while providing enhanced interpretability and more robust predictors. These findings suggest that the choice of dimensionality reduction method should align with the specific analytical goal—whether prediction accuracy, biological interpretability, or data visualization.
Table 4: Essential Computational Tools for Advanced PCA Applications
| Tool/Resource | Function | Implementation Considerations |
|---|---|---|
| SPCA Algorithms | Sparse loading estimation | Available in R (elasticnet, PMA) and Python (scikit-learn) |
| Kernel PCA with RBF | Nonlinear dimensionality reduction | Bandwidth selection critical; use cross-validation |
| Integrative SPCA | Multi-dataset joint analysis | Requires homogeneous sparsity assumption |
| NMF Algorithms | Parts-based decomposition | Sequential NMF enhances stability for genomic data |
| Model-Based PCA | Count data dimensionality reduction | Poisson/NB distributions for sequencing data |
The evolution of PCA from a general dimensionality reduction technique to specialized variants has significantly enhanced its utility for genomic research. Sparse PCA addresses the interpretability limitations of standard PCA by producing loading vectors with biological meaningful gene subsets. Kernel PCA extends the methodological framework to capture nonlinear patterns prevalent in gene regulatory networks. Model-based alternatives specifically designed for count data provide more appropriate statistical foundations for modern sequencing technologies.
Future developments will likely focus on hybrid approaches that combine the strengths of multiple techniques, such as sparse kernel methods for interpretable nonlinear dimensionality reduction. The integration of dimensionality reduction with deep learning architectures, particularly through advanced autoencoder designs, shows promise for capturing hierarchical biological structures. As genomic technologies continue to evolve toward multi-omics integration, specialized dimensionality reduction methods that can jointly analyze heterogeneous data types while maintaining biological interpretability will become increasingly essential for extracting meaningful insights from complex biological systems.
Navigating PCA Pitfalls: Ensuring Robust and Biologically Meaningful Results
In the analysis of high-dimensional genomic data, particularly gene expression data from technologies like single-cell RNA sequencing (scRNA-Seq), researchers routinely face datasets where the number of genes (features) vastly exceeds the number of samples (cells). This \(P \gg N\) scenario, known as the "curse of dimensionality," presents substantial challenges for computational analysis, visualization, and biological interpretation [20]. Gene expression matrices often contain expressions of >20,000 genes across fewer than 100 samples, creating a high-dimensional space where data points become sparse and computational costs escalate [20].
Principal Component Analysis (PCA) serves as a foundational technique to address these challenges by transforming the original high-dimensional gene expression data into a lower-dimensional space while retaining its essential biological information. This orthogonal linear transformation creates new, uncorrelated variables called principal components (PCs), which are linear combinations of genes and capture decreasing proportions of the total variance in the original dataset [19]. The central challenge for researchers lies in determining how many of these principal components to retain—a critical choice that balances information preservation against noise reduction. Selecting too few components risks discarding biologically meaningful signals, while retaining too many incorporates noise and undermines the benefits of dimensionality reduction [68] [19].
Quantitative Metrics for Dimension Selection
Multiple quantitative approaches exist for determining the optimal number of principal components to retain in gene expression studies. These methods provide objective, data-driven guidance for this critical parameter.
Table 1: Core Metrics for Selecting the Number of Principal Components
| Method | Description | Interpretation in Gene Expression Context | Advantages | Limitations |
|---|---|---|---|---|
| Scree Test | Visual inspection of the eigenvalue curve [11] [68] | Identify the "elbow" point where eigenvalues drop sharply [19] | Intuitive visualization of variance distribution | Subjective; elbow may not be clearly defined [19] |
| Kaiser's Criterion | Retain components with eigenvalues >1 [68] | Based on the notion that a component should explain at least the variance of one standardized variable | Simple objective threshold | Often retains too many components in genomic data |
| Variance Threshold | Retain components that collectively explain a target percentage of total variance (e.g., 95%) [11] | Ensures sufficient biological signal is preserved | Directly controls information retention | May include noise components; threshold is arbitrary |
| Cross-Validation | Evaluate model performance with different component numbers [68] | Assess how well components generalize to new biological data | Statistical rigor; prevents overfitting | Computationally intensive for large gene expression matrices |
The scree plot visualizes the eigenvalues associated with each principal component in descending order, allowing researchers to identify the point where the curve flattens—the "elbow"—indicating components beyond this point contribute minimal additional variance [68] [19]. In practice, the scree plot's elbow corresponds to the transition from components capturing biological signal to those representing technical noise or stochastic gene expression variation.
For a more objective approach, researchers often employ the variance explained criterion, which involves calculating the cumulative proportion of total variance explained by successive components [11]. A common threshold in genomic studies is retaining enough components to explain 80-95% of total variance, though this depends on the specific biological question and data characteristics.
Diagram 1: Workflow for determining the optimal number of principal components (k) in gene expression analysis.
Experimental Protocols for Method Validation
Benchmarking Framework for Dimension Selection
Robust validation of dimension selection strategies requires a systematic benchmarking approach that evaluates how well the reduced-dimensional space preserves biological information. A comprehensive protocol should include:
Data Preparation: Begin with a quality-controlled gene expression matrix (cells × genes). Standardize the data by centering each gene to zero mean and scaling to unit variance to prevent highly expressed genes from dominating the analysis [68].
Parameter Sweep: Apply PCA and extract principal components across a range of values for
k(e.g., from 1 to 100 components).Downstream Evaluation: For each value of
k, assess the quality of the reduced-dimensional representation using biologically meaningful downstream tasks:- Clustering Analysis: Apply clustering algorithms (e.g., hierarchical clustering, spherical k-means) to the PCA-reduced data and compare the resulting cell groups to known cell type annotations using accuracy metrics like the Hungarian algorithm and Mutual Information for labeled datasets [34].
- Cluster Separation Metrics: For unlabeled data, compute internal validation measures such as the Dunn Index and Gap Statistic to quantify how well-separated the clusters are in the reduced space [34].
- Within-Cluster Sum of Squares (WCSS): Evaluate cluster compactness, which indicates how well the reduction preserves the natural grouping structure of cells [34].
- Locality Preservation: Assess how well local neighborhoods of cells are maintained in the PC space compared to the original high-dimensional space [34].
Comparative Analysis: Compare the performance of different dimension selection criteria against this benchmarking framework to identify the optimal strategy for a given dataset type.
Case Study: scRNA-Seq Analysis Pipeline
In scRNA-Seq analysis, PCA is typically applied after quality control and normalization but before clustering and visualization [19]. A standardized protocol includes:
Diagram 2: Integration of dimension selection within the standard scRNA-Seq analysis workflow.
Input: A normalized gene expression matrix with cells as rows and genes as columns, typically filtered to include only highly variable genes to improve signal-to-noise ratio.
PCA Implementation:
- Center the data by subtracting the mean of each gene.
- Compute the covariance matrix to capture gene-gene relationships.
- Perform eigenvalue decomposition of the covariance matrix:
\(C = V\Lambda V^T\), where eigenvectors represent principal components and eigenvalues indicate the amount of variance each PC captures [69].
Dimension Selection: Apply multiple criteria (Table 1) to determine the optimal number of components. In scRNA-Seq analysis, the "elbow" method applied to scree plots is particularly common, though subjective [19].
Validation: Project the data onto the selected PCs and evaluate whether known cell types separate in the reduced space. Use domain knowledge to verify biological relevance.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for PCA in Gene Expression Analysis
| Tool/Library | Application Context | Key Functionality | Implementation Considerations |
|---|---|---|---|
| scikit-learn (Python) | General-purpose ML | Robust PCA implementation with explained variance metrics | Easy integration with pandas dataframes; supports large-scale data [11] [68] |
| Scanpy (Python) | scRNA-Seq analysis | PCA specialized for single-cell data with neighbors graph construction | Built-in preprocessing and visualization tailored to genomic data |
| Seurat (R) | scRNA-Seq analysis | Comprehensive PCA integration in single-cell workflow | Handles sparse matrices efficiently; includes dimension selection guides |
| Cell Ranger | 10x Genomics scRNA-Seq | Automated pipeline including PCA | Proprietary software optimized for specific sequencing technology [19] |
| Boosting Autoencoder (BAE) | Interpretable dimension reduction | Sparse dimensionality reduction with gene selection | Identifies specific genes driving each latent dimension [70] [7] |
Advanced Considerations in Genomic Applications
Beyond Linear Assumptions: Addressing PCA Limitations
While PCA remains a cornerstone technique for dimensionality reduction in gene expression analysis, it carries important limitations that researchers must consider when interpreting results. PCA assumes linear relationships among variables, yet biological systems often exhibit nonlinear interactions that PCA cannot capture [69] [34]. Additionally, PCA is sensitive to outliers and technical artifacts common in genomic data, which can disproportionately influence principal components [69] [34].
When PCA proves insufficient for capturing complex biological patterns, several advanced approaches show particular promise for genomic data:
Kernel PCA (KPCA): Extends PCA to capture nonlinear structures using kernel functions, effectively mapping data to a higher-dimensional space where linear separation becomes possible [69]. While computationally intensive (
\(O(n^3)\)complexity), KPCA can reveal biological patterns invisible to standard PCA.Sparse Kernel PCA: Addresses KPCA's computational limitations by selecting a subset of representative training points, significantly reducing memory requirements while preserving nonlinear modeling capabilities [69].
Boosting Autoencoders (BAE): Combine the representation learning power of neural networks with the interpretability of boosting methods to identify small sets of genes that explain latent dimensions [7]. This approach is particularly valuable for pinpointing specific ligand-receptor interactions or marker genes associated with cell clusters [70].
Emerging Alternatives: Random Projections
Recent benchmarking studies indicate that Random Projection (RP) methods rival or even surpass PCA in certain genomic applications, particularly for large-scale scRNA-Seq datasets [34]. Both Sparse Random Projection (SRP) and Gaussian Random Projection (GRP) offer significant computational advantages over PCA while approximately preserving pairwise distances between data points [34].
These methods are particularly valuable when computational efficiency is paramount or when the assumptions of PCA (linearity, sensitivity to outliers) are severely violated. The choice between PCA and RP depends on the specific analytical goals: PCA maximizes variance retention, while RP optimizes distance preservation with greater computational efficiency.
The selection of appropriate dimensions in PCA represents a critical methodological decision that directly influences the biological insights derived from gene expression data. No single approach universally outperforms others across all datasets and research questions. Rather, the optimal strategy integrates multiple quantitative metrics—scree plots, variance explained, and cross-validation—with rigorous benchmarking using biologically meaningful outcomes.
As genomic technologies evolve toward increasingly high-dimensional measurements, sophisticated dimension selection strategies will grow ever more essential. Future methodologies will likely combine the computational efficiency of random projections, the nonlinear modeling capacity of kernel methods, and the interpretability of sparse approaches like boosting autoencoders. By carefully evaluating dimension selection strategies within their specific biological context, researchers can ensure that their analytical choices enhance rather than obscure the complex biological signals embedded in high-throughput genomic data.
Mitigating Overfitting and Instability in High-Dimensional Settings
In the analysis of high-dimensional genomic data, particularly gene expression data, researchers frequently encounter the "curse of dimensionality," where the number of features (genes) vastly exceeds the number of samples. This environment creates fertile ground for overfitting, a phenomenon where models learn not only the underlying biological signal but also the noise and random variations specific to the training dataset [71] [72]. An overfitted model performs exceptionally well on its training data but fails to generalize its predictions to new, unseen data, compromising its utility in real-world applications like drug development and diagnostic biomarker discovery [72].
The implications of overfitting extend beyond mere statistical inconvenience. In academic and clinical research, it can lead to misguided conclusions, wasted resources, and a dangerous erosion of trust in data-driven findings [72]. For instance, a overfitted model might identify a set of genes as perfect biomarkers for a disease, only for subsequent studies to find them useless, thereby misdirecting research efforts and clinical resources [71] [72]. Therefore, mitigating overfitting is not just a technical step in model tuning but a fundamental requirement for producing robust, reliable, and reproducible scientific knowledge.
Principal Component Analysis (PCA) as a Dimensionality Reduction Technique
Theoretical Foundation of PCA
Principal Component Analysis (PCA) is a classical statistical technique for dimensionality reduction. It operates by transforming the original high-dimensional data into a new set of variables called principal components (PCs) [73] [27]. These components are linear combinations of the original genes (features) and are designed to capture the directions of maximum variance in the data [74]. The first principal component (PC1) accounts for the largest possible variance, PC2 for the second largest, and so on, with all PCs being mutually orthogonal [27].
The mathematical formulation begins with a data matrix ( X ) with ( n ) samples (e.g., patients) and ( p ) features (e.g., gene expression levels). After standardizing the data to have a mean of zero and unit variance, PCA involves computing the covariance matrix and performing an eigen decomposition [74]. The eigenvectors of this covariance matrix form the principal components, and the corresponding eigenvalues indicate the amount of variance each PC explains [27] [74]. The data is then projected onto this new subspace defined by the top ( k ) PCs, resulting in a transformed dataset ( Z = XW ), where ( W ) is the matrix of the selected eigenvectors [74].
How PCA Mitigates Overfitting in Gene Expression Data
PCA addresses overfitting in high-dimensional gene expression data through several key mechanisms. First, by reducing the number of features from thousands of genes to a few dozen or fewer PCs, it drastically reduces model complexity [71] [73]. A simpler model with fewer parameters is less capable of "memorizing" noise in the training data, thereby enhancing its ability to generalize [72].
Second, the principal components are constructed to capture the dominant, coordinated patterns of variation across many genes [27]. In genomic studies, these patterns often represent robust biological signals, such as a pathway activation or a major cellular response. In contrast, random noise and measurement errors are typically uncoordinated across genes and are consequently relegated to the lower-order, less important PCs, which are then discarded [27]. This process effectively acts as a noise filter, stripping away irrelevant variations that could lead a model to overfit [73].
Finally, the reduced dimensionality achieved by PCA decreases computational cost and training time, while also making subsequent analyses like clustering or regression more stable and interpretable [71] [75]. By focusing on a small number of meta-features (the PCs), researchers can build models that are both more robust and more transparent.
Limitations and Considerations for PCA
Despite its utility, PCA is not a universal solution and has important limitations. Its primary assumption is linearity; it seeks linear combinations of genes that maximize variance. It may fail to capture complex, non-linear relationships present in biological systems [18]. Furthermore, the resulting principal components can be difficult to interpret biologically, as they are mathematical constructs that amalgamate hundreds or thousands of genes [27]. The sample composition of the dataset can also heavily influence the PCs. For example, if a dataset contains an overrepresentation of samples from a specific tissue (e.g., liver), this can dominate one of the early principal components, potentially obscuring other important biological signals [16].
Table 1: Key Characteristics of PCA in Gene Expression Analysis
| Characteristic | Description | Implication for Overfitting |
|---|---|---|
| Input Data | Original feature matrix (e.g., gene expression values) [18] | Works directly with the high-dimensional data to find structure. |
| Linearity Assumption | Identifies linear directions of maximum variance [18] | May not capture complex non-linear interactions, a potential source of remaining instability. |
| Variance Focus | Prioritizes components with highest variance [27] | Effectively filters out low-variance noise, reducing the risk of fitting to noise. |
| Output | A reduced set of orthogonal principal components [27] | Provides a simpler, less correlated set of features for downstream modeling. |
| Interpretability | PCs can be hard to interpret; require examination of gene loadings [27] | While the model is stabilized, the biological explanation becomes less direct. |
A Practical Workflow for Implementing PCA on Gene Expression Data
Implementing PCA correctly is critical for its effectiveness in mitigating overfitting. The following workflow provides a step-by-step protocol for a typical gene expression analysis.
Data Preprocessing and Standardization
The initial and crucial step is data preprocessing. Gene expression data must be standardized before applying PCA. This involves centering each variable (gene) to have a mean of zero and scaling it to have a standard deviation of one [76]. Standardization is essential because PCA is sensitive to the variances of the initial variables. Without it, genes with naturally higher expression ranges would dominate the first principal component, not necessarily because they are more biologically informative, but simply due to their scale [76].
Executing PCA and Determining Component Number
Once the data is standardized, PCA can be executed using standard software libraries like scikit-learn in Python [76] [74]. A key decision is choosing the number of principal components, ( k ), to retain. A common method is to use a scree plot, which visualizes the proportion of total variance explained by each consecutive PC [76]. Researchers often look for an "elbow" in the plot—a point where the marginal gain in explained variance drops significantly. The goal is to select a number of components that captures the majority of the biological signal while discarding dimensions that likely represent noise. Another rule of thumb is to retain enough components to explain a high percentage (e.g., 80-95%) of the total cumulative variance [27].
Visualization and Biological Interpretation
The principal components can be used to visualize the global structure of the data in a low-dimensional space. By plotting the samples on a scatter plot using the first two PCs (PC1 vs. PC2), researchers can identify clusters, outliers, and batch effects [76] [74]. For example, in a dataset containing wild-type and knock-out samples, a clear separation along PC1 would indicate that the largest source of variation in the data is driven by the genetic difference between these groups [76].
Biological interpretation involves examining the loadings (weights) of the genes on each PC. Genes with the highest absolute loadings for a given PC are the strongest drivers of that component's variation. Researchers can then perform functional enrichment analysis on these top-weighted genes to understand the biological processes, pathways, or functions that the PC might represent [76]. This step translates the mathematical output of PCA into actionable biological insights.
Diagram 1: PCA analysis workflow for gene expression data.
Advanced and Hybrid Strategies for Enhanced Stability
While PCA is powerful, it can be complemented with other advanced dimensionality reduction and feature selection techniques to further combat overfitting and instability.
Supervised, Sparse, and Kernel PCA
Several advanced variants of PCA have been developed to address its limitations:
- Supervised PCA: This method incorporates the relationship between gene expression data and an outcome variable (e.g., patient survival) to guide the identification of principal components, making them more relevant for prediction [27].
- Sparse PCA: This technique incorporates sparsity constraints to produce principal components that are linear combinations of only a small subset of genes. This enhances biological interpretability by making it easier to pinpoint which specific genes are driving the variation, effectively performing feature selection within the dimensionality reduction process [27] [74].
- Kernel PCA: For capturing non-linear relationships, Kernel PCA uses the "kernel trick" to project data into a higher-dimensional space where non-linear patterns can be separated linearly before applying PCA. This is useful for complex biological data where gene interactions are not purely additive [18] [74].
Hybrid Feature Selection Frameworks
Combining PCA with robust feature selection algorithms can create a powerful hybrid defense against overfitting. Recent research has focused on hybrid AI-driven frameworks that use metaheuristic algorithms to select an optimal subset of features before or after dimensionality reduction [75]. These include:
- Two-phase Mutation Grey Wolf Optimization (TMGWO): Enhances exploration and exploitation in the search for the best feature subset [75].
- Improved Salp Swarm Algorithm (ISSA): Uses adaptive inertia weights and local search techniques to improve convergence accuracy [75].
- Binary Black Particle Swarm Optimization (BBPSO): A velocity-free PSO variant that simplifies the search for an optimal feature subset [75].
Studies have shown that such hybrid approaches (e.g., TMGWO followed by a Support Vector Machine classifier) can achieve high accuracy (e.g., 96%) using only a very small number of features, thereby maximizing generalization [75].
Comparison of Dimensionality Reduction Methods
PCA is one of several dimensionality reduction techniques available to bioinformaticians. The choice of method depends on the data characteristics and research question.
Table 2: Comparison of Dimensionality Reduction Techniques for Biological Data
| Method | Input Data | Key Strength | Key Weakness | Ideal Use Case |
|---|---|---|---|---|
| PCA [18] [27] | Original feature matrix | Computationally efficient; excellent for linear data. | Assumes linearity; difficult interpretation. | Initial exploratory analysis, visualization of linear structures. |
| PCoA [18] | Distance matrix (e.g., Bray-Curtis) | Flexible with any distance metric; good for ecology. | Computationally heavy for large datasets. | Beta-diversity analysis in microbiome studies. |
| NMDS [18] | Distance matrix | Suited for non-linear data; preserves rank-order. | Results can vary; requires iterative refinement. | Analyzing complex datasets where only relative sample order matters. |
Experimental Validation and the Scientist's Toolkit
Validating Stability and Generalizability
Applying PCA or any other complexity-reduction technique does not automatically guarantee a robust model. It is imperative to validate the stability of the feature set and the generalizability of the model using rigorous experimental protocols. A critical practice is cross-validation. In this process, the dataset is repeatedly split into training and testing sets. The entire analytical pipeline—including feature selection (or PCA) and model training—is applied only to the training set. The final model is then evaluated on the held-out testing set to estimate its performance on unseen data [72]. A significant performance drop from training to testing is a classic indicator of overfitting.
Furthermore, the stability of feature importance rankings should be assessed. In the context of PCA, this could involve checking the consistency of gene loadings on principal components across different data subsamples. High sensitivity to minor changes in the dataset is a sign of instability, often linked to overfitting [71]. Finally, the gold standard for validation is testing the model on a completely independent, external dataset [72]. This confirms that the findings are not artifacts of a specific dataset but represent a true biological signal.
The following table details key software tools and resources essential for implementing the methodologies discussed in this guide.
Table 3: Research Reagent Solutions for Genomic Analysis
| Tool/Resource | Function | Application Context |
|---|---|---|
| Scikit-learn [77] | A comprehensive machine learning library for Python. | Provides implementations for PCA, model training, and cross-validation. |
| R Statistical Environment [27] | A language and environment for statistical computing and graphics. | Offers extensive packages for PCA (prcomp) and advanced statistical analysis. |
| TensorFlow/PyTorch [72] | Open-source libraries for deep learning. | Used for building complex models and implementing regularization like Dropout. |
| Affymetrix Microarrays [16] | A high-throughput technology for gene expression profiling. | A common data source for generating the gene expression matrices analyzed by PCA. |
| Synthetic Data Generation [72] | A technique to artificially create new data points. | Used for data augmentation to improve model robustness and for method validation. |
Diagram 2: Multi-faceted strategy for mitigating overfitting.
In the challenging landscape of high-dimensional genomic research, overfitting presents a significant threat to the validity and translational potential of scientific findings. Principal Component Analysis stands as a foundational and powerful technique to mitigate this risk by reducing dimensionality, filtering noise, and revealing robust underlying biological structures. However, as we have explored, a modern approach should not rely on PCA alone. The integration of advanced PCA variants, hybrid feature selection frameworks, and rigorous validation protocols creates a comprehensive defense strategy. By systematically applying these principles, researchers and drug development professionals can enhance the stability and generalizability of their models, ensuring that their discoveries are not merely artifacts of a dataset but represent true, actionable biological insights that can reliably inform future research and clinical application.
Handling Technical Noise, Sparsity, and Data Heteroscedasticity
Gene expression datasets, derived from technologies like RNA-Seq and single-cell RNA-Seq (scRNA-Seq), typically measure tens of thousands of genes across multiple samples, creating a high-dimensional space where the number of features (genes) vastly exceeds the number of observations (samples). Principal Component Analysis (PCA) serves as a fundamental dimensionality reduction technique within this context, transforming correlated gene expressions into a smaller set of uncorrelated principal components (PCs) that capture the predominant patterns of variation [78] [28]. The first PC specifies the direction of the largest variability in the data, with subsequent components capturing the next highest orthogonal variances [78]. This linear transformation allows researchers to project high-dimensional data into a two or three-dimensional space for visualization, quality control, and identification of sample clusters or batch effects [28].
However, the application of PCA to gene expression data presents significant challenges related to technical noise, data sparsity, and heteroscedasticity. Technical noise arises from variations in experimental conditions, such as library preparation and sequencing depth, which can confound biological signals [79]. Data sparsity is particularly pronounced in scRNA-Seq data, where >90% of entries may be zero counts due to both biological and technical factors [79]. Heteroscedasticity—the non-constant variability across different expression levels—is inherent to count-based sequencing data, where variance typically increases with mean expression. These characteristics violate key assumptions of standard PCA, which operates most effectively on continuous, normally-distributed data with constant variance [79] [27]. This technical guide examines specialized PCA methodologies and preprocessing techniques that address these challenges within gene expression research, providing practical frameworks for maintaining analytical rigor while extracting biologically meaningful patterns from complex transcriptomic data.
Mathematical Foundations of PCA in Gene Expression Context
Core Algorithm and Geometric Interpretation
PCA functions by identifying the principal directions of maximum variance in high-dimensional data through eigen decomposition of the covariance matrix. Given a gene expression matrix X with p genes (columns) and n samples (rows), where each column has mean zero, the sample variance-covariance matrix is computed as Σ = (XᵀX)/(n-1). The principal components are obtained by solving the eigen equation Σvᵢ = λᵢvᵢ, where λᵢ represents the eigenvalue (magnitude of variance) and vᵢ represents the eigenvector (direction) for the i-th principal component [27]. The resulting PCs are ordered by decreasing variance explained (λ₁ ≥ λ₂ ≥ ... ≥ λₚ), with each PC representing a normalized linear combination of the original genes: Zᵢ = Φ₁ᵢX₁ + Φ₂ᵢX₂ + ... + ΦₚᵢXₚ, where Φᵢ is the loading vector constrained to a sum of squares equal to 1 [42].
Geometrically, PCA performs a rotation of the coordinate system to align with the directions of maximal variance. The first PC (Z¹) defines the line closest to the data in terms of average squared Euclidean distance, with each subsequent component capturing the remaining orthogonal variance [42]. This transformation allows the majority of information contained in the original p-dimensional gene expression space to be represented in a much lower-dimensional subspace defined by the first k principal components, where k << p [27]. The effectiveness of this low-dimensional representation depends critically on the correlation structure among genes, with higher correlation enabling greater dimensionality reduction without substantial information loss [80].
Low-Dimensionality as a Biological Property Enabling PCA
The theoretical foundation for PCA's effectiveness in gene expression analysis rests on the biological property of low-dimensionality inherent in transcriptional systems. While genomes contain thousands of genes, they operate in coordinated transcriptional modules due to co-regulation, meaning the effective dimensionality is determined by the number of regulatory programs rather than the number of genes [80]. This co-regulation produces covariation in gene expression patterns, constraining the data to lie on a low-dimensional manifold within the high-dimensional measurement space.
Perturbation theory analyses demonstrate that the stability of principal components against noise depends on the spectral properties of the covariance matrix. The error in estimating the i-th principal component under measurement noise can be approximated by ‖pcᵢ - pĉᵢ‖ ≈ √[∑_{j≠i}((pcᵢᵀ(Ĉ - C)pcⱼ)/(λᵢ - λⱼ))²], where C and Ĉ are the true and noisy covariance matrices, and λᵢ are the eigenvalues [80]. This equation reveals that principal component stability increases with larger gaps between consecutive eigenvalues (λᵢ - λⱼ), which occurs when variance is concentrated in fewer dominant components. This low-dimensional structure explains why transcriptional programs can be accurately identified even with substantial technical noise and shallow sequencing depths [80].
Handling Technical Noise and Batch Effects
Normalization Strategies Prior to PCA
Technical noise in gene expression data arises from multiple sources, including variation in RNA capture efficiency, library preparation protocols, and sequencing depth. The standard PCA pipeline for RNA-Seq incorporates specific normalization steps to address these technical artifacts before dimensionality reduction:
Log-CPM Transformation: Counts per Million (CPM) values are calculated using effective library sizes as determined by TMM (Trimmed Mean of M-values) normalization, followed by log₂ transformation to stabilize variance [78]. This approach adjusts for differences in sequencing depth across samples.
Z-score Normalization: After log-CPM transformation, each gene is mean-centered and scaled to unit variance across samples [78] [81]. This ensures that genes with different expression levels contribute more equally to the principal components, preventing highly expressed genes from dominating the variance structure simply due to their measurement scale.
Gene Filtering: Genes with zero expression across all samples or containing invalid values (NaN or +/- Infinity) are removed prior to PCA, as they contribute no meaningful biological information [78].
For single-cell RNA-Seq data with Unique Molecular Identifiers (UMIs), the multinomial distribution provides a more appropriate model for the count data, avoiding the need for zero-inflated models required for read counts [79]. The standard practice of adding a pseudocount before log transformation can introduce subtle biases, as the choice of pseudocount is arbitrary and can artificially influence variance estimates [79].
Advanced Dimensionality Reduction Methods for Noise Removal
Beyond standard normalization, specialized dimensionality reduction techniques can explicitly separate technical noise from biological signal:
Robust PCA (RPCA): This variant decomposes the gene expression matrix into a low-rank component (representing biological signal) and a sparse component (representing noise and outliers). In benchmarking studies on CRISPR screen data from the Cancer Dependency Map, RPCA combined with "onion" normalization outperformed classical PCA for enhancing functional gene networks by effectively removing dominant mitochondrial-associated signals that masked more subtle cancer-specific dependencies [82].
Autoencoder Networks: These neural network-based approaches learn nonlinear mappings to compress and reconstruct gene expression data, potentially capturing more complex patterns of biological variation than linear methods. In comparative analyses, autoencoders most efficiently captured and removed mitochondrial-associated technical bias from functional genomics data [82].
Table 1: Comparison of Dimensionality Reduction Methods for Handling Technical Noise
| Method | Mechanism | Advantages | Limitations | Best-Suited Applications |
|---|---|---|---|---|
| Classical PCA | Linear transformation to orthogonal components | Computationally efficient, interpretable components | Assumes linear relationships, sensitive to outliers | Initial data exploration, quality assessment |
| Robust PCA | Matrix decomposition into low-rank + sparse components | Resistant to outliers, separates signal from noise | Higher computational complexity | Functional network analysis, data with technical artifacts [82] |
| Autoencoders | Neural network with bottleneck layer | Captures non-linear relationships, flexible architecture | Black-box nature, requires extensive tuning | Large datasets with complex latent structure [82] |
| GLM-PCA | Generalized linear model framework | Directly models count distribution, no need for normalization | Computationally intensive for large datasets | scRNA-Seq with UMI counts [79] |
Addressing Data Sparsity in Single-Cell RNA-Seq
The Multinomial Model for UMI Counts
Single-cell RNA-Seq data with Unique Molecular Identifiers (UMIs) presents exceptional challenges due to the extreme sparsity of the resulting count matrices. Analytical frameworks based on read counts from bulk RNA-Seq often assume zero-inflated distributions, but empirical evidence demonstrates that UMI counts follow multinomial sampling distributions without zero inflation [79]. The apparent excess zeros in read counts result from PCR duplicates rather than true biological absence, which UMIs effectively correct.
The multinomial model provides a statistically rigorous foundation for analyzing scRNA-Seq UMI counts. Consider a cell i containing tᵢ total mRNA transcripts, from which nᵢ UMIs are successfully sequenced (nᵢ << tᵢ). The probability of observing a specific count vector xᵢ = (xᵢ₁, ..., xᵢₚ) for *p* genes follows a multinomial distribution: xᵢ ~ Multinomial(nᵢ, πᵢ), where πᵢ = (πᵢ₁, ..., πᵢₚ) represents the true relative expression proportions [79]. This model properly accounts for the compositionality of UMI data and the dependency between gene counts within each cell.
Generalized PCA for Sparse Count Data
Standard PCA applied to normalized scRNA-Seq data can produce distorted low-dimensional embeddings, frequently where the first principal component primarily captures the fraction of zeros in each cell rather than biological variation [79]. To address this limitation, generalized PCA (GLM-PCA) extends the PCA framework to exponential family likelihoods, enabling direct operation on raw UMI counts without problematic normalization steps:
GLM-PCA Algorithm: This approach replaces the Gaussian likelihood assumption of standard PCA with a binomial or multinomial likelihood, better suited for count data. The model estimates parameters that maximize the likelihood of the observed counts under a low-rank constraint [79].
Deviance and Pearson Residuals: As a computationally efficient approximation to GLM-PCA, deviance residuals or Pearson residuals from a multinomial model can be calculated and used as input to standard PCA. These residuals effectively normalize the count data while preserving the statistical properties of the multinomial distribution [79].
Benchmarking evaluations using ground truth datasets demonstrate that GLM-PCA and PCA on deviance residuals outperform standard PCA applied to log-normalized counts in downstream clustering accuracy, particularly for recovering known cell type identities [79].
Managing Heteroscedasticity and Variance Structure
Variance-Stabilizing Transformations
Heteroscedasticity in gene expression data refers to the non-constant variability across different expression levels, where highly expressed genes typically exhibit greater absolute variance than lowly expressed genes. This property violates the homoscedasticity assumption of standard PCA, which treats all variables as having equal statistical weight. To address this, several variance-stabilizing approaches are employed:
Logarithmic Transformation: The most common approach applies log(1 + x) transformation to counts or normalized expressions, which approximately stabilizes variance across the dynamic range of expression [78] [81]. However, the choice of pseudocount is arbitrary and can influence results [79].
Feature Selection via Highly Variable Genes: Prior to PCA, selection of genes with high cell-to-cell variation enriches for biologically informative features. The Seurat workflow identifies 2,000-3,000 highly variable genes based on a variance-to-mean relationship, focusing the PCA on genes that contribute most to meaningful sample separation [81].
Z-score Standardization: Scaling each gene to have mean zero and unit variance across samples (Z-score normalization) ensures that genes with different expression levels contribute more equally to the principal components [81] [42]. This is particularly important when genes are measured on different scales or have intrinsically different variances.
Systematic Benchmarking of Dimensionality Reduction Performance
Comparative evaluations of dimensionality reduction methods provide guidance for selecting appropriate techniques based on data characteristics and analytical goals. A comprehensive benchmarking study evaluated 30 dimensionality reduction methods across four experimental conditions using the Connectivity Map (CMap) drug-induced transcriptome dataset [83]. The study assessed performance using internal cluster validation metrics (Davies-Bouldin Index, Silhouette score, Variance Ratio Criterion) and external validation metrics (Normalized Mutual Information, Adjusted Rand Index) comparing clusters to known biological labels [83].
Table 2: Performance of Dimensionality Reduction Methods in Transcriptomic Applications
| Method Category | Representative Methods | Preservation of Local Structure | Preservation of Global Structure | Detection of Dose-Dependent Responses | Computational Efficiency |
|---|---|---|---|---|---|
| Linear Methods | PCA, GLM-PCA | Moderate | Strong | Weak | High |
| Neighbor-Graph Methods | UMAP, t-SNE | Strong | Moderate | Moderate | Moderate |
| Distance-Based Methods | PaCMAP, TRIMAP | Strong | Strong | Moderate | Moderate |
| Diffusion Methods | PHATE | Moderate | Strong | Strong | Moderate |
The benchmarking results indicated that methods including t-SNE, UMAP, PaCMAP, and TRIMAP outperformed others in preserving both local and global biological structures, particularly in separating distinct drug responses and grouping compounds with similar molecular targets [83]. However, for detecting subtle dose-dependent transcriptomic changes, Spectral, PHATE, and t-SNE showed stronger performance. Notably, PCA performed relatively poorly in preserving biological similarity compared to these more modern techniques, highlighting its limitations for certain analytical tasks [83].
Experimental Protocols and Workflows
Standardized PCA Protocol for Bulk RNA-Seq Data
For bulk RNA-Seq analysis, the following workflow provides a robust approach for dimensionality reduction:
Data Preprocessing:
- Calculate log-CPM values using TMM-normalized effective library sizes
- Filter out genes with zero expression across all samples
- Perform Z-score normalization for each gene across samples [78]
PCA Implementation:
- Compute principal components using singular value decomposition (SVD)
- Retain top k components explaining >80% cumulative variance [84]
- Export component coordinates for downstream analysis
Visualization and Interpretation:
Specialized Protocol for Single-Cell RNA-Seq Data
For scRNA-Seq with UMI counts, this modified protocol addresses sparsity:
Count Modeling:
- Start with raw UMI counts without logarithmic transformation
- Calculate deviance or Pearson residuals from a multinomial model [79]
- Alternatively, apply GLM-PCA directly to raw counts
Feature Selection:
- Identify highly variable genes using deviance-based statistics [79]
- Select 1,000-5,000 features showing significant biological variation
Dimensionality Reduction:
- Perform PCA on residuals or selected features
- Determine optimal number of components using elbow method [81]
- Use PCs as input to downstream clustering and trajectory analysis
Diagram Title: scRNA-Seq PCA Workflow Addressing Sparsity
Table 3: Research Reagent Solutions for PCA in Gene Expression Studies
| Resource Category | Specific Tools/Platforms | Primary Function | Application Context |
|---|---|---|---|
| Computational Frameworks | Seurat [81], Scater [81], FactoMineR [84] | Integrated pipelines for normalization, PCA, and visualization | Bulk and single-cell RNA-Seq analysis |
| Normalization Methods | TMM [78], CPM [78], Multinomial Residuals [79] | Adjust for technical variation in sequencing depth | Study-specific based on data type and sparsity |
| PCA Implementations | GLM-PCA [79], Robust PCA [82], prcomp [84] | Dimensionality reduction adapted to data characteristics | Choice depends on noise structure and data distribution |
| Benchmarking Frameworks | FLEX [82] | Evaluate functional information in reduced dimensions | Method selection and validation |
| Visualization Packages | factoextra [84], ggplot2 [81] | Create publication-quality PCA plots | Result communication and exploratory analysis |
The effective application of Principal Component Analysis to gene expression data requires careful consideration of technical noise, sparsity, and heteroscedasticity inherent in transcriptomic measurements. While standard PCA with appropriate normalization provides a solid foundation for exploratory analysis, specialized approaches including GLM-PCA for sparse UMI counts and Robust PCA for noise removal offer enhanced performance for specific data types and research questions. The low-dimensionality intrinsic to gene expression data, resulting from coordinated transcriptional regulation, enables these methods to extract biologically meaningful patterns even in the presence of substantial technical artifacts. By selecting dimensionality reduction techniques matched to their experimental systems and analytical goals, researchers can leverage PCA and its extensions to illuminate transcriptional programs underlying fundamental biological processes and therapeutic responses.
Principal Component Analysis (PCA) serves as a foundational technique for dimensionality reduction in gene expression studies, transforming high-dimensional genomic data into a lower-dimensional space while preserving critical biological information. This process identifies orthogonal principal components (PCs) that capture maximum variance in the dataset, with the first PC representing the direction of highest variance, followed by subsequent components [61]. In genomic research, where datasets often contain thousands of genes across limited samples, PCA enables researchers to visualize sample relationships, identify patterns, and reduce computational complexity before applying downstream classification or clustering algorithms [16] [85].
The effectiveness of PCA and subsequent analytical procedures depends heavily on parameter optimization across three critical domains: k-nearest neighbor (kNN) thresholds for classification, spatial smoothing for transcriptomic data, and penalty terms for variable selection. Each domain presents unique challenges in balancing model complexity with biological fidelity. This technical guide examines current methodologies for optimizing these parameters within the context of gene expression analysis, providing structured protocols and benchmarking data to inform research design and implementation in genomic studies and drug development.
Principal Component Analysis in Gene Expression Research
Fundamental Principles and Workflow
PCA operates through a standardized mathematical procedure that transforms original variables into new orthogonal variables called principal components. The algorithm requires standardized data with a mean of zero and standard deviation of one to ensure all variables contribute equally regardless of their original scale [61]. The computational workflow involves:
- Standardization: Transforming the dataset to have zero mean and unit variance
- Covariance Matrix Calculation: Capturing relationships between all variables
- Eigenvalue Decomposition: Identifying eigenvalues and eigenvectors of the covariance matrix
- Component Selection: Choosing top components based on highest eigenvalues
- Data Projection: Transforming original data into the new principal component space
Biological Applications and Limitations
In gene expression analysis, PCA enables visualization of sample relationships, identification of batch effects, and detection of overarching biological patterns. Studies applying PCA to large-scale gene expression datasets have revealed that the first three principal components often separate major biological categories such as hematopoietic cells, neural tissues, and cell lines [16]. However, the apparent low intrinsic dimensionality of gene expression data – where early components explain approximately 36% of variability – masks significant biological information contained in higher-order components, particularly tissue-specific expression patterns [16].
PCA exhibits several limitations in genomic applications. As a linear method, it struggles to capture complex nonlinear relationships in gene expression [86]. The technique is sensitive to outliers and requires careful normalization to prevent technical artifacts from dominating biological signals [86]. Perhaps most significantly, principal components represent linear combinations of all original genes, complicating biological interpretation and identification of specific marker genes [16].
Optimizing k-Nearest Neighbor Parameters for Genomic Classification
Critical kNN Parameters and Performance Impact
The k-nearest neighbor algorithm represents a simple yet powerful classification approach that assigns labels to unknown samples based on the majority vote of its k most similar training examples [87]. Its application to genomic data requires optimization of multiple parameters that significantly impact performance:
- Feature Ranking Method: Selection of biologically relevant genes
- Number of Features: Optimal feature subset size balancing information and noise
- Distance Metric: Measurement of sample similarity (Euclidean, Manhattan, cosine)
- Number of Neighbors (k): Balance between overfitting and oversmoothing
- Vote Weighting: Uniform versus distance-weighted voting
- Decision Threshold: Classification cutoff for binary endpoints
The MAQC-II project demonstrated that systematic optimization of these parameters across 463,320 models generated substantial performance variations in cancer classification, underscoring the importance of methodical parameter selection [87].
Protocol for kNN Optimization in Gene Expression Classification
Experimental Protocol: kNN Parameter Grid Search
Data Preparation
- Partition gene expression data into training (70%), validation (15%), and test (15%) sets
- Standardize data to zero mean and unit variance
- Perform initial feature filtering based on variance or significance
Parameter Space Definition
- k values: 1, 3, 5, 7, 10, 15
- Distance metrics: Euclidean, Manhattan, cosine, correlation
- Feature ranking methods: t-test, fold change, mutual information
- Feature numbers: 50, 100, 200, 500, 1000
- Weighting schemes: uniform, distance-weighted
Cross-Validation Optimization
- Perform 5-fold cross-validation on training set for each parameter combination
- Evaluate using balanced accuracy for imbalanced datasets
- Select top 10 parameter sets based on validation performance
Final Model Evaluation
- Apply best parameters to independent test set
- Compare performance with clinical standards or existing classifiers
- Validate biological interpretability of selected features
Table 1: kNN Performance Variation Across Parameter Combinations (MAQC-II Project)
| Parameter | Values Tested | Performance Range (AUC) | Optimal Setting |
|---|---|---|---|
| Number of Neighbors (k) | 1-15 | 0.65-0.89 | 3-7 |
| Distance Metric | Euclidean, Manhattan, Cosine | 0.71-0.86 | Cosine |
| Feature Number | 50-1000 genes | 0.62-0.91 | 100-500 |
| Feature Ranking | t-test, fold change, Wilcoxon | 0.69-0.88 | t-test |
| Vote Weighting | Uniform, distance-based | 0.75-0.83 | Distance-based |
Spatial Smoothing Techniques for Transcriptomic Data
Spatial Dimension Reduction Methods
Spatial transcriptomics technologies measure gene expression while preserving spatial localization information, generating datasets where neighboring locations exhibit inherent correlation [56]. This spatial dependency structure enables specialized dimension reduction approaches that incorporate spatial coordinates alongside expression values:
- SpatialPCA: Extends probabilistic PCA by modeling spatial correlation through kernel matrices, preserving spatial relationships in low-dimensional components [56]
- RASP (Randomized Spatial PCA): Employs randomized two-stage PCA with configurable spatial smoothing via kNN thresholding for computational efficiency [48]
- BayesSpace: Uses explicit spatial arrangement modeling for domain detection with resolution enhancement [56]
- SpaGCN: Applies graph convolutional networks with spatial adjacency matrices [56]
These spatially-aware methods outperform conventional PCA in spatial domain detection accuracy by incorporating tissue organization patterns into the dimension reduction process [56].
Spatial Smoothing Parameter Optimization
Experimental Protocol: Spatial Parameter Optimization
Spatial Neighborhood Definition
- Calculate pairwise distances between spatial locations
- Construct k-nearest neighbor graph (k typically 4-10 for grid-based data)
- Define spatial kernel (Gaussian, exponential, uniform)
Smoothing Intensity Calibration
- Test bandwidth parameters for spatial kernel (β = 1, 2, 3)
- Evaluate kNN thresholds for graph sparsification (k = 5, 10, 15, 20)
- Assess smoothing effect on cluster coherence and separation
Validation Metrics
- Spatial autocorrelation (Moran's I)
- Cluster accuracy against ground truth (Adjusted Rand Index)
- Spatial continuity (CHAOS score)
Table 2: Benchmarking Spatial Dimension Reduction Methods
| Method | ARI Score | Runtime | Spatial Preservation | Key Parameters |
|---|---|---|---|---|
| SpatialPCA | 0.68-0.94 | Moderate | High | Kernel bandwidth, spatial coordinates |
| RASP | 0.65-0.89 | Fast | Medium-High | kNN threshold, β power |
| BayesSpace | 0.37-0.77 | Slow | High | Spatial neighborhood, clustering resolution |
| PCA | 0.23-0.58 | Fast | Low | Number of components |
| SpaGCN | 0.28-0.63 | Moderate | Medium | Adjacency weight, convolution layers |
Penalty Function Selection for High-Dimensional Genomic Data
Penalty Functions for Variable Selection
High-dimensional genomic datasets, particularly those from genome-wide association studies (GWAS), present significant variable selection challenges where the number of covariates (SNPs) far exceeds sample sizes. Penalized regression approaches address this through various penalty functions that perform simultaneous variable selection and effect estimation [88]:
- Lasso (L1): Provides sparse solutions but requires irrepresentable conditions for consistency
- SCAD (Smoothly Clipped Absolute Deviation): Reduces bias in large coefficients while maintaining sparsity
- MCP (Minimax Concave Penalty): Offers unbiased estimation for large coefficients with convexity properties
- SICA (Smooth Integration of Counting and Absolute deviation): Bridges L0 and L1 penalties with less restrictive dimensionality requirements
- Log Penalty: Functions as a folded-concave penalty with superior high-dimensional performance
Theoretical analyses indicate that penalty functions bridging L0 and L1 penalties (SICA, Log) require less restrictive conditions on dimensionality and minimum effect sizes to achieve optimal variable selection and unbiased estimation [88].
Protocol for Penalty Function Optimization
Experimental Protocol: Penalized Regression for Genomic Data
Preprocessing and Screening
- Perform quality control on genomic variants
- Apply sure independence screening to reduce feature space
- Standardize genotypes and phenotypes
Penalty Function Comparison
- Implement multiple penalty functions (Lasso, SCAD, MCP, SICA, Log)
- Use coordinate descent algorithms for efficient optimization
- Employ extended Bayesian Information Criterion (eBIC) for model selection
Tuning Parameter Selection
- Perform k-fold cross-validation for regularization parameters
- For two-parameter penalties (SCAD, MCP), optimize both parameters jointly
- Select parameters minimizing prediction error or maximizing selection accuracy
Validation and Interpretation
- Evaluate selected models on independent validation sets
- Assess biological interpretability of selected features
- Compare with known biological pathways and mechanisms
Table 3: Comparison of Penalty Functions in High-Dimensional Genomic Studies
| Penalty Function | Variable Selection Consistency | Unbiased Estimation | Dimensionality Tolerance | Tuning Parameters |
|---|---|---|---|---|
| Lasso (L1) | Requires irrepresentable condition | Biased for large coefficients | Low | 1 |
| SCAD | Yes | Asymptotically unbiased | Medium | 2 |
| MCP | Yes | Unbiased for large coefficients | Medium | 2 |
| SICA | Yes | Near unbiased | High | 2 |
| Log | Yes | Near unbiased | High | 2 |
Integrated Workflow for Genomic Data Analysis
Comprehensive Analytical Pipeline
A robust genomic analysis pipeline integrates dimensionality reduction, spatial smoothing where applicable, and regularized classification to maximize biological insight while maintaining statistical rigor. The sequential workflow includes:
Quality Control and Normalization
- Remove low-quality samples and features
- Apply appropriate normalization for technology platform
- Address batch effects and technical covariates
Initial Dimension Reduction
- Apply PCA to visualize overall data structure
- Identify potential outliers and technical artifacts
- Determine intrinsic data dimensionality
Specialized Analysis Phase
- For spatial data: implement spatial dimension reduction (SpatialPCA, RASP)
- For high-dimensional features: apply penalty function selection
- For classification: optimize kNN parameters
Biological Validation
- Assess enrichment of biological pathways
- Validate findings in independent datasets
- Correlate with clinical outcomes where available
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Computational Tools for Dimensionality Reduction Optimization
| Tool/Resource | Function | Application Context |
|---|---|---|
| Seurat | PCA and clustering | Single-cell and spatial transcriptomics |
| Scanpy | PCA and visualization | Single-cell RNA sequencing analysis |
| SpatialPCA R Package | Spatially-aware dimension reduction | Spatial transcriptomics |
| RASP Algorithm | Randomized spatial PCA | Large-scale spatial datasets |
| MAQC-II Protocols | kNN optimization framework | Genomic classifier development |
| Boosting Autoencoder (BAE) | Interpretable dimension reduction | Marker gene identification |
Optimizing parameters for kNN thresholds, spatial smoothing, and penalty terms represents a critical frontier in extracting meaningful biological signals from high-dimensional genomic data. Methodical optimization of kNN parameters significantly enhances classification accuracy in gene expression studies, with systematic evaluation revealing optimal configurations typically employing 3-7 neighbors, cosine distance metrics, and 100-500 carefully selected features [87]. Spatial smoothing techniques, particularly SpatialPCA and RASP, demonstrate that incorporating spatial correlation structures improves domain detection accuracy in transcriptomic data while maintaining computational feasibility [48] [56]. Penalty function selection substantially impacts variable selection efficacy in high-dimensional genomic applications, with bridge penalties like SICA and Log offering superior performance under less restrictive conditions [88].
Future methodological development should focus on integrated approaches that simultaneously optimize across multiple parameter domains, leverage biological constraints to guide statistical learning, and enhance computational efficiency for increasingly large-scale genomic datasets. As spatial technologies advance toward single-cell resolution and genomic studies continue expanding in sample size, refined parameter optimization strategies will remain essential for translating high-dimensional molecular measurements into clinically actionable insights.
Addressing Scalability for Datasets with Millions of Cells
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the profiling of gene expression at the level of individual cells. However, this technological advancement has introduced significant computational challenges, particularly as datasets now routinely encompass millions of cells [89] [19]. The high-dimensional nature of scRNA-seq data, where each cell is represented by expressions of thousands of genes, necessitates robust dimensionality reduction techniques before any meaningful downstream analysis can be performed. Principal Component Analysis (PCA) serves as a critical first step in this pipeline, transforming the sparse, high-dimensional count matrix into a lower-dimensional representation that captures the dominant sources of biological variation [19]. Within the broader thesis of how PCA reduces dimensionality in gene expression research, this technical guide addresses the specific computational constraints and methodological innovations required to apply PCA to datasets with millions of cells, ensuring analyses remain computationally tractable without sacrificing biological signal.
The core challenge stems from the immense scale of modern scRNA-seq data. A dataset with one million cells and 20,000 genes produces a matrix with 20 billion entries, creating substantial memory and processing demands for standard statistical software [90]. Furthermore, scRNA-seq data are characterized by extreme sparsity, with a large proportion of zero counts due to both biological and technical factors, including the pervasive "dropout" phenomenon where expressed genes fail to be detected [91] [89]. This guide provides researchers, scientists, and drug development professionals with the technical foundation and practical protocols to implement scalable PCA, thereby unlocking the biological insights contained within these massive datasets.
Computational Foundations of Scalable PCA
The Mathematics of PCA in High Dimensions
Principal Component Analysis is an orthogonal linear transformation that projects high-dimensional data into a new coordinate system defined by principal components (PCs). These PCs are ordered such that the first component captures the maximum variance in the data, with each subsequent component capturing the remaining variance in decreasing order [92] [93]. Mathematically, for a data matrix X with n cells (columns) and p genes (rows), PCA can be represented as:
[ X = TP^T + E ]
where T is the score matrix (the transformed data in the PC space), P is the loading matrix (the principal components), and E is the residual matrix [92]. The principal components are the eigenvectors of the covariance matrix of X, and the eigenvalues represent the amount of variance explained by each component [93].
In the context of large-scale scRNA-seq data, the traditional approach of computing the full covariance matrix becomes computationally prohibitive. Instead, scalable implementations leverage the connection between PCA and Singular Value Decomposition (SVD). For a centered data matrix X, the SVD is given by X = UΣV^T, where the columns of V are the PCs (loadings), and the columns of UΣ are the scores [90]. This formulation allows for the use of iterative, approximate algorithms that can efficiently compute only the top k components, bypassing the need for the full, computationally expensive decomposition.
Critical Data Preprocessing Steps
Proper data preprocessing is a prerequisite for successful and scalable PCA. The following steps are essential:
Normalization: The raw UMI counts for each cell are normalized to account for differences in sequencing depth. A common approach is to normalize to counts per million (CPM) followed by a log transformation (
log1p)```r
R code for normalization
countstocpm <- function(A, norm = median(Matrix::colSums(A))) { A@x <- A@x / rep.int(Matrix::colSums(A), diff(A@p)) A@x <- norm * A@x return(A) } logcpm <- log1p(countstocpm(counts_matrix))
- Standardization: Prior to PCA, the data is typically standardized such that each gene has a mean of zero and a unit variance. This prevents genes with high expression levels from dominating the variance calculation [93]. However, for very large-scale data, the scaling step can be memory-intensive and is sometimes omitted when using regularized or model-based approaches [89].
Scalable PCA Algorithms and Implementations
Benchmarking PCA Algorithms for Runtime and Memory Efficiency
The standard prcomp function in R becomes infeasible for datasets with hundreds of thousands to millions of cells. Benchmarking studies have compared several specialized implementations for their performance on large scRNA-seq data [90]. The table below summarizes the key characteristics and performance metrics of these algorithms.
Table 1: Benchmark of Scalable PCA Implementations for scRNA-seq Data
| Function / Package | Core Algorithm | Key Feature | Best Suited For | Relative Speed (vs. prcomp) |
|---|---|---|---|---|
stats::prcomp |
Full SVD | Base R implementation | Small datasets (<10k cells) | 1.0x (baseline) |
irlba::prcomp_irlba |
IRLBA | Fast partial SVD | Medium-large datasets | ~5-10x faster |
RSpectra::svds |
Krylov subspace methods | High accuracy for top components | Large datasets where accuracy is critical | ~5-10x faster |
rsvd::rpca |
Randomized SVD | Extreme speed, configurable precision | Very large datasets (millions of cells) | ~10-50x faster |
scGBM |
Poisson bilinear model | Models count data directly; quantifies uncertainty | Data where technical noise is a major concern | Varies (model-based) |
The performance gains from algorithms like irlba and rsvd are achieved through iterative methods that compute only a pre-specified number of leading principal components, avoiding the computational cost of calculating the full decomposition [90]. The scGBM method represents a different, model-based approach that fits a Poisson bilinear model directly to the count matrix, addressing the limitations of standard PCA on sparse, discrete data [89].
A Practical Workflow for Large-Scale PCA
The following workflow outlines the steps for performing PCA on a dataset of millions of cells, integrating the algorithms and preprocessing steps discussed.
Figure 1: A Scalable PCA Workflow for Large scRNA-seq Datasets. This workflow outlines the key stages of data preprocessing, efficient PCA computation, and result validation.
Detailed Protocol:
Data Preprocessing:
- Input: A sparse count matrix (e.g., a
dgCMatrixobject in R). - Normalization: Normalize the counts using the median total count per cell as a scaling factor, then apply the
log1ptransformation [90]. This stabilizes variance and makes the data more amenable to linear methods. - Feature Selection: Identify Highly Variable Genes (HVGs). A robust method involves fitting a loess curve to model the relationship between the log10(mean) and log10(standard deviation) of each gene's expression and selecting genes with the highest positive residuals [90]. This typically retains 10-20% of all genes, drastically reducing the problem's dimensionality.
- Input: A sparse count matrix (e.g., a
PCA Computation with a Scalable Algorithm:
- Algorithm Selection: For datasets exceeding 50,000 cells, prefer
rsvd::rpcaorirlba::prcomp_irlbaover the baseprcompfunction [90]. - Code Example (R):
- Parameter Tuning: The
p(oversampling) andq(power iterations) parameters inrsvdcontrol the trade-off between speed and accuracy. Increasing them improves accuracy at a computational cost [90].
- Algorithm Selection: For datasets exceeding 50,000 cells, prefer
Result Validation and Component Selection:
- Determine the optimal number of components (k): Use a combination of the scree plot (looking for the "elbow") and the percent cumulative variance. A common practice is to retain enough PCs to explain 80-90% of the total variance, though this can be subjective [94].
- Output: The primary outputs are the PC scores (a
n_cells x kmatrix representing cells in the low-dimensional space) and the PC loadings (an_genes x kmatrix showing the contribution of each gene to each PC). These scores are used for downstream analyses like clustering and visualization.
Advanced Methodologies and Alternative Approaches
Model-Based Dimensionality Reduction
While standard PCA on transformed data is widely used, it can induce spurious heterogeneity in scRNA-seq data. Model-based approaches directly model the count nature of the data, which can be more robust. The scGBM (single-cell Generalized Bilinear Model) method, for instance, uses a Poisson bilinear model:
[ \text{Count}{ij} \sim \text{Poisson}(\exp(\mui + \betaj + \langle Ui, V_j \rangle)) ]
where (Ui) and (Vj) are low-dimensional latent representations of cells and genes, respectively [89]. This approach avoids the pitfalls of transformations like log(1+x) and naturally handles the mean-variance relationship of count data. Furthermore, scGBM introduces a fast estimation algorithm based on iteratively reweighted singular value decompositions, allowing it to scale to millions of cells while also quantifying uncertainty in each cell's latent position [89].
Compositional Data Analysis (CoDA) Framework
Another emerging approach recognizes that scRNA-seq data is inherently compositional—the counts for genes in a cell are relative, summing to a total count per cell. The Compositional Data Analysis (CoDA) framework applies log-ratio transformations to the raw counts before analysis. The Centered-Log-Ratio (CLR) transformation is defined as:
[ \text{CLR}(x) = \left[ \log\left(\frac{x1}{g(x)}\right), \dots, \log\left(\frac{xp}{g(x)}\right) \right] ]
where (g(x)) is the geometric mean of the count vector for a cell [91]. Applying PCA to CLR-transformed data has shown advantages in producing more distinct cell clusters and improving trajectory inference by mitigating the impact of dropout events [91]. Specialized count addition schemes (e.g., the SGM method) have been developed to handle the zeros that are incompatible with the CLR transformation, making CoDA applicable to high-dimensional, sparse scRNA-seq matrices [91].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Software Tools and Packages for Scalable scRNA-seq Analysis
| Tool / Package | Primary Function | Application in Scalable PCA | Reference/Link |
|---|---|---|---|
| Seurat / Scanpy | End-to-end scRNA-seq analysis | Provides wrappers for PCA; default normalization and scaling. | [Satija et al. 2015] |
| scGBM R Package | Model-based dim. reduction | Directly models UMI counts for more robust PCA. | https://github.com/phillipnicol/scGBM |
| CoDAhd R Package | Compositional data transformation | Applies CLR and other log-ratio transforms for PCA input. | https://github.com/GO3295/CoDAhd |
| Irlba / Rsvd / RSpectra | Fast SVD/PCA algorithms | Core computational engines for efficient PCA on large matrices. | [90] |
| Benchmarking Code | Performance comparison | Allows researchers to test algorithm performance on their data. | https://slowkow.com/notes/pca-benchmark/ |
The scalability of PCA for datasets with millions of cells is no longer a theoretical concern but a practical necessity in modern single-cell genomics. This guide has outlined a multi-faceted approach to this challenge, emphasizing that there is no single "best" solution but rather a set of strategies that must be chosen based on the specific dataset and research goals. The path forward involves a combination of computational efficiency, achieved through randomized and iterative algorithms, and statistical robustness, offered by model-based and compositionally-aware methods.
As dataset sizes continue to grow, the integration of these scalable PCA methodologies into standard analysis pipelines will be crucial for extracting biologically meaningful insights. Researchers are encouraged to move beyond default parameters and implementations, to experiment with the advanced tools and frameworks presented here, and to always validate their low-dimensional embeddings through downstream biological interpretation. By doing so, the field can continue to leverage the full power of scRNA-seq technology to unravel cellular heterogeneity at an unprecedented scale.
Benchmarking PCA Performance: Validation Metrics and Method Comparisons
In high-dimensional biological research, particularly gene expression analysis, validating clustering results is paramount for deriving meaningful biological insights. Techniques like Principal Component Analysis (PCA) are indispensable for reducing data dimensionality, while metrics like the Adjusted Rand Index (ARI) provide a standardized framework for quantifying clustering accuracy against known benchmarks. This technical guide details the theoretical foundations and practical methodologies for employing these tools to assess cluster quality and biological coherence. Framed within the context of a broader thesis on PCA's role in gene expression data research, this whitepaper provides researchers, scientists, and drug development professionals with protocols for rigorous, quantifiable validation of their computational analyses.
Single-cell RNA sequencing (scRNA-Seq) and other high-throughput genomic technologies generate data where the number of features (genes) far exceeds the number of observations (cells). This high-dimensionality poses significant challenges for clustering algorithms, including increased computational cost and the "curse of dimensionality," where distance measures become less meaningful [19]. Principal Component Analysis (PCA) addresses this by performing an orthogonal linear transformation of the data onto a new coordinate system [27]. The principal components (PCs) are linear combinations of the original genes, often called "metagenes" or "eigengenes," and are ordered such that the first PC captures the largest possible variance in the data, the second PC captures the next largest variance while being orthogonal to the first, and so on [27] [22]. By retaining only the top PCs, researchers can project samples into a lower-dimensional space that concentrates biological signal and removes noise, creating a more manageable and informative input for downstream clustering analyses [54].
Principal Component Analysis: A Dimensionality Reduction Primer
Mathematical Foundation of PCA
PCA is an orthogonal linear transformation that converts high-dimensional data into a new coordinate system. Given a data matrix X of dimensions ( n \times p ) (with ( n ) samples and ( p ) genes), the goal is to compute a set of principal components that are linear combinations of the original genes. The first weight vector ( \mathbf{w}{(1)} ) must satisfy [22]: [ \mathbf{w}{(1)}= \arg \max{\|\mathbf{w}\|=1} \left{ \sumi \left( \mathbf{x}_{(i)} \cdot \mathbf{w} \right)^2 \right} ] This translates to finding the eigenvectors of the covariance matrix of X [22]. The resulting transformation is ( \mathbf{T} = \mathbf{X} \mathbf{W} ), where T is the matrix of PC scores, and W is the matrix of eigenvectors [22].
PCA in Bioinformatics and Gene Expression Analysis
In bioinformatics, PCA is a cornerstone technique for handling the "large d, small n" problem characteristic of genomic studies [27]. When applied to gene expression data, PCA offers several critical properties [27]:
- Orthogonality: Different PCs are orthogonal to each other, effectively solving collinearity problems in regression analysis.
- Dimensionality Reduction: The number of non-zero PCs is at most the rank of the data matrix, often much lower than the original number of genes.
- Variance Explanation: The first few PCs typically explain the majority of the variation in the dataset, allowing for efficient data summarization.
The PCs serve as "metagenes" that can be used for exploratory analysis, data visualization, clustering of genes or samples, and as covariates in regression models to predict clinical outcomes [27].
Quantifying Clustering Accuracy with the Adjusted Rand Index (ARI)
Definition and Mathematical Formulation
The Adjusted Rand Index (ARI) is a foundational metric for the quantitative evaluation of clustering results. It measures the similarity between two data clusterings by correcting the Rand Index for chance agreement [95]. For two partitions ( U ) and ( V ) of ( n ) data points, the ARI is computed using a contingency table where ( n{ij} ) represents the number of objects in cluster ( i ) of ( U ) and cluster ( j ) of ( V ), with marginals ( ai = \sumj n{ij} ) and ( bj = \sumi n_{ij} ). The formula is [95]:
[ \text{ARI} = \frac{ \sum{ij} \binom{n{ij}}{2} - \frac{ \sumi \binom{ai}{2} \sumj \binom{bj}{2} }{ \binom{n}{2} } }{ \frac{1}{2} \left[ \sumi \binom{ai}{2} + \sumj \binom{bj}{2} \right] - \frac{ \sumi \binom{ai}{2} \sumj \binom{bj}{2} }{ \binom{n}{2} } } ]
This measures the fraction of object pairs classified together or apart in both clusterings, adjusted for expected agreement under a random permutation model [95].
Table 1: Interpretation of ARI Values
| ARI Value | Interpretation |
|---|---|
| ARI = 1 | Perfect match between two clusterings (modulo cluster labels). |
| ARI ≈ 0 | Agreement is equivalent to what would be expected by random assignment. |
| ARI < 0 | Less agreement than expected by chance (indicating discordant partitions). |
ARI strengths, limitations, and comparative analysis
Strengths:
- Corrects for Chance: Unlike the Rand Index, ARI accounts for baseline similarity induced by chance, making values comparable across different scenarios and algorithms [95].
- Symmetry and Invariance: ARI is symmetric and invariant to cluster label permutations [95].
- Generalizability: Extensions of ARI allow its application to overlapping, fuzzy, and network-based clusters [95].
Limitations:
- Interpretability: The absolute magnitude of ARI is not directly interpretable as an error rate [95].
- Structural Dependence: Its value can be influenced by the structure and balance of the clusters, requiring careful interpretation in unbalanced settings [95].
Comparison with Other Metrics:
- Rand Index (RI): Does not adjust for chance, which can lead to inflated scores, especially with unbalanced clusters [95].
- Adjusted Mutual Information (AMI): Another adjusted metric that may be preferred when clusters are small or highly unbalanced [95].
A Practical Protocol for Clustering Validation
This section provides a detailed, step-by-step experimental protocol for performing dimensionality reduction and validating cell clustering in scRNA-seq data, using ARI as a key metric.
Experimental Workflow and Data Preprocessing
Step 1: Data Acquisition and Normalization
- Obtain a gene expression matrix (cells x genes) from an scRNA-seq platform (e.g., 10x Genomics). The raw data undergoes quality control, adapter sequence removal, alignment to a reference genome, and feature counting [19].
- Normalize the data to account for technical variations (e.g., sequencing depth). A common approach is to apply a log-transformation to the counts or unique molecular identifier (UMI) counts after normalization [54].
Step 2: Feature Selection
- Identify Highly Variable Genes (HVGs) to reduce noise and computational load. Restricting downstream analysis to HVGs helps prevent the earlier PCs from capturing high-dimensional random noise instead of biological structure [54]. The number of HVGs is often arbitrary but typically ranges from 1,000 to 5,000.
Step 3: Principal Component Analysis (PCA)
- Perform PCA on the normalized and scaled expression matrix of the selected HVGs. This is computationally achieved via singular value decomposition (SVD) [27] [54].
- The output is a new matrix of reduced dimensions (cells x PCs), where each PC is a linear combination of all HVGs.
Step 4: Selection of Principal Components
- Determine the number of top PCs (( d )) to retain for downstream analysis. This choice represents a bias-variance trade-off.
- Arbitrary Selection: A common practice is to choose a "reasonable" but arbitrary value between 10 and 50 [54].
- "Elbow" Method: Plot the proportion of variance explained by each PC and look for an "elbow" point where the marginal gain in explained variance drops sharply [19].
- Data-Driven Methods: More advanced strategies involve using the technical noise structure or based on the assumption that biological signal is concentrated in the earlier PCs [54].
Step 5: Cell Clustering
- Apply a clustering algorithm (e.g., k-means, hierarchical clustering, or graph-based methods) to the cell embeddings in the reduced PC space (e.g., the top 50 PCs). The Euclidean distance in this space approximates the distance in the original high-dimensional gene expression space [54].
Step 6: Validation using ARI
- If a ground truth label is available (e.g., known cell types from a reference dataset or manually annotated labels), compute the ARI between the algorithm-derived clusters and the ground truth labels [95].
- A higher ARI indicates a clustering result that more accurately recapitulates the known biological classification.
The Scientist's Toolkit: Essential Reagents and Computational Solutions
Table 2: Key Research Reagent Solutions for scRNA-seq Clustering Validation
| Item Name | Function/Explanation | Example Sources/Tools |
|---|---|---|
| Single-Cell Isolation Kit | Isolates individual cells from tissue for sequencing. Essential for generating input data. | 10x Genomics Chromium System [19] |
| scRNA-seq Library Prep Kit | Prepares sequencing libraries from isolated single cells. | Various commercial platforms |
| Alignment & Quantification Software | Maps sequencing reads to a reference genome and generates the gene-cell count matrix. | Cell Ranger (for 10x Genomics data) [19] |
| Normalization Algorithm | Corrects for technical variation (e.g., sequencing depth) to make counts comparable across cells. | Procedures in scran/scater (R/Bioconductor) [54] |
| HVG Selection Method | Identifies genes that drive biological heterogeneity, improving PCA performance. | Strategies described in OSCA [54] |
| PCA Implementation | Performs the linear dimensionality reduction at the core of the workflow. | prcomp (R), scran::fixedPCA() (R) [27] [54] |
| Clustering Algorithm | Groups cells based on their expression profiles in the reduced PC space. | k-means, hierarchical clustering, graph-based methods |
| Validation Metric Software | Computes the ARI to compare clusterings. Available in standard data science libraries. | sklearn.metrics.adjusted_rand_score (Python) |
The integration of robust dimensionality reduction techniques like PCA with rigorous validation metrics such as the Adjusted Rand Index is fundamental for success in high-dimensional biological research. PCA provides an effective, theoretically sound method for transforming vast gene expression datasets into a lower-dimensional space that retains biological signal and facilitates clustering. The ARI then offers a critical, chance-corrected measure for quantifying the accuracy of these clusters against a known benchmark. As single-cell technologies and other high-throughput methods continue to advance, adhering to detailed experimental protocols and utilizing the essential computational tools outlined in this guide will empower researchers and drug development professionals to draw reliable, quantifiable, and biologically coherent conclusions from their data.
Within the field of gene expression data research, dimensionality reduction is an indispensable step for extracting meaningful biological knowledge from high-dimensional, noisy datasets. This technical guide provides an in-depth comparison of three foundational linear dimensionality reduction techniques: Principal Component Analysis (PCA), Non-negative Matrix Factorization (NMF), and Linear Discriminant Analysis (LDA). We explore their mathematical principles, application workflows in genomics, and comparative performance. Framed within a broader thesis on how PCA reduces dimensionality of gene expression data, this review highlights how the choice of algorithm directly influences the biological interpretability of results, with direct implications for biomarker discovery and therapeutic development.
High-throughput transcriptomic technologies, such as RNA sequencing (RNA-seq), generate data where the number of genes (features) vastly exceeds the number of samples (observations). This "curse of dimensionality" poses significant challenges for statistical analysis and interpretation. Dimensionality reduction techniques address this by mapping data to a lower-dimensional space, discarding redundant variance and noise to reveal underlying structure.
Principal Component Analysis (PCA) is a cornerstone of exploratory data analysis in genomics. However, its limitations, particularly regarding biological interpretability, have led to the adoption of alternative methods like NMF and LDA. NMF's non-negativity constraints often yield more interpretable, parts-based representations, making it attractive for identifying distinct biological components. LDA, a supervised technique, excels when the goal is maximal separation between predefined classes, such as different cancer subtypes. Understanding the relative strengths and applications of these methods is crucial for modern genomic researchers and drug development professionals.
Theoretical Foundations and Mathematical Principles
Principal Component Analysis (PCA)
PCA is an orthogonal linear transformation that projects high-dimensional data onto a new coordinate system defined by the principal components (PCs). These PCs are the eigenvectors of the data's covariance matrix, ordered such that the first PC accounts for the largest possible variance in the data, each succeeding component in turn has the highest variance possible under the constraint that it is orthogonal to the preceding components.
Given a centered data matrix X of dimensions n × m (with n samples and m genes), PCA solves the eigen decomposition problem: C = X^TX = VΛV^T, where C is the covariance matrix, V is the matrix of eigenvectors (principal components), and Λ is a diagonal matrix of eigenvalues (explained variances). The projected data in the lower-dimensional space is simply Z = XV.
Non-negative Matrix Factorization (NMF)
In contrast to PCA, NMF factorizes a non-negative data matrix V (e.g., gene expression counts) into two lower-rank, non-negative matrices W (the basis matrix) and H (the coefficient matrix) such that V ≈ WH [96]. The non-negativity constraint forces the model to learn a parts-based representation of the data, where each sample is represented as an additive combination of non-negative basis elements.
The factorization is achieved by solving the optimization problem: min‖V - WH‖² with W, H ≥ 0. This is typically solved using iterative update rules. A significant extension, stretchedNMF, introduces a stretching factor matrix to account for signal variability along an independent variable's axis, proving beneficial for analyzing data such as powder diffraction at varying temperatures, and by analogy, certain time-series or condition-series gene expression experiments [97].
Linear Discriminant Analysis (LDA)
LDA is a supervised method that finds a linear combination of features that best separates two or more classes of samples. Unlike PCA, which maximizes variance, LDA maximizes the ratio of between-class variance to within-class variance.
For a multi-class problem with classes C₁, C₂, ..., Cₖ, LDA seeks the projection matrix W that maximizes the Fisher-Rao criterion: J(W) = W^TSBW / W^TSWW, where SB is the between-class scatter matrix and SW is the within-class scatter matrix. The eigenvectors of SW^{-1}SB corresponding to the largest eigenvalues form the discriminant directions.
Algorithm Selection Workflow: A decision pathway for choosing between PCA, NMF, and LDA based on analytical goals and data structure.
Application to Gene Expression Data Analysis
PCA in Genomic Workflows
PCA is ubiquitously applied in the quality control and exploratory analysis of gene expression data. It helps identify major sources of variation, which could be biological (e.g., tumor vs. normal samples, different cell lineages) or technical (e.g., batch effects, RNA quality). For instance, in large-scale studies like The Cancer Genome Atlas (TCGA), PCA has been used to reveal that the dominant patterns of variation in pan-cancer data often relate to the cell cycle, mitochondrial function, gender, interferon response, and immune infiltration, rather than the cancer type itself [98].
NMF for Molecular Pattern Discovery
The non-negative and parts-based nature of NMF makes it particularly suited for deconvoluting complex biological mixtures and identifying metagenes—groups of co-expressed genes representing distinct molecular programs. In cancer research, NMF has been successfully used to discover molecular subtypes from transcriptomic data. A notable application is in the identification of consensus molecular subtypes in colorectal cancer, which has direct clinical implications. The stretchedNMF variant demonstrates how the core algorithm can be adapted to handle specific data artifacts or biological phenomena, such as continuous shifts in signal profiles [97].
LDA for Diagnostic and Prognostic Classification
LDA's strength lies in building robust classifiers for disease subtyping. In a study aiming to classify cancer types from RNA-seq data, an SVM model ultimately achieved the highest accuracy (99.87%), but LDA and QDA (Quadratic Discriminant Analysis) remain fundamental tools in the genomic classification toolkit [99]. LDA is often employed after a feature selection step (e.g., using genes with the highest ANOVA F-statistic between classes) to create a diagnostic model that can assign new samples to known categories, such as distinguishing between acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) [100].
Comparative Analysis: Performance and Applications
Table 1: Comparative Summary of PCA, NMF, and LDA
| Feature | PCA | NMF | LDA |
|---|---|---|---|
| Core Objective | Maximize variance of projected data | Approximate data with additive parts | Maximize class separation |
| Model Type | Unsupervised | Unsupervised | Supervised |
| Constraints | Orthogonality | Non-negativity | Linear class boundaries |
| Output | Principal Components (PCs) | Basis (W) and Coefficient (H) matrices | Discriminant functions |
| Interpretability | Global structure; can be mixed-sign | Parts-based; often more intuitive | Directly related to class labels |
| Data Requirements | Any numeric data | Non-negative data (e.g., counts) | Requires class labels |
| Handling Sparsity | Standard | Excellent (sparse solutions common) | Standard |
| Use Case in Genomics | QC, Batch effect detection, visualization | Metagene discovery, molecular subtyping | Diagnostic classification, biomarker prioritization |
Table 2: Empirical Performance in Genomic Studies
| Method | Reported Accuracy/Performance | Dataset | Key Finding |
|---|---|---|---|
| NMF (stretched) | Outperformed conventional NMF for data with signal stretching | Simulated PXRD & PDF data [97] | Better extraction of physically meaningful components when signals undergo expansion/contraction. |
| SVM | 99.87% accuracy (5-fold CV) | PANCAN RNA-seq (5 cancer types) [99] | Demonstrated high potential of ML on RNA-seq data; used as benchmark. |
| LDA/QDA with nonlinear pre-processing | High robustness, reduced false positives | Acute Leukemia & SRBCT datasets [100] | Hybrid models (nonlinear projection + LDA/QDA) improve handling of class overlap and biological heterogeneity. |
Experimental Protocols and Workflows
Standard Protocol for PCA on RNA-seq Data
- Data Preprocessing: Load the gene expression count matrix (samples × genes). Perform quality control, normalization (e.g., TPM, FPKM, or variance-stabilizing transformation), and log-transformation if necessary. Center the data to have a mean of zero for each gene.
- Covariance Matrix Computation: Calculate the covariance matrix or directly perform Singular Value Decomposition (SVD) on the preprocessed data matrix.
- Eigen Decomposition: Compute the eigenvectors (principal components) and eigenvalues (explained variances).
- Projection: Project the original data onto the selected principal components to obtain the lower-dimensional representation (scores).
- Visualization & Analysis: Plot the first few PCs against each other to visualize sample relationships. Examine the loadings of each PC to identify genes contributing most to the variance.
Standard Protocol for NMF on RNA-seq Data
- Data Preparation: Begin with a non-negative input matrix (e.g., raw counts or transformed non-negative values). It is often crucial to select a subset of highly variable genes to improve performance.
- Rank Selection: Determine the factorization rank k (number of components). This is a critical step, often using metrics like cophenetic correlation coefficient or residual sum of squares, combined with biological plausibility.
- Factorization: Apply an NMF algorithm (e.g., multiplicative update, coordinate descent) to factorize the data matrix V into W and H. Multiple runs with different random seeds are recommended for stability.
- Component Interpretation: Analyze the W matrix to identify metagenes (basis components) and the H matrix to see the contribution of these metagenes to each sample. Use this to cluster samples and assign molecular subtypes.
Standard Protocol for LDA on RNA-seq Data
- Feature Selection: Given the high dimensionality, reduce the number of genes prior to LDA. This can be done using a filter method (e.g., selecting genes with the highest variance or ANOVA F-statistic across the known classes).
- Model Training: Compute the within-class and between-class scatter matrices for the training set. Solve the generalized eigenproblem for SW^{-1}SB to obtain the discriminant vectors.
- Dimensionality Projection: Project both the training and test data onto the discriminant vectors.
- Classification: Assign class labels to new samples in the projected space based on a classification rule (e.g., maximum Mahalanobis distance, or by coupling with a classifier like k-NN in the reduced space).
Gene Expression Analysis Workflow: A standard pipeline for applying PCA, NMF, and LDA to RNA-seq data, from raw counts to downstream biological insights.
Table 3: Key Research Reagents and Computational Tools
| Resource | Type | Function/Description | Example Source/Catalog |
|---|---|---|---|
| RNA-seq Library Prep Kit | Wet-lab Reagent | Prepares sequencing libraries from RNA samples; critical for data generation. | Illumina TruSeq, NEBNext Ultra II |
| TCGA / GEO Datasets | Data Resource | Public repositories of curated gene expression data for method validation and discovery. | National Cancer Institute; NCBI GEO |
| Seurat / Scanpy | Software Package | Comprehensive toolkits for single-cell and bulk RNA-seq analysis, including PCA and NMF. | CRAN/Bioconductor (R); Python PyPI |
| NMF R/Python Package | Software Package | Dedicated implementations of multiple NMF algorithms and utilities. | CRAN: NMF; Python: Nimfa [96] |
| FactoMineR / scikit-learn | Software Package | Libraries providing robust implementations of PCA, LDA, and other multivariate methods. | CRAN (R); Python PyPI |
| Cluster Validation Metrics | Computational Method | Algorithms and indices (e.g., silhouette score, ARI) to evaluate clustering results from dimensionality reduction. | Built into major analysis packages |
The choice between PCA, NMF, and LDA for analyzing gene expression data is not a matter of identifying a single superior algorithm, but rather of selecting the right tool for the specific biological question and data structure. PCA remains the workhorse for unsupervised exploration and quality control due to its computational efficiency and mathematical simplicity. NMF offers a powerful alternative when the goal is to identify coherent, parts-based biological patterns or molecular subtypes, thanks to its intuitive, non-negative constraints. LDA provides a robust framework for supervised classification tasks where the objective is to build a model that optimally discriminates between known classes.
The ongoing development of enhanced versions of these algorithms, such as sparse-stretchedNMF [97] and the integration of nonlinear kernel methods with LDA [100], indicates a vibrant field of research. For the practicing genomic scientist, a hybrid strategy that leverages the complementary strengths of these techniques—using PCA for initial data exploration, NMF for novel pattern discovery, and LDA for final diagnostic model building—will often yield the most comprehensive and biologically actionable insights, thereby advancing the broader thesis of how we effectively reduce dimensionality to uncover meaning in complex genomic data.
In the analysis of high-dimensional biological data, such as gene expression profiles from single-cell RNA sequencing (scRNA-seq) or spatial transcriptomics, dimensionality reduction is an indispensable preprocessing step. It mitigates the curse of dimensionality, reduces computational complexity, and enables the visualization of data structures that are otherwise inaccessible to the human eye [101] [54]. While Principal Component Analysis (PCA) has served as the de facto standard linear technique for decades, recent advances have popularized a suite of non-linear methods, including t-Distributed Stochastic Neighbor Embedding (t-SNE), Uniform Manifold Approximation and Projection (UMAP), and Autoencoders (AEs) [102] [103].
The central thesis of this whitepaper is that the choice of dimensionality reduction technique profoundly influences the biological insights extracted from gene expression data. PCA provides an efficient, linear baseline that maximizes variance, but its performance is constrained when biological processes exhibit complex, non-linear relationships. In contrast, non-linear techniques are capable of uncovering intricate, clustered structures often representative of true biological heterogeneity, though they come with their own trade-offs in computational demand, reproducibility, and interpretability [101] [102] [104]. This guide provides an in-depth technical comparison of these methods, benchmarking their performance using quantitative metrics and providing detailed protocols for their application in a research setting, with a particular focus on the field of genomics and drug development.
Core Concepts and Mathematical Foundations
Principal Component Analysis (PCA): The Linear Benchmark
PCA is a linear transformation technique that identifies the orthogonal directions of maximum variance in the high-dimensional data. These directions, the principal components, form a new coordinate system where the first component captures the greatest variance, the second the next greatest, and so on [101] [54].
Mathematical Foundation: PCA is typically solved via the eigen-decomposition of the covariance matrix of the data. Given a centered data matrix X with n samples (cells) and d features (genes), PCA solves the optimization problem:
Here, W is an orthonormal matrix whose columns are the principal components, and Z = XW is the low-dimensional projection (the embedding) [103]. The principal components are ordered by the amount of variance they explain, which is reflected in the magnitude of their corresponding eigenvalues.
Non-Linear Challengers: t-SNE, UMAP, and Autoencoders
t-Distributed Stochastic Neighbor Embedding (t-SNE)
t-SNE is a non-linear technique specialized for visualization and local structure preservation. It operates by modeling pairwise similarities between data points in both high and low dimensions [101] [54].
- High-Dimensional Space: Similarities between points are calculated using a Gaussian distribution, conditional on a user-specified perplexity parameter, which dictates the effective number of local neighbors.
- Low-Dimensional Space: Similarities are modeled using the heavier-tailed Student's t-distribution. This allows the algorithm to "crowd" points more effectively in the low-dimensional map, mitigating the so-called "crowding problem."
- The algorithm then iteratively minimizes the Kullback-Leibler (KL) divergence between the two similarity distributions [101].
Uniform Manifold Approximation and Projection (UMAP)
UMAP is a newer non-linear technique grounded in manifold learning and topological data analysis. Its theoretical foundations offer several practical advantages over t-SNE [101] [104].
- Manifold Assumption: UMAP assumes the data lies on a topological manifold (a locally Euclidean space).
- Graph Construction: It constructs a weighted
k-neighbor graph in high-dimensional space to represent the local structure of the data. - Optimization: The low-dimensional embedding is found by creating a similar graph in the target space and minimizing the cross-entropy between the two fuzzy topological structures. This process allows UMAP to better preserve global data structure alongside local neighborhoods [101] [104].
Autoencoders (AEs) and Variational Autoencoders (VAEs)
Autoencoders are neural network-based models that learn a compressed, low-dimensional representation (the "bottleneck" layer) of the input data in an unsupervised manner [103] [82].
- Standard Autoencoder (AE): An AE consists of an encoder network
f_θthat maps inputXto a latent codeZ, and a decoder networkg_φthat reconstructs the input fromZ. It is trained to minimize the reconstruction error: - Variational Autoencoder (VAE): A VAE introduces a probabilistic twist. The encoder outputs parameters of a distribution (e.g., mean and variance), and the latent code
Zis sampled from this distribution. The training objective is the Evidence Lower Bound (ELBO), which includes a reconstruction term and a KL divergence term that regularizes the latent space to follow a prior distribution (e.g., a standard Gaussian) [103]. This promotes a smooth, well-structured latent space.
Performance Benchmarking and Quantitative Comparison
A systematic evaluation of dimensionality reduction techniques requires a multifaceted approach, assessing not just computational efficiency but, more critically, the biological fidelity of the resulting low-dimensional embeddings.
Table 1: Core Algorithmic Characteristics and Performance
| Characteristic | PCA | t-SNE | UMAP | Autoencoders (AE/VAE) |
|---|---|---|---|---|
| Linearity | Linear [102] | Non-linear [102] | Non-linear [102] | Non-linear [103] |
| Structure Preservation | Global variance [54] | Strong local structure [101] [54] | Balanced local & global [101] [104] | Data-driven; VAE encourages continuous latent space [103] |
| Determinism | Deterministic [102] | Stochastic (requires random seed) [102] | Stochastic (requires random seed) [102] | Stochastic (initialization & training) |
| Computational Scalability | Fast; efficient for large datasets [102] | Slow; not ideal for large datasets (>10k cells) [102] | Fast; better for large datasets than t-SNE [101] [102] | High training cost; inference is fast [103] |
| Key Hyperparameters | Number of components | Perplexity, learning rate [54] | Number of neighbors, min_dist [104] | Network architecture, latent dimension, regularization |
| Primary Application in Gene Analysis | Data preprocessing, noise reduction, initial exploration [101] [54] | Cluster visualization, fine-grained population discovery [101] [54] | General-purpose visualization for large datasets, preserving hierarchy [102] | Feature extraction, denoising, integration with other deep learning models [103] [82] |
Recent benchmarking studies on spatial transcriptomics data have introduced novel, biologically-grounded metrics for a more meaningful comparison. These include Cluster Marker Coherence (CMC), which measures the fraction of cells in a cluster expressing its designated marker genes, and Marker Exclusion Rate (MER), which quantifies the fraction of cells that would be better assigned to a different cluster based on marker expression [103].
Table 2: Benchmarking on Spatial Transcriptomics Data (Illustrative Results)
| Method | Reconstruction Error (MSE) | Clustering Quality (ARI) | Biological Coherence (CMC) | Interpretability |
|---|---|---|---|---|
| PCA | Baseline | Moderate (e.g., ~0.556 ARI) [47] | Low to Moderate | High (components are linear combinations of genes) [103] |
| NMF | Moderate | Lower in some benchmarks (e.g., ~0.185 ARI) [47] | High (additive parts-based representation) [103] | Very High (non-negative, sparse factors) |
| UMAP | N/A (not a reconstructive method) | High | High | Low (distances not directly interpretable) [102] |
| VAE | Low | High (e.g., ~0.78 ARI with GraphPCA) [47] | High | Moderate (latent dimensions can be interrogated) [103] |
These benchmarks reveal distinct performance profiles. PCA provides a fast, interpretable baseline. NMF excels in producing interpretable, biologically relevant components, often maximizing marker gene enrichment. Deep learning methods like AEs and VAEs offer a powerful balance between accurate reconstruction and clustering quality, while UMAP consistently performs well for visualization tasks that require a clear view of both local and global structure [103] [47].
Experimental Protocols and Methodologies
Standardized Workflow for Dimensionality Reduction
A robust experimental pipeline for applying and benchmarking these techniques involves several key stages, as visualized below.
Diagram 1: Experimental workflow for dimensionality reduction
Detailed Protocol: Applying and Benchmarking Techniques
The following protocol is adapted from best practices in scRNA-seq and spatial transcriptomics analysis [54] [103] [47].
Step 1: Data Preprocessing and Normalization
- Input: Raw count matrix (cells x genes).
- Quality Control: Filter out low-quality cells based on metrics like mitochondrial gene percentage and total unique molecular identifier (UMI) counts.
- Normalization: Normalize for sequencing depth (e.g., counts per 10,000) and apply a log transformation (
log1p). - Feature Selection: Identify Highly Variable Genes (HVGs). Restricting analysis to HVGs reduces noise and computational load [54].
Step 2: Applying Dimensionality Reduction
- PCA:
- Standardize the normalized HVG matrix to have zero mean and unit variance.
- Apply PCA using a scalable implementation (e.g.,
scikit-learnorScanpy). Compute the top 50 principal components as a default. - Determine the number of significant components using an elbow plot of explained variance or a more data-driven method [54].
- t-SNE:
- It is highly recommended to use the top PCs (e.g., the first 50) from the previous step as input, rather than the full gene expression matrix. This denoises the data and speeds up computation [54].
- Set a random seed for reproducibility.
- Optimize key parameters: perplexity (typically between 5-50), which influences the granularity of clusters, and learning rate.
- UMAP:
- Similarly, use the top PCs as input for efficiency.
- Set a random seed.
- Key parameters are
n_neighbors(balances local/global view; default ~15) andmin_dist(controls clustering tightness; default ~0.1).
- Autoencoder:
- Design the network architecture. A simple AE might have symmetric encoder/decoder with fully connected layers.
- For a VAE, ensure the encoder outputs mean and variance vectors.
- Train the model using the normalized expression data, with the mean squared error as a common reconstruction loss.
Step 3: Downstream Analysis and Benchmarking
- Clustering: Apply a clustering algorithm (e.g., Leiden, Louvain) on the low-dimensional embeddings.
- Validation:
- Quantitative: Calculate metrics like Adjusted Rand Index (ARI) against known labels, Silhouette Score for cluster cohesion, and novel biological metrics like CMC and MER [103].
- Qualitative: Visually inspect 2D/3D projections to assess cluster separation and overall data structure.
The Scientist's Toolkit: Essential Research Reagents and Computational Tools
Table 3: Key Reagents and Tools for Dimensionality Reduction Analysis
| Item Name | Function / Description | Example Sources / Packages |
|---|---|---|
| Single-Cell RNA-seq Data | The primary high-dimensional input data for analysis. | 10X Genomics, Smart-seq2 |
| Spatial Transcriptomics Data | Gene expression data with spatial coordinates, used for spatially-aware methods. | 10X Visium, Slide-seq, MERFISH [47] |
| Normalization Software | Corrects for technical variation (e.g., sequencing depth). | Scanpy (sc.pp.normalize_total), Seurat |
| Highly Variable Gene Selector | Identifies informative genes for downstream analysis, reducing dimensionality and noise. | Scanpy (sc.pp.highly_variable_genes), Seurat |
| PCA Implementation | Computes linear principal components. | Scikit-learn (decomposition.PCA), Scanpy |
| t-SNE Implementation | Computes non-linear t-SNE embeddings. | Scikit-learn (manifold.TSNE), Scanpy |
| UMAP Implementation | Computes non-linear UMAP embeddings. | UMAP-learn, Scanpy |
| Deep Learning Framework | Provides environment to build and train autoencoders. | PyTorch, TensorFlow |
| Clustering Algorithm | Identifies cell groups in low-dimensional space. | Leiden, Louvain (in Scanpy/Seurat) |
| Benchmarking Metrics | Quantifies performance of reduction and clustering. | ARI, NMI, CMC, MER [103] |
Advanced Applications and Future Directions
The field of dimensionality reduction is rapidly evolving, with new methods being developed to address specific challenges in genomic research.
Spatially-Aware Dimensionality Reduction: Methods like GraphPCA, SpatialPCA, and STAGATE explicitly incorporate spatial coordinates of cells/spots into the dimensionality reduction process. For example, GraphPCA integrates a graph constraint derived from spatial neighborhoods into the PCA framework, significantly improving the detection of spatially coherent domains in tissues [47]. The diagram below illustrates its core innovation.
Diagram 2: GraphPCA integrates gene expression and spatial information
Normalization via Dimensionality Reduction: An advanced application involves "flipping the script" on traditional assumptions. In the analysis of CRISPR screen data (e.g., the Cancer Dependency Map, DepMap), dominant low-dimensional signals can represent technical biases or overwhelming biological processes (e.g., mitochondrial function). Techniques like Robust PCA (RPCA) and Autoencoders can be used to capture and remove these dominant signals, thereby enhancing the visibility of subtler, cancer-specific genetic dependencies [82].
The benchmarking of PCA against modern non-linear techniques reveals a landscape defined by trade-offs. PCA remains a powerful, fast, and interpretable method for initial data compaction, noise reduction, and as a preprocessing step for non-linear algorithms. Its linear nature, however, is a fundamental limitation when analyzing the complex, non-linear manifolds that often underlie gene expression programs.
Non-linear methods like t-SNE, UMAP, and Autoencoders excel at uncovering the nuanced, clustered structure of cellular heterogeneity. UMAP, in particular, has emerged as a favored choice for visualization due to its speed and superior preservation of global data relationships. For tasks demanding the highest fidelity in feature extraction and integration with deep learning pipelines, Autoencoders, especially VAEs, offer immense flexibility and power.
The choice of method must be guided by the specific biological question. Is the goal rapid exploration, publication-quality visualization, or the extraction of maximally informative features for downstream prediction? As the field progresses, the development of specialized, interpretable, and biologically-validated algorithms like GraphPCA signals a move beyond one-size-fits-all solutions towards a more nuanced toolkit, empowering researchers and drug developers to extract deeper, more meaningful insights from the ever-growing volumes of genomic data.
Modern transcriptomic studies, which analyze the complete set of RNA transcripts in a biological sample, generate vast, high-dimensional datasets. In fields like virology and oncology, this high dimensionality—where the number of measured genes (features) far exceeds the number of samples—presents significant challenges for analysis and interpretation. Dimensionality reduction has therefore become an essential computational step, transforming high-dimensional gene expression data into lower-dimensional spaces that are manageable for visualization and downstream analysis while preserving critical biological information [19].
Among various techniques, Principal Component Analysis (PCA) is a foundational linear dimensionality reduction method. PCA operates by identifying orthogonal directions of maximum variance in the original data, known as principal components (PCs). It transforms the dataset into a new set of linearly uncorrelated variables, ordered so that the first few retain most of the variation present in the original features [2] [18]. In transcriptomics, each cell or sample is a data point in a high-dimensional space defined by gene expression levels. PCA reduces these dimensions by creating a smaller set of "latent genes," which are linear combinations of the original genes, facilitating efficient clustering of samples and identification of biological patterns [19].
This whitepaper explores the application of PCA as a validation tool through case studies in COVID-19 and cancer research, detailing specific methodologies, visualizing workflows, and providing resources for researchers.
PCA is a statistical procedure that uses an orthogonal transformation to convert a set of observations of possibly correlated variables into a set of values of linearly uncorrelated variables called principal components. This transformation is defined such that the first principal component accounts for the largest possible variance in the data, and each succeeding component, in turn, has the highest variance possible under the constraint that it is orthogonal to the preceding components.
In the context of gene expression data, which is typically organized as a matrix where rows represent genes and columns represent samples, PCA reduces the dimensionality by projecting each sample from its original high-dimensional gene space onto a new, smaller subspace of principal components. Formally, given a gene expression matrix ( X ) with dimensions ( n \times d ) (where ( n ) is the number of samples and ( d ) is the number of genes), PCA transforms it into a new matrix ( Y ) with dimensions ( n \times k ) (where ( k \ll d )), while striving to preserve global variance [2].
The number of principal components to retain is a critical decision. Common approaches include selecting components that explain an arbitrarily selected percentage of the total variability (e.g., 90%) or using the "elbow" method on a scree plot to identify a point where the explained variance drops markedly [19]. The resulting lower-dimensional representation is crucial for visualizing sample relationships, identifying outliers, batch effects, and informing subsequent analyses like clustering and differential expression.
PCA Workflow for Transcriptomic Data
The following diagram illustrates the standard PCA workflow for transcriptomic data, from raw counts to biological interpretation:
Case Study 1: Unraveling Long COVID Through Endothelial Cell Transcriptomics
Experimental Background and Rationale
Endothelial dysfunction is a key pathophysiological feature of both acute COVID-19 and Long COVID, characterized by persistent symptoms lasting over 12 weeks post-infection. Vascular endothelium, which lines blood vessels, is vital for regulating vascular tone, barrier integrity, inflammation, and coagulation. SARS-CoV-2 can directly infect endothelial cells or indirectly cause injury via a systemic inflammatory response, triggering a pro-inflammatory and pro-coagulant state [105].
Endothelial Colony-Forming Cells (ECFCs), a type of endothelial progenitor cell with strong proliferative potential, serve as key biomarkers for vascular damage and cardiovascular risk. This study applied transcriptomic profiling to ECFCs isolated from post-COVID-19 patients to elucidate the molecular mechanisms of sustained endothelial impairment [105].
Detailed Experimental Protocol
1. Sample Collection and Preparation:
- Patient Cohort: ECFCs were isolated from peripheral blood of post-COVID-19 patients at 3- and 6-months post-infection. A control group of healthy, non-COVID individuals was included for baseline comparison. Subjects were meticulously matched for age, gender, BMI, and comorbidities [105].
- Cell Isolation and Culture: ECFCs were isolated from peripheral blood mononuclear cells (PBMCs) and cultured in endothelial-specific growth medium. Cells with high proliferative potential were selected and expanded for analysis [105].
2. RNA Sequencing and Data Generation:
- Library Preparation: Total RNA was extracted from ECFCs. RNA quality was assessed (e.g., RIN > 8). High-throughput RNA sequencing libraries were prepared and sequenced on an Illumina platform to generate transcriptome-wide data [105].
- Data Pre-processing: Raw sequencing reads (FASTQ files) underwent quality control (adapter trimming, quality filtering). Filtered reads were aligned to a human reference genome. Gene-level counts were generated using unique molecular identifiers (UMIs) to account for amplification bias [105] [19].
3. Data Pre-processing for Dimensionality Reduction:
- Count Transformation: Raw gene counts were normalized and transformed using a Variance Stabilizing Transformation (VST) implemented in the DESeq2 package. This step stabilizes variance across the mean, making the data more suitable for linear-based methods like PCA [106].
- Batch Effect Correction: Known technical batch effects were modeled as covariates in the DESeq2 design formula. For unknown batch effects, Surrogate Variable Analysis (SVA) was employed to identify and remove unwanted variation [106].
- Outlier Removal: Principal Component Analysis (PCA) was performed on the VST-transformed data to identify and remove outlier samples that could skew downstream analysis [106].
4. Differential Expression and Pathway Analysis:
- Differential Gene Expression (DGE): Analysis was performed using DESeq2. Genes with an absolute log2 fold change (|Log2FC|) ≥ 1 and an adjusted p-value (FDR) ≤ 0.05 were classified as differentially expressed genes (DEGs) [105].
- Functional Enrichment: DEG lists were subjected to pathway enrichment analysis using databases like KEGG and GO to identify biological processes and signaling pathways significantly disrupted in post-COVID ECFCs [105].
Key Findings and PCA Validation
PCA was instrumental in validating the core finding of sustained endothelial dysfunction. The analysis revealed a clear separation in gene expression profiles between ECFCs from healthy controls and those from post-COVID-19 patients at both 3 and 6 months. This distinct clustering confirmed a significant and persistent transcriptomic shift following infection [105].
Furthermore, when patients were stratified based on a history of pulmonary embolism (PE) during initial infection, PCA showed distinct clustering between PE and non-PE groups. This indicated unique, long-lasting transcriptional signatures associated with thromboembolic complications [105].
Table 1: Key Differentially Expressed Pathways in Post-COVID ECFCs
| Contrast | Key Upregulated Pathways | Key Downregulated Pathways | Implication |
|---|---|---|---|
| 3-month vs. Control | IL-17 signaling, Leukocyte transendothelial migration, VEGF signaling [105] | PI3K-Akt signaling, cGMP-PKG signaling [105] | Sustained inflammation & impaired vascular homeostasis |
| 6-month vs. Control | IL-17 signaling, Leukocyte transendothelial migration [105] | PI3K-Akt signaling, Ribosome structure [105] | Persistent inflammatory & metabolic dysregulation |
| PE vs. Non-PE | N/A | Thrombosis-related genes (PTGS2, ACKR3) [105] | Unique signature of impaired thrombosis resolution |
The minimal gene expression differences observed between the 3- and 6-month time points within the same cohort, as evidenced by only three DEGs, reinforced the conclusion that endothelial dysfunction is not a transient phenomenon but persists for at least six months post-infection [105].
Case Study 2: Integrated Multi-Sample Analysis of COVID-19 Host Responses
Experimental Background and Rationale
Early COVID-19 transcriptomic studies, while insightful, showed considerable heterogeneity in their findings due to diverse study designs, sampling strategies, and analytical pipelines. To overcome the limitations of individual studies and identify robust, conserved biological signals, a comprehensive meta-analysis of publicly available RNA-Seq datasets was conducted across multiple sample types: swabs (viral entry site), blood (systemic response), and tissues (site of pathogenesis) [106].
Detailed Experimental Protocol
1. Data Acquisition and Cohort Definition:
- Dataset Sourcing: Ten RNA-Seq datasets from Illumina platforms were sourced from the Gene Expression Omnibus (GEO) database. Datasets required raw gene counts and a minimum of 10 samples each for both COVID-19 patients and healthy controls [106].
- Sample Grouping: Datasets were categorized into three meta-groups based on sample type and biological context:
- Swab: Nasopharyngeal (NP) and oropharyngeal (OP) swabs.
- Blood: Peripheral blood mononuclear cells (PBMCs) and whole blood.
- Tissue: Bronchoalveolar lavage (BAL) fluid, lung, and kidney tissues [106].
2. Standardized Bioinformatics Pipeline:
- Pre-processing and QC: A uniform pipeline was applied to all datasets. Raw gene counts were filtered for low reads, transformed with VST, and assessed for outliers via PCA. Batch effects were corrected using the design formula in DESeq2 or SVA [106].
- Differential Expression Analysis: DGE analysis for each dataset was performed independently using DESeq2 with a standardized threshold of |log2FC| > 2.0 and adjusted p-value < 0.05 [106].
- Rank Combination Meta-Analysis: To integrate findings across datasets, a rank combination meta-analysis was performed using the RankProd method. This non-parametric method ranks genes by their fold changes and calculates the geometric mean of ranks across studies, identifying consistent DEGs robust to data heterogeneity [106].
The following diagram outlines this integrated analysis workflow:
Key Findings and System-Level Insights
The meta-analysis successfully delineated the host response to SARS-CoV-2 across different biological compartments, with PCA and other methods providing consistent sample separation and quality control.
Table 2: Conserved Transcriptomic Signatures Across COVID-19 Sample Types
| Sample Type | Key Upregulated Genes/Pathways | Key Downregulated Genes/Pathways | Biological Interpretation |
|---|---|---|---|
| Swab | ISGs (e.g., IFIT2); Innate immune pathways [106] | Interleukins [106] | Dominant local innate antiviral response at viral entry point |
| Blood | Diverse immune & neurological regulators [106] | N/A | Complex systemic immune and neurological interplay |
| Tissue | Immunoglobulin genes, Extracellular matrix genes [106] | Metabolic genes (GPD1, CYP4A11) [106] | Adaptive immunity, tissue remodeling, and metabolic dysregulation |
This systems biology approach portrayed the molecular progression of COVID-19 from a localized innate response in the respiratory tract to systemic effects in the blood, and finally to tissue-specific adaptive immunity and remodeling in affected organs [106].
Case Study 3: Single-Cell RNA Sequencing in Cancer Research
Experimental Background and Rationale
Single-cell RNA sequencing (scRNA-Seq) has revolutionized cancer biology by enabling the high-resolution study of gene expression at the individual cell level within complex tissues like tumors. This reveals hidden cellular diversity, identifies rare cell populations (e.g., cancer stem cells), and characterizes the tumor microenvironment [19].
However, scRNA-Seq data poses significant computational challenges. Data are characterized by high dimensionality (thousands of cells and genes) and high sparsity due to an abundance of zero counts ("dropout events") from low mRNA capture [19]. Dimensionality reduction is thus an indispensable step in the scRNA-Seq pipeline to overcome these issues, reduce data size for computational efficiency, and facilitate biological interpretation through visualization and clustering [19].
Detailed scRNA-Seq Protocol and PCA Application
1. Single-Cell Sequencing and Data Generation:
- Cell Isolation: Single cells are dissociated from a tumor tissue sample using enzymatic or mechanical methods. Cells are isolated via microfluidics (e.g., 10x Genomics) or other platforms.
- Library Prep and Sequencing: Cells are lysed, mRNA is barcoded, and libraries are prepared for sequencing. The output is FASTQ files containing nucleotide sequences and quality scores [19].
- Expression Matrix Generation: Raw data is processed through a pipeline (e.g., Cell Ranger for 10x Genomics data). Steps include quality control, alignment to a reference genome, and feature counting using UMIs, resulting in a gene (rows) by cell (columns) count matrix [19].
2. Data Pre-processing and Dimensionality Reduction:
- Quality Control and Normalization: Low-quality cells and genes are filtered out. Counts are normalized to account for sequencing depth and other technical biases.
- Feature Selection: Highly variable genes (HVGs) that drive biological heterogeneity are identified.
- PCA on scRNA-Seq Data: PCA is applied to the normalized and scaled expression data of the HVGs. Each cell is a data point in a high-dimensional space defined by gene expression. PCA performs an orthogonal linear transformation, creating a new set of variables—principal components (PCs)—that are linear combinations of the original genes (latent genes). The top PCs, which capture the most biological variance, are retained for downstream analysis [19].
3. Downstream Analysis:
- The low-dimensional PC space is used as input for clustering algorithms (e.g., Louvain, Leiden) to identify distinct cell populations.
- These populations are then visualized using non-linear visualization techniques like t-SNE or UMAP, which are often initialized with the PCA results [19].
- Differential expression analysis between clusters reveals marker genes that define cell types and states within the tumor.
The standard scRNA-Seq analysis pipeline, highlighting the critical role of PCA, is shown below:
Key Insights in Cancer Biology
By applying PCA and subsequent clustering to scRNA-Seq data from tumors, researchers have made pivotal discoveries:
- Tumor Heterogeneity: Uncovered previously unappreciated diversity within cancer cell populations, revealing subclones with different mutation and expression profiles [19].
- Tumor Microenvironment (TME): Deconvoluted the complex mixture of cells within a tumor, including immune cells (T cells, B cells, macrophages), cancer-associated fibroblasts (CAFs), and endothelial cells, and characterized their interactions [19].
- Therapy Resistance: Identified rare cell populations that exhibit pre-existing or acquired resistance to treatments, such as chemotherapy or targeted therapy, providing new targets for combination therapies [19].
Table 3: Key Research Reagent Solutions for Transcriptomic Studies
| Reagent / Resource | Function | Example Use Case |
|---|---|---|
| DESeq2 | A software package for differential gene expression analysis based on a negative binomial model. It performs normalization, dispersion estimation, and statistical testing [106]. | Standardized DGE analysis in COVID-19 meta-analysis [106]. |
| RankProd | A tool for rank product meta-analysis, used to identify consistently differentially expressed genes across multiple independent studies in a robust manner [106]. | Integrating DEG findings from multiple COVID-19 datasets [106]. |
| Weighted Gene Co-expression Network Analysis (WGCNA) | Used to find clusters (modules) of highly correlated genes, identify hub genes, and relate modules to external sample traits [106]. | Identifying key hub genes (e.g., GPD1, CYP4A11) in tissue samples [106]. |
| Cell Ranger | A standardized software pipeline provided by 10x Genomics for processing scRNA-Seq data, from raw sequencing reads to a gene-cell count matrix [19]. | Processing scRNA-Seq data from tumor samples to generate expression matrices [19]. |
| Unique Molecular Identifiers (UMIs) | Short nucleotide barcodes that label individual mRNA molecules before PCR amplification, allowing for accurate quantification by correcting for PCR bias [19]. | Accurate gene expression quantification in droplet-based scRNA-Seq (e.g., 10x Genomics) [19]. |
| Variance Stabilizing Transformation (VST) | A data transformation method applied to normalized count data to stabilize variance across the mean, making it suitable for linear dimensionality reduction techniques like PCA [106]. | Pre-processing step before PCA in RNA-Seq analysis pipelines [106]. |
PCA remains an indispensable, robust, and interpretable method for reducing the dimensionality of high-throughput transcriptomic data. As demonstrated in the case studies of COVID-19 and cancer research, it serves as a critical validation tool, enabling researchers to confirm hypotheses about sample separation, identify batch effects and outliers, and create a foundation for more complex analyses. Its application, from bulk RNA-Seq of patient cohorts to the intricate analysis of single-cell tumor ecosystems, underscores its fundamental role in transforming vast genomic data into biologically meaningful insights. By following standardized protocols and leveraging integrated analysis workflows, scientists can continue to utilize PCA to unlock the complexities of disease pathogenesis, paving the way for novel diagnostic and therapeutic strategies.
Principal Component Analysis (PCA) stands as a cornerstone multivariate technique in biological data analysis, particularly for reducing the complexity of high-dimensional gene expression datasets. However, its widespread application often overlooks a critical distinction: principal components (PCs) are mathematical constructs designed to maximize variance capture, not necessarily to reflect biological meaning. This guide details the methodological caveats of PCA, provides evidence-based protocols for robust implementation, and outlines advanced interpretive frameworks to help researchers discriminate true biological signals from statistical artifacts, ensuring conclusions are driven by biology rather than analytical output alone.
In the analysis of high-dimensional biological data, such as gene expression from RNA-seq or microarrays, researchers frequently face the "curse of dimensionality" [20]. A typical transcriptomic dataset involves measuring the expression levels of thousands of genes (P variables) across a much smaller number of samples or individuals (N observations), creating an N ≪ P scenario that poses significant challenges for visualization, analysis, and mathematical operations [20]. PCA addresses this by performing a linear transformation that converts the original correlated variables (genes) into a set of uncorrelated principal components (PCs) that successively capture the maximum possible variance in the data [11]. The outcome is a reduced-dimension dataset that can be visualized in 2D or 3D scatterplots, ideally with minimal information loss.
However, the central thesis for researchers is this: PCA is an unsupervised, assumption-free mathematical tool [23]. It will always produce components that explain variance, but this variance may stem from technical artifacts, batch effects, or biologically irrelevant sources rather than the experimental conditions or phenotypes of interest. Distinguishing between biologically relevant variance and technical or irrelevant variance is the fundamental challenge in interpreting PCA outputs.
The Fundamental Challenges and Biases of PCA
Mathematical Artifacts versus Biological Meaning
The application of PCA to biological data is fraught with interpretative risks. A comprehensive analysis of twelve common test cases using an intuitive color-based model demonstrated that PCA results can be artifacts of the data and can be easily manipulated to generate desired outcomes [23]. This finding raises profound concerns about the validity of a significant portion of the scientific literature in genetics and related fields.
- Sensitivity to Data Composition: The inclusion or exclusion of specific populations or samples in the input data can drastically alter the orientation of principal components, leading to different and potentially contradictory clustering patterns [23].
- Variance Does Not Equal Biological Importance: PCA prioritizes directions of maximum variance, but the largest sources of variance in a dataset may be related to batch effects, age, sample processing, or other confounding factors rather than the biological process under investigation [107].
- The Linearity Assumption: PCA captures only linear relationships between variables. Biological systems, however, are replete with non-linear interactions, which PCA cannot adequately model [11].
Documented Instances of PCA Bias Across Disciplines
The biases of PCA are not theoretical; they have been empirically demonstrated across biological disciplines:
- Population Genetics: A 2022 study in Scientific Reports concluded that PCA results may not be reliable, robust, or replicable as the field assumes, potentially requiring the reevaluation of 32,000-216,000 genetic studies [23]. The study showed that PCA could be manipulated to support multiple opposing arguments in the same debate.
- Physical Anthropology and Morphometrics: Research published in eLife found that PCA outcomes in geometric morphometrics are artefacts of the input data, challenging evolutionary insights derived from this method [108]. The standard approach of Generalized Procrustes Analysis (GPA) followed by PCA was shown to be problematic for taxonomic classification and phylogenetic inference.
Table 1: Documented Limitations of PCA in Biological Research
| Research Domain | Documented PCA Limitation | Potential Impact |
|---|---|---|
| Population Genetics [23] | Results are sensitive to sample selection and can be manipulated to generate desired outcomes. | Conclusions about ancestry, relatedness, and population structure may be artifactual. |
| Transcriptomics [107] | May fail if biological question is unrelated to the highest variance or data follows non-Gaussian distribution. | Key differentially expressed pathways or conditions may be obscured. |
| Geometric Morphometrics [108] | PCA of landmark data after Procrustes fitting can yield unreliable taxonomic and phylogenetic clusters. | Misclassification of specimens and incorrect evolutionary inferences. |
Methodological Framework: Ensuring Biologically Relevant PCA
Pre-Processing and Data Quality Assessment
The foundation of a biologically relevant PCA begins long before the analysis is run.
- Data Standardization: For gene expression data, features (genes) must be standardized to have zero mean and unit variance before applying PCA. This prevents highly expressed genes from dominating the variance structure simply due to their scale [11].
- Outlier Detection and Management: Identify technical outliers that may disproportionately influence the components. However, exercise caution, as putative "outliers" could represent genuine biological extremes.
- Batch Effect Correction: Use methods like ComBat or surrogate variable analysis (SVA) to correct for known technical batches before PCA. This prevents batch effects from becoming the primary source of variation captured in the first few PCs.
Robust Implementation and Validation Protocols
To mitigate the risk of drawing conclusions from artifacts, adhere to the following experimental protocol:
- Sub-Sampling and Sensitivity Analysis: Repeatedly run PCA on randomly sub-sampled datasets. If the observed sample clustering is robust and biologically meaningful, it should persist across most sub-samples. Inconsistent clustering suggests the structure is an artifact of a specific data composition [23].
- Benchmarking with Supervised Methods: Validate PCA clustering patterns using supervised machine learning classifiers. If a classifier can reliably assign samples to groups based on gene expression, it corroborates that the groups are biologically distinct [108].
- Variance Correlation Analysis: Systematically correlate PC scores with known technical and biological covariates (e.g., sequencing batch, RIN value, age, sex, treatment dose). This quantifies how much of each component's variance is explained by these factors.
Table 2: Key Research Reagents and Computational Tools for Robust PCA
| Tool/Reagent Category | Example | Function in PCA Workflow |
|---|---|---|
| Computational Environment | R/Python | Provides the statistical and programming foundation for data pre-processing, analysis, and visualization. |
| PCA Implementation | SmartPCA (EIGENSOFT), PLINK, scikit-learn | Software packages that perform the core PCA calculations, each with algorithm-specific nuances. |
| Alternative Algorithms | Independent Component Analysis (ICA), t-SNE, UMAP | Non-linear or independence-optimizing techniques used to validate or complement PCA findings. |
| Benchmarking Data | Intuitive color-based models [23], known positive controls | Datasets where the "ground truth" is unambiguous, used to test the accuracy and limitations of PCA. |
Advanced and Sparse PCA Variations
When standard PCA fails to yield biologically interpretable results, consider these advanced variations:
- Independent Principal Component Analysis (IPCA): This hybrid approach combines PCA with Independent Component Analysis (ICA). PCA is used first for dimension reduction, followed by ICA as a denoising process on the loading vectors. IPCA has been shown to offer better data visualization with fewer components than PCA alone and better clustering of biological samples [107].
- Sparse PCA (sPCA): Techniques like sIPCA perform an internal variable selection by applying soft-thresholding on the loading vectors [107]. This forces the loadings for less important genes to zero, resulting in components defined by a smaller, more biologically relevant set of features, which enhances interpretability.
The following diagram illustrates the decision-making workflow for implementing and validating PCA to ensure biological relevance:
Best Practices for Interpretation and Reporting
To enhance the rigor and reproducibility of PCA-based findings, researchers should adopt the following reporting standards:
- Full Disclosure of Inputs: Explicitly report the exact sample composition, the number of variables (genes) used, and any pre-filtering steps applied. Acknowledge how these choices might influence the results [23].
- Quantify and Report Component Stability: Use measures like the Tracy-Widom statistic to assess the significance of components, but do not rely on them exclusively. Report the results of sensitivity analyses [23].
- Avoid Over-Interpretation of Clustering: Do not equate proximity on a PCA plot with shared ancestry or biological identity without robust, independent validation. Clustering can be driven by hidden confounders [108].
- Integrate Multiple Lines of Evidence: Never base a biological conclusion solely on a PCA scatterplot. Use PCA as a hypothesis-generating tool within a broader analytical framework that includes phylogenetic methods, genome-wide association studies (GWAS), or functional enrichment analysis [23].
Principal Component Analysis is a powerful tool for navigating the high-dimensional seas of biological data. However, its mathematical elegance belies a significant interpretive peril. The principal components it generates are not inherently biological; they are statistical constructs that require rigorous, critical validation. By adopting the methodologies and best practices outlined in this guide—including robust pre-processing, sensitivity analyses, the use of benchmark data, and the integration of complementary analytical techniques—researchers can fortify their interpretations. The ultimate goal is to ensure that the stories we tell are truly those written in the language of biology, not the silent, and potentially misleading, artifacts of statistical computation.
Conclusion
Principal Component Analysis remains a cornerstone technique for dimensionality reduction in gene expression analysis, offering an unparalleled combination of computational efficiency, interpretability, and proven utility. Its foundational principle—summarizing multidimensional data into a 'best-fitting' lower-dimensional representation—is more relevant than ever in the age of high-throughput transcriptomics and spatial biology. While standard PCA provides a robust baseline, its specialized variants and spatially-aware adaptations are pushing the boundaries of biomedical data exploration. Future directions point towards greater integration with probabilistic modeling, enhanced scalability for million-cell datasets, and hybrid approaches that combine PCA's strengths with nonlinear deep learning methods. For researchers in drug development and clinical translation, mastering PCA and its modern applications is not merely a technical skill but a critical step towards unlocking the complex, correlation-driven patterns that underlie disease mechanisms and therapeutic responses.
References
- 1. an R/Bioconductor package for interacting with RNA-seq ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6567655/]
- 2. Comprehensive review of dimensionality reduction algorithms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12453773/]
- 3. Multiple Mean Comparison for Clusters of Gene ... [https://lifescienceglobal.com/pms/index.php/ijsmr/article/view/10023]
- 4. Analysis and validation of characteristic genes in RNA ... [https://pubmed.ncbi.nlm.nih.gov/40933310/]
- 5. Efficient and easy gene expression and genetic variation ... [https://www.nature.com/articles/s41598-025-93067-5]
- 6. ideal: an R/Bioconductor package for interactive differential ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03819-5]
- 7. Infusing structural assumptions into dimensionality ... [https://www.nature.com/articles/s42003-025-07872-9]
- 8. A Comprehensive Guide to Dimensionality Reduction [https://medium.com/@adnan.mazraeh1993/a-comprehensive-guide-to-dimensionality-reduction-from-basic-to-super-advanced-techniques-4-dabcd33eff8d]
- 9. The application of principal component analysis to drug ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644617300181]
- 10. ( Principal ): Explained Step-by-Step | Built In Component Analysis PCA [https://builtin.com/data-science/step-step-explanation-principal-component-analysis]
- 11. 27 Principal ( Component ) & Analysis PCA Dimensionality Reduction [https://medium.com/@kiranvutukuri/27-principal-component-analysis-pca-dimensionality-reduction-b7ed1b724a02]
- 12. on TCGA bulk RNAseq data | Chatomics PCA analysis [https://divingintogeneticsandgenomics.com/post/pca-tcga/]
- 13. Normalization of gene counts affects principal components ... [https://www.sciencedirect.com/science/article/pii/S1874939924000543]
- 14. A comparison of RNA-Seq data preprocessing pipelines for ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05801-x]
- 15. | Nature Reviews Methods Primers Principal component analysis [https://www.nature.com/articles/s43586-022-00184-w?error=cookies_not_supported&code=218556ba-b752-451c-ae16-c911736a1830]
- 16. and the reported low intrinsic... Principal components analysis [https://www.nature.com/articles/srep25696?error=cookies_not_supported]
- 17. The applications of PCA in QSAR studies [https://pubmed.ncbi.nlm.nih.gov/28656625/]
- 18. PCA vs PCoA vs NMDS: Key Differences, Applications, and ... [https://www.cd-genomics.com/blog/pca-pcoa-nmds-differences-applications/]
- 19. Single‑cell RNA sequencing data (Review) dimensionality reduction [https://www.spandidos-publications.com/10.3892/wasj.2025.315]
- 20. Exploring Principal Component Analysis (PCA) [https://bioinformaticamente.com/2025/03/11/exploring-principal-component-analysis-pca/]
- 21. Introduction to Dimensionality - GeeksforGeeks Reduction [https://www.geeksforgeeks.org/machine-learning/dimensionality-reduction/]
- 22. Principal component analysis [https://en.wikipedia.org/wiki/Principal_component_analysis]
- 23. Principal Component Analyses (PCA)-based findings in ... [https://www.nature.com/articles/s41598-022-14395-4]
- 24. Benchmarking of dimensionality reduction methods to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12402208/]
- 25. A Genealogical Interpretation of Principal Components ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2757795/]
- 26. How PCA helps multi-criteria decision making for feature ... [https://www.sciencedirect.com/science/article/pii/S1110016825009913]
- 27. Principal component analysis based methods in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3220871/]
- 28. Chapter 7 Dimension Reduction - Shirley Liu Lab [https://liulab-dfci.github.io/bioinfo-combio/dr.html]
- 29. Interpret all statistics and graphs for Principal Components ... [https://support.minitab.com/en-us/minitab/help-and-how-to/statistical-modeling/multivariate/how-to/principal-components/interpret-the-results/all-statistics-and-graphs/]
- 30. Understanding Scores and Loadings • LearnPCA [https://bryanhanson.github.io/LearnPCA/articles/Vig_04_Scores_Loadings.html]
- 31. Benchmarking of dimensionality reduction methods to ... [https://pubmed.ncbi.nlm.nih.gov/40890192/]
- 32. Protein Dynamics and Drug Discovery: elevate your project ... [https://cresset-group.com/science/science-resources/drug-discovery-principal-component-analysis/]
- 33. Principal component analysis: a review and recent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4792409/]
- 34. A Benchmarking Study of Random Projections and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11838541/]
- 35. Single‑cell RNA sequencing data (Review) dimensionality reduction [https://www.spandidos-publications.com/10.3892/wasj.2025.315?text=fulltext]
- 36. Decoding Dimensionality : Reduction , UMAP, and... | Medium PCA [https://medium.com/@naila.srivastava/decoding-dimensionality-reduction-pca-umap-and-t-sne-in-bioinformatics-38a7f93b1af1]
- 37. Principal component regression [https://en.wikipedia.org/wiki/Principal_component_regression]
- 38. Top 12 Dimensionality Reduction Techniques for Machine ... [https://encord.com/blog/dimentionality-reduction-techniques-machine-learning/]
- 39. Comparison and Evaluation of Classical Dimensionality ... [https://drpress.org/ojs/index.php/HSET/article/view/13890]
- 40. Principal Component Analysis Made Easy: A Step-by- ... [https://medium.com/data-science/principal-component-analysis-made-easy-a-step-by-step-tutorial-184f295e97fe]
- 41. What Is Principal Component Analysis (PCA)? [https://www.ibm.com/think/topics/principal-component-analysis]
- 42. Principal Component Analysis (PCA) [https://www.analyticsvidhya.com/blog/2016/03/pca-practical-guide-principal-component-analysis-python/]
- 43. Why do we need to normalize data before principal ... [https://stats.stackexchange.com/questions/69157/why-do-we-need-to-normalize-data-before-principal-component-analysis-pca]
- 44. A Guide to Principal Component Analysis (PCA) for ... [https://www.keboola.com/blog/pca-machine-learning]
- 45. What is Principal Component Analysis (PCA)? 5-Step Guide [https://julius.ai/articles/principal-component-analysis-pca]
- 46. Benchmarking principal component analysis for large-scale ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1900-3]
- 47. GraphPCA: a fast and interpretable dimension reduction ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03429-x]
- 48. Randomized Spatial PCA (RASP): a computationally efficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11875318/]
- 49. Means and Issues for Adjusting Principal Component ... [https://www.mdpi.com/1999-4893/18/3/129]
- 50. A brief procedure for big data analysis of gene expression [https://pmc.ncbi.nlm.nih.gov/articles/PMC6388068/]
- 51. Exploring transcriptome-wide changes using PCA [https://tavareshugo.github.io/data-carpentry-rnaseq/03_rnaseq_pca.html]
- 52. The Application of Principal Component Analysis on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8438425/]
- 53. The application of principal component analysis to drug ... [https://www.sciencedirect.com/science/article/pii/S1359644617300181]
- 54. Chapter 4 Dimensionality reduction | Basics of Single-Cell ... [https://bioconductor.org/books/3.13/OSCA.basic/dimensionality-reduction.html]
- 55. Across the space: applications of spatial transcriptomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583089/]
- 56. Spatially aware dimension reduction for ... [https://www.nature.com/articles/s41467-022-34879-1]
- 57. SMOPCA: spatially aware dimension reduction integrating ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03576-9]
- 58. Randomized Spatial PCA (RASP): a computationally efficient ... [https://pubmed.ncbi.nlm.nih.gov/40034439/]
- 59. KSRV: a Kernel PCA-Based framework for inferring spatial ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1695803/full]
- 60. Integrative Sparse Principal Component Analysis of Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5912177/]
- 61. of Overview - CD Genomics Principal Component Analysis [https://bioinfo.cd-genomics.com/resource-overview-of-principal-component-analysis.html]
- 62. 10 Data Insights: PCA in Modern Biotechnology Research [https://www.numberanalytics.com/blog/pca-biotech-data-insights]
- 63. Unleashing the Power of Kernel | by KHWAB KALRA | Medium PCA [https://medium.com/@khwabkalra1/unleashing-the-power-of-kernel-pca-bce7f4d2923d]
- 64. New bandwidth selection criterion for Kernel : Approach to... PCA [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-137]
- 65. - Wikipedia Dimensionality reduction [https://en.wikipedia.org/wiki/Dimensionality_reduction]
- 66. Dimensionality reduction by UMAP reinforces sample ... [https://www.sciencedirect.com/science/article/pii/S2211124721008597]
- 67. Exploring combinations of dimensionality reduction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11046904/]
- 68. Mastering PCA for Cognitive Insights [https://www.numberanalytics.com/blog/mastering-pca-cognitive-insights]
- 69. A Survey: Potential Dimensionality Reduction Methods For Data... [https://arxiv.org/html/2502.11036v2]
- 70. Sparse dimensionality reduction for analyzing single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12527234/]
- 71. How Overfitting Ruins Your Feature Selection - Hex [https://hex.tech/blog/overfitting-model-impact/]
- 72. Overfitting In Academic Research [https://www.meegle.com/en_us/topics/overfitting/overfitting-in-academic-research]
- 73. ( PCA ) | ABI Bioinformatics Guide 2024 Principal Component Analysis [https://edu.abi.am/statistics-theory/dimentionality-reduction/pca-principal-component-analysis]
- 74. Mastering PCA for Data Analysis [https://www.numberanalytics.com/blog/mastering-pca-data-analysis]
- 75. Optimizing high dimensional data classification with a ... [https://www.nature.com/articles/s41598-025-08699-4]
- 76. GitHub - farhanrn/DNA-Dimentionality- Reduction - PCA [https://github.com/farhanrn/DNA-Dimentionality-Reduction-PCA]
- 77. How to Mitigate Overfitting by Creating Ensembles [https://www.geeksforgeeks.org/machine-learning/how-to-mitigate-overfitting-by-creating-ensembles/]
- 78. PCA for RNA-Seq [https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/2200/index.php?manual=PCA_RNA_Seq.html]
- 79. Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model | Genome Biology | Full Text [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1861-6]
- 80. Low-dimensionality in gene expression data enables the accurate extraction of transcriptional programs from shallow sequencing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4856162/]
- 81. Dimensionality Reduction [https://nbisweden.github.io/workshop-scRNAseq/labs/seurat/seurat_02_dimred.html]
- 82. Dimensionality reduction methods for extracting functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10632734/]
- 83. Benchmarking of dimensionality reduction methods to ... [https://www.nature.com/articles/s41598-025-12021-7]
- 84. PCA - Principal Component Analysis Essentials - Articles [https://www.sthda.com/english/articles/31-principal-component-methods-in-r-practical-guide/112-pca-principal-component-analysis-essentials/]
- 85. Optimized hybrid investigative based dimensionality reduction ... [https://journalofbigdata.springeropen.com/articles/10.1186/s40537-021-00415-z]
- 86. Potential Dimensionality Reduction Methods For Data ... [https://arxiv.org/html/2502.11036]
- 87. k-Nearest neighbor models for microarray gene expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2920072/]
- 88. Designing penalty functions in high dimensional problems [https://pmc.ncbi.nlm.nih.gov/articles/PMC5628772/]
- 89. Model-based dimensionality reduction for single-cell RNA- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12342792/]
- 90. Benchmark principal component analysis (PCA) of scRNA- ... [https://slowkow.com/notes/pca-benchmark/]
- 91. Compositional data modeling of high-dimensional single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12539173/]
- 92. Unlocking Neuroscience with PCA [https://www.numberanalytics.com/blog/principal-component-analysis-neuroscience-guide]
- 93. Understanding Principal ( Component )... | Medium Analysis PCA [https://medium.com/@f2021376111/introduction-unveiling-the-power-of-dimensionality-reduction-e39123c1f41f]
- 94. Optimizing PCA for Health and Care Research : A Reliable Approach to... [https://arxiv.org/html/2503.24248v1]
- 95. Adjusted Rand Index (ARI) Overview [https://www.emergentmind.com/topics/adjusted-rand-index-ari]
- 96. The rise of nonnegative matrix factorization: Algorithms and ... [https://www.sciencedirect.com/science/article/abs/pii/S0306437924000371]
- 97. Stretched non-negative matrix factorization [https://www.nature.com/articles/s41524-024-01377-5]
- 98. Dimension reduction techniques for the integrative analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4945831/]
- 99. Machine learning approach to identify significant genes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12547461/]
- 100. Comparative study of nonlinear hard and soft ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25030826]
- 101. Dimensionality Reduction: Dive into PCA, t-SNE, and UMAP [https://medium.com/@prathik.codes/dimensionality-reduction-dive-into-pca-t-sne-and-umap-9af37f6d9cb3]
- 102. PCA vs UMAP vs t-SNE [https://biostatsquid.com/pca-umap-tsne-comparison/]
- 103. Benchmarking Dimensionality Reduction Techniques ... [https://arxiv.org/html/2509.13344v1]
- 104. Research on a multivariate measurement system for ... [https://www.nature.com/articles/s41598-025-23236-z]
- 105. Transcriptomic profiling of endothelial progenitor cells in post ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12595092/]
- 106. Integrated multi-sample transcriptomic analysis of COVID- ... [https://www.nature.com/articles/s41598-025-03640-1]
- 107. Independent Principal Component Analysis for biologically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3298499/]
- 108. Biases of Principal Component Analysis (PCA) in Physical ... [https://elifesciences.org/reviewed-preprints/94685]