Improving Loop Modeling Accuracy in Homology Models: A Guide for Biomedical Researchers
Accurate loop modeling remains a critical challenge in homology modeling, directly impacting the reliability of protein structures used in drug design and functional analysis.
Improving Loop Modeling Accuracy in Homology Models: A Guide for Biomedical Researchers
Abstract
Accurate loop modeling remains a critical challenge in homology modeling, directly impacting the reliability of protein structures used in drug design and functional analysis. This article provides a comprehensive guide for researchers and drug development professionals, exploring the foundational principles behind loop modeling difficulties and presenting a detailed examination of current methodologies, from data-based fragment assembly to advanced machine learning techniques. We offer practical troubleshooting strategies for refining models and a rigorous framework for validation and comparative assessment, synthesizing the latest advances in the field to empower scientists in building more trustworthy structural models for their biomedical research.
Why Loops Are the Critical Challenge in Protein Structure Prediction
The Critical Role of Loops in Protein Function and Drug Binding
Frequently Asked Questions (FAQs)
FAQ 1: Why is loop prediction so critical in structure-based drug design?
Flexible loop regions are often directly involved in ligand binding and molecular recognition. Using an incorrect loop configuration in a structural model can be detrimental to drug design studies if that loop is capable of interacting with the ligand. The inherent flexibility of loops means they can adopt alternative configurations upon ligand binding, a process described by the conformational selection model. This model posits that the apo (ligand-free) form of the protein samples higher energy conformations, and the ligand selectively binds to and stabilizes a holo-like conformation [1].
FAQ 2: What are the main challenges in accurately modeling loop regions?
Accurately modeling loops remains difficult for several key reasons [1]:
- Inherent Flexibility: Loops often lack regular secondary structure, making their conformation hard to predict.
- Scoring Function Limitations: No single scoring function consistently ranks native-like loop configurations accurately across different protein systems. The optimal choice often depends on the specific system being studied.
- Ligand-Induced Changes: Modeling how loop conformations change upon ligand binding adds a layer of complexity.
- Data Gaps: Due to their flexibility, loop regions are frequently missing from experimentally solved protein structures, creating gaps in homology models.
FAQ 3: How can I improve the ranking of native-like loop configurations in my predictions?
Research indicates that physics-based scoring functions can offer improvements. Studies comparing scoring methods have found that single snapshot Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) scoring often provides better ranking of native-like loop configurations compared to statistically-based functions like DFIRE. Furthermore, re-ranking predicted loops in the presence of the bound ligand can sometimes yield more accurate results [1].
FAQ 4: Are there new computational methods that help with modeling flexible loops in specific conformations?
Yes, recent advances allow the integration of experimental data or biological hypotheses to guide loop and protein conformation. Distance-AF is one such method built upon AlphaFold2. It allows users to specify distance constraints between residues, which are incorporated into the loss function during structure prediction. This is particularly useful for [2]:
- Fitting protein structures into cryo-electron microscopy density maps.
- Modeling specific functional states (e.g., active vs. inactive conformations of GPCRs).
- Generating ensembles that satisfy NMR-derived distance restraints.
Troubleshooting Guides
Issue 1: Low Accuracy in Predicted Loop Conformations
Problem: Your homology model has a loop region that is predicted with low accuracy, and you suspect it is impacting your analysis of a binding site.
Solution:
- Generate an Ensemble: Use a specialized loop prediction program like CorLps to generate an ensemble of hundreds to thousands of possible loop conformations [1].
- Apply Multiple Scoring Functions: Do not rely on a single scoring function. The original DFIRE score in CorLps may not rank the best loop highly. Re-rank the top loop decoys using alternative functions like MM/GBSA or the optimized MM/GBSA-dsr [1].
- Consider the Ligand: If the ligand is known, re-score the loop ensembles in the presence of the ligand to identify conformations that are complementary to the binder.
- Incorporate Experimental Data: If available, use experimental constraints (e.g., from cross-linking mass spectrometry, NMR, or cryo-EM maps) with tools like Distance-AF to guide the prediction towards a biologically relevant conformation [2].
Issue 2: Modeling Loop-Ligand Interactions
Problem: You are studying a protein-ligand complex where a flexible loop is known to interact with the ligand, but your static model doesn't capture this interaction well.
Solution:
- Adopt an Ensemble Docking Approach: Instead of docking into a single rigid structure, use an ensemble of low-energy loop conformations generated from the apo protein structure. This helps mimic the conformational selection mechanism of binding [1].
- Explore De Novo Loop Design: For creating entirely new binders, consider designs that incorporate structured, buttressed loops. Recent methods allow for the de novo design of proteins with multiple long loops (9-14 residues) that are stabilized by extensive hydrogen-bonding networks. These designed loops can form one side of an extended binding pocket for peptides or other ligands [3].
- Identify "Hot Loops": Use comprehensive analysis tools like LoopFinder to identify interface loops that contribute significantly to binding energy (so-called "hot loops"). Mimicking these critical loops with synthetic macrocycles or constrained peptides is a promising strategy for inhibitor design [4].
Experimental Protocols
Protocol 1: Loop Prediction and Ranking Using CorLps
This protocol outlines the steps for predicting and selecting loop conformations for a region of interest [1].
1. Input Preparation:
- Obtain your protein structure (e.g., a homology model) and identify the loop residues to be predicted.
- Remove all co-factors, water molecules, and co-crystallized ligands from the structure file.
2. Loop Conformation Generation:
- Use the CorLps program, which employs the ab initio predictor loopy.
- Initialize the run to generate a large ensemble of loop conformations (e.g., 10,000).
- Output the top several hundred energetically favorable loops.
3. Clustering and Uniqueness:
- Use a Quality Threshold (QT) clustering algorithm to remove redundant conformations.
- Set a maximum cluster diameter (e.g., 1-2 Å) to produce a set of several hundred unique loop conformations.
4. Side-Chain Optimization:
- Repack the side chains of residues within a 6 Å zone around the predicted loop region using a tool like SCAP.
5. Initial Ranking:
- Rank the top 100 predicted loop configurations using the DFIRE scoring function. This is your DFIRE ensemble.
6. Advanced Re-ranking:
- Re-rank the DFIRE ensemble using more sophisticated scoring functions like MM/GBSA or Optimized MM/GBSA-dsr.
- Perform this re-ranking both with and without the bound ligand present to identify the best candidate.
Protocol 2: Identifying Critical Binding Loops with LoopFinder
This protocol describes how to use LoopFinder to systematically identify loops that are critical for protein-protein interactions, which can be prime targets for inhibitor design [4].
1. Data Collection:
- Acquire multi-chain protein structures from the PDB, filtered by resolution (e.g., ≤ 4Å) and sequence identity (e.g., < 90%).
2. Loop Identification:
- Run LoopFinder with customizable parameters. A standard setup includes:
- Loop length: 4 to 8 consecutive amino acids.
- Interface proximity: At least 80% of loop residues must have an atom within 6.5 Å of the binding partner.
- Loop geometry: A distance cutoff between Cα atoms of the loop termini (e.g., 6.2 Å) to ensure a true loop conformation and exclude extended secondary structures.
3. Energy Analysis:
- Perform computational alanine scanning on all identified loops using Rosetta or PyRosetta.
- Calculate the predicted ΔΔG for each residue upon mutation to alanine.
4. "Hot Loop" Selection:
- Identify "hot loops" by applying filters such as:
- Loops containing two consecutive hot spot residues (ΔΔG ≥ 1 kcal/mol).
- Loops with at least three hot spot residues.
- Loops where the average ΔΔG per residue is greater than 1 kcal/mol.
Data Presentation
Table 1: Comparison of Scoring Functions for Loop Prediction
This table summarizes the performance of different scoring functions evaluated for ranking native-like loop configurations in a scientific study [1].
| Scoring Function | Type | Key Principle | Reported Performance in Loop Ranking |
|---|---|---|---|
| DFIRE | Statistical | Knowledge-based potential derived from known protein structures. | Did not accurately rank native-like loops in tested systems. |
| MM/GBSA | Physics-based | Molecular Mechanics combined with Generalized Born and Surface Area solvation. | Provided the best ranking of native-like loop configurations in general. |
| Optimized MM/GBSA-dsr | Physics-based | MM/GBSA optimized for decoy structure refinement. | Performance was system-dependent; not consistently superior to standard MM/GBSA. |
Table 2: Research Reagent Solutions for Loop Modeling
This table lists key computational tools and resources essential for researching protein loops.
| Tool/Resource Name | Type | Primary Function in Loop Research |
|---|---|---|
| CorLps [1] | Software Suite | Performs ab initio loop prediction and generates ensembles of loop conformations. |
| Rosetta/PyRosetta [4] | Software Suite | Used for computational alanine scanning and energy calculations to identify critical "hot spot" residues in loops. |
| AlphaFold2 & Variants [5] [2] | AI Structure Prediction | Provides highly accurate initial models; variants like Distance-AF can incorporate constraints for modeling specific loop conformations. |
| LoopFinder [4] | Analysis Algorithm | Comprehensively identifies and analyzes peptide loops at protein-protein interfaces within the entire PDB. |
| PDB | Database | Source of experimental protein structures for analysis and use as templates in homology modeling [1] [6]. |
| ATLAS, GPCRmd | Specialized Database | Databases of molecular dynamics trajectories for analyzing loop and protein dynamics [6]. |
Workflow and Relationship Visualizations
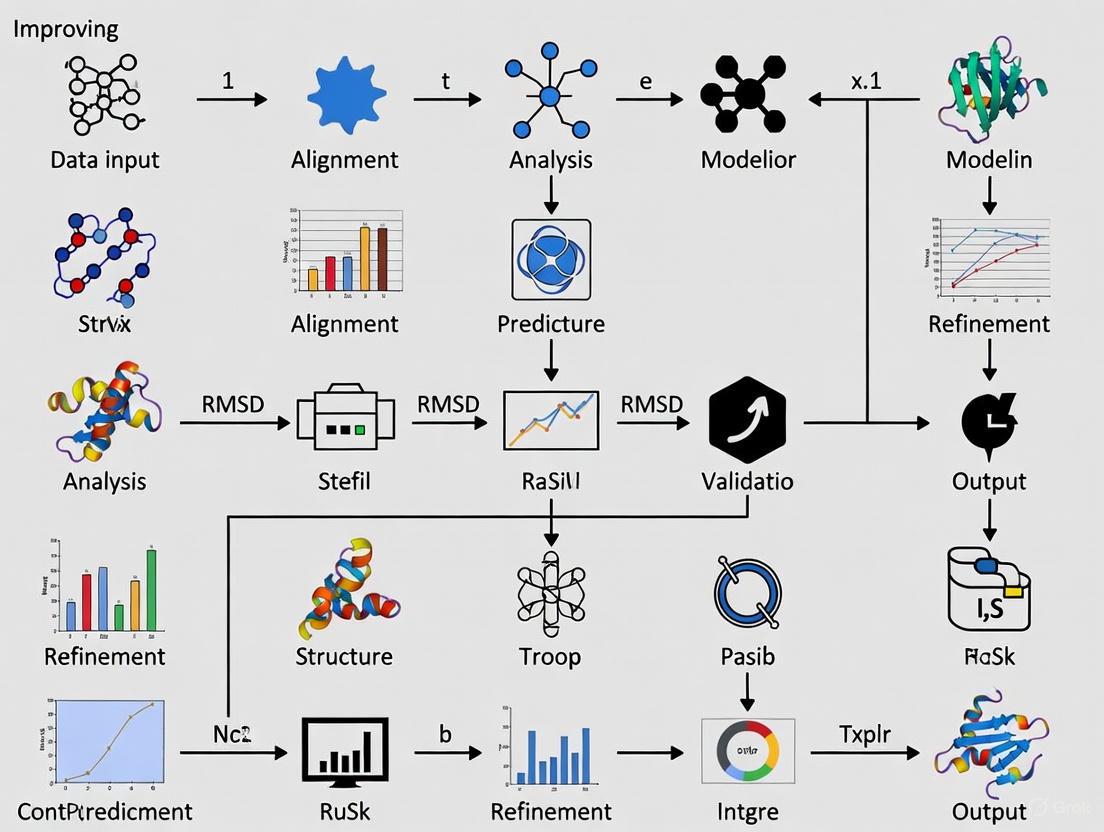
Loop Prediction and Validation Workflow
Scoring Function Decision Guide
In protein structure prediction, the "conformational sampling problem" refers to the significant challenge of efficiently exploring the vast number of possible three-dimensional structures that flexible protein regions can adopt. This problem is particularly acute in loop modeling, where even short loops can exhibit remarkable flexibility, making them difficult to predict accurately in homology models. Loops often play critical functional roles in ligand binding, catalysis, and molecular recognition, making their accurate modeling essential for reliable structure-based drug design [7] [8].
The core of the problem lies in the astronomical number of possible conformations a protein loop can sample. With each residue having multiple torsion angles that can rotate, the conformational space grows exponentially with loop length. Computational methods must navigate this high-dimensional energy landscape to identify biologically relevant structures from among countless possibilities [9]. While molecular dynamics (MD) simulations can, in principle, characterize conformational states and transitions, the energy barriers between states can be high, preventing efficient sampling without substantial computational resources [9].
Frequently Asked Questions (FAQs)
Q1: Why is loop modeling particularly challenging in homology modeling?
Loop modeling represents a "mini protein folding problem" under geometric constraints. The challenge arises from the high flexibility of loops compared to structured secondary elements, the exponential increase in possible conformations with loop length, and the difficulty in accurately scoring which conformations are biologically relevant. Even in high-identity homology modeling, loops often differ significantly between homologous proteins, necessitating ab initio prediction methods [7] [8].
Q2: What are the key limitations in current conformational sampling methods?
The primary limitations include:
- Computational expense: Comprehensive sampling requires significant resources, especially for longer loops (>8 residues)
- Force field accuracy: Inaccurate energy functions may favor non-native conformations
- Sampling barriers: High energy barriers between conformational states impede efficient exploration
- Timescale gap: Biologically relevant conformational changes often occur on timescales (microseconds to milliseconds) that are challenging for standard molecular dynamics [7] [9]
Q3: How can I improve sampling for long loops (≥12 residues)?
For longer loops, consider:
- Implementing enhanced sampling techniques like replica-exchange molecular dynamics
- Using specialized loop modeling software like PLOP or LoopBuilder that combine extensive sampling with sophisticated scoring
- Incorporating experimental restraints from NMR or cryo-EM when available
- Applying collective variable biasing to focus sampling on relevant conformational subspaces [7] [9] [10]
Q4: What is the relationship between conformational sampling and scoring functions?
Sampling and scoring are intrinsically linked—extensive sampling is useless without accurate scoring to identify native-like conformations, while perfect scoring functions cannot compensate for poor sampling that misses near-native conformations. The most successful protocols apply hierarchical filtering, using fast statistical potentials initially followed by more computationally expensive all-atom force fields for final ranking [7].
Troubleshooting Common Sampling Issues
Table 1: Common Conformational Sampling Problems and Solutions
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| Sampling stuck in local minima | High energy barriers; Inadequate sampling algorithm | Implement replica-exchange MD; Use torsion angle dynamics; Apply biasing methods [9] [10] |
| Poor loop closure | Insufficient closure algorithms; Steric clashes | Employ advanced closure methods like Direct Tweak; Check for loop-protein interactions during closure [7] |
| Long computation times for sampling | Inefficient sampling method; Too many degrees of freedom | Use hierarchical filtering with DFIRE potential; Implement internal coordinate methods; Focus sampling on relevant collective variables [7] [11] [9] |
| Accurate sampling but poor final selection | Inadequate scoring function; Insufficient final refinement | Apply molecular mechanics force field minimization (e.g., OPLS); Use multiple scoring functions; Include solvation effects [7] |
| Difficulty sampling specific loop conformations | Limited template diversity; Incorrect initial alignment | Blend sequence- and structure-based alignments; Utilize multiple templates; Account for crystal contacts when available [7] [12] |
Table 2: Typical Loop Modeling Accuracy by Method and Loop Length
| Loop Length | LOOPY (Å) | RAPPER (Å) | Rosetta (Å) | PLOP (Å) | PLOP II (with crystal contacts) (Å) |
|---|---|---|---|---|---|
| 8 residues | 1.45 | 2.28 | 1.45 | 0.84 | NA |
| 10 residues | 2.21 | 3.48 | NA | 1.22 | NA |
| 12 residues | 3.42 | 4.99 | 3.62 | 2.28 | 1.15 |
Table 3: Performance Comparison of Enhanced Sampling Methods
| Method | Sampling Approach | Best Application | Computational Efficiency |
|---|---|---|---|
| Torsion Angle MD (GNEIMO) | Freezes bond lengths/angles; samples torsion space | Domain motions; conformational transitions | High (allows 5 fs timesteps) [10] |
| Replica-Exchange MD | Parallel simulations at different temperatures | Overcoming energy barriers; exploring substates | Moderate to high (parallelizable) [10] |
| Collective Variable Biasing | Bias potential along defined coordinates | Focused sampling of specific transitions | Variable (depends on CV quality) [9] |
| Metadynamics | History-dependent bias potential | Free energy calculations; barrier crossing | Moderate (requires careful parameterization) [9] |
Experimental Protocols for Enhanced Conformational Sampling
LoopBuilder Protocol for High-Accuracy Loop Modeling
The LoopBuilder protocol employs a hierarchical approach combining extensive sampling with sophisticated filtering and refinement:
Initial Sampling Phase:
- Use the Direct Tweak algorithm to generate sterically feasible backbone conformations
- Check for interactions between the loop and the rest of the protein during closure
- Generate an ensemble of at least 10,000 initial conformations for an 8-residue loop
Statistical Potential Filtering:
- Apply the DFIRE statistical potential to select a subset (typically 5-10%) of conformations closest to native structure
- This filtering step significantly reduces computational time for subsequent refinement
All-Atom Refinement:
- Perform energy minimization using the OPLS/SBG-NP force field
- Include generalized Born solvation terms
- Rank final conformations based on force field energy
This protocol has been shown to achieve prediction accuracies of 0.84 Å RMSD for 8-residue loops and maintains reasonable accuracy (<2.3 Å) for loops up to 12 residues [7].
Torsion Angle Molecular Dynamics with GNEIMO
The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method enhances sampling by focusing on torsional degrees of freedom:
System Preparation:
- Partition protein into rigid clusters connected by hinges
- Freeze bond lengths and bond angles via holonomic constraints
- Use implicit solvation model (Generalized Born) with interior dielectric constant of 4.0
Simulation Parameters:
- Integration timestep: 5 fs (compared to 1-2 fs for Cartesian MD)
- Temperature: 310 K maintained with Nosé-Hoover thermostat
- Nonbonded forces: switched off at 20 Å cutoff
- For enhanced sampling: Combine with replica-exchange (REXMD)
Analysis:
- Monitor torsional angle distributions
- Calculate RMSD to known experimental structures
- Identify conformational substates using cluster analysis
This approach has successfully sampled conformational transitions in flexible proteins like fasciculin and calmodulin that are rarely observed in conventional Cartesian MD simulations [10].
Multi-Template Hybridization for Low-Identity Homology Modeling
For modeling loops when template identity is below 40%:
Template Selection and Alignment:
- Select multiple templates (typically 3-5) covering different conformational states
- Create structure-based alignments focusing on conserved structural motifs
- Manually curate loop alignments based on Cα to Cβ vectors and secondary structure
Hybrid Model Building:
- Maintain all templates in defined global geometry
- Randomly swap template segments using Monte Carlo sampling
- Simultaneously incorporate peptide fragments from PDB-derived library
Loop Refinement:
- Close loops simultaneously during template swapping
- Apply specialized loop scoring terms
- Use iterative refinement cycles with increasing spatial restraints
This protocol has enabled accurate modeling of GPCRs using templates as low as 20% sequence identity [12].
Essential Research Reagent Solutions
Table 4: Key Software Tools for Conformational Sampling
| Tool Name | Primary Function | Application in Loop Modeling |
|---|---|---|
| PLOP | Systematic dihedral-angle build-up with OPLS force field | High-accuracy loop prediction; Long loops with crystal contacts [7] |
| LoopBuilder | Hierarchical sampling, filtering, and refinement | Balanced accuracy and efficiency for medium-length loops [7] |
| GNEIMO | Torsion angle molecular dynamics | Enhanced sampling of conformational transitions; Domain motions [10] |
| Rosetta | Fragment assembly with Monte Carlo | Multiple template hybridization; Low-identity homology modeling [12] |
| MDAnalysis | Trajectory analysis and toolkit development | Analysis of sampling completeness; Custom analysis scripts [13] |
| Modeller | Comparative modeling with spatial restraints | Standard homology modeling; Multiple template approaches [12] |
Table 5: Force Fields and Scoring Functions for Loop Modeling
| Scoring Method | Type | Best Use Case |
|---|---|---|
| DFIRE | Statistical potential | Initial filtering of loop conformations [7] |
| OPLS/SBG-NP | All-atom force field with implicit solvation | Final ranking and refinement [7] |
| AMBER ff99SB | All-atom force field | Cartesian and torsion dynamics simulations [10] |
| Rosetta Scoring Function | Mixed statistical/physico-chemical | Template hybridization and fragment assembly [12] |
| RAPDF | Statistical potential | Rapid screening of loop ensembles [7] |
Workflow and Pathway Visualizations
Loop Modeling Sampling Workflow
Enhanced Sampling Method Classification
Sampling Problem Troubleshooting Guide
Frequently Asked Questions (FAQs)
1. What is a "template gap" in homology modeling? A template gap occurs when there are regions in your target protein sequence, often loops or flexible domains, that have no structurally similar or homologous counterpart in the available template structures. This is most problematic in low-homology regions where sequence identity is low, leading to alignment errors and inaccurate models [14] [15].
2. Why are loops, especially the H3 loop in antibodies, particularly hard to model? Loops are often surface-exposed and flexible, exhibiting great diversity in length, sequence, and structure. The H3 loop in antibodies is especially challenging because its conformation is not determined by a robust canonical structure model like the other five complementary determining region (CDR) loops. Its high variability makes finding suitable templates difficult, and ab initio methods are hampered by an incomplete understanding of the physicochemical principles governing its structure [16].
3. When should I consider using multiple templates? Using multiple templates can be beneficial when no single template provides good coverage for all regions of your target protein. It can help model different domains from their best structural representatives and extend model coverage. However, use it judiciously, as automatic inclusion of multiple templates does not guarantee improvement and can sometimes produce models worse than the best single-template model [14].
4. How can I improve the accuracy of my loop models? For critical loops, especially in applications like antibody engineering, consider specialized methods. One effective approach uses machine learning (e.g., Random Forest) to select structural templates based not only on sequence similarity but also on structural features and the likelihood of specific interactions between the loop and the rest of the protein framework [16].
5. What are the key steps in a standard homology modeling workflow? The classical steps are: (1) Template identification and selection, (2) Target-Template alignment, (3) Model building, (4) Loop modeling, (5) Side-chain modeling, (6) Model optimization, and (7) Model validation [15].
Troubleshooting Guides
Problem: Poor Model Quality in Specific Loop Regions
Symptoms: High root-mean-square deviation (RMSD) in loop regions when comparing your model to a later-determined experimental structure; steric clashes or unusual torsion angles in loops.
Solution: Implement a specialized loop modeling protocol.
Experimental Protocol: Machine Learning-Based Template Selection for Loops
This methodology is adapted from a successful approach for antibody H3 loops and can be generalized for other difficult loops [16].
Create a Non-Redundant Loop Structure Library
- Action: Scan the PDB for all proteins (or a specific subset like antibodies) using tools like HMMER with profile Hidden Markov Models (HMMs) [16] [15].
- Quality Control: Filter structures for resolution (e.g., better than 3.0 Å) using a server like PISCES [16].
- Clustering: Cluster the remaining loops based on sequence identity (e.g., 90% threshold) using a tool like CD-HIT. Select one representative structure from each cluster to create a non-redundant dataset [16].
Feature Extraction for Machine Learning
- Action: For a target loop, calculate features against all loops in your library. Key features include:
- Sequence similarity scores.
- Loop length compatibility.
- Presence of key residue interactions (e.g., with the framework).
- Environment-specific substitution scores [16].
- Action: For a target loop, calculate features against all loops in your library. Key features include:
Train a Random Forest Model for Prediction
- Action: Use the R package
randomForestto train a regression model. The input features (from Step 2) are used to predict the 3D structural similarity (e.g., TM-score) between loop pairs [16]. - Output: The model will predict the most structurally similar loop(s) from your library for your target sequence.
- Action: Use the R package
Build and Rank Models
- Action: Use the top-ranked template loops to build your loop model.
- Validation: Rank the resulting models using a scoring function that evaluates intramolecular interactions. The quality estimate provided by the Random Forest model can also be a reliable indicator [16].
Problem: Deciding When to Use Multiple Templates
Symptoms: Your single-template model has regions with no structural coordinates ("gaps") or regions that are known to be inaccurate.
Solution: A strategic multi-template approach.
Experimental Protocol: Strategic Multi-Template Modeling
Generate High-Quality Input Alignments
Build and Evaluate Single-Template Models
Build Multi-Template Models
- Action: Feed the alignments from Step 1 into a modeling program capable of using multiple templates (e.g., Modeller). Limit the number of templates to 2 or 3, as performance often degrades with more templates [14].
Compare and Select the Final Model
- Action: Critically compare the multi-template model to your best single-template model.
- Core Residue Analysis: Compare the quality only for the residues that were present in your best single-template model. This removes the "trivial" improvement from simply extending the model length and shows if the multi-template approach genuinely improved the core structure [14].
- Final Selection: Use an MQAP like ProQ to rank the models. If the multi-template model scores significantly higher, select it. If not, the single-template model may be your best option [14].
The following workflow diagram summarizes the decision process for addressing template gaps:
Quantitative Data on Multi-Template Modeling
The table below summarizes findings from a systematic study on the impact of using multiple templates on model quality, measured by TM-score (a measure of structural similarity where 1.0 is a perfect match) [14].
Table 1: Impact of Multiple Templates on Model Quality (TM-score)
| Number of Templates | Modeling Program | Average TM-Score Change (All Residues) | Average TM-Score Change (Core Residues Only) | Notes |
|---|---|---|---|---|
| 1 | Nest | Baseline | Baseline | Slightly better single-template models than Modeller. |
| 2-3 | Modeller | +0.01 | Slight Improvement | Optimal range for Modeller. Most improved cases produced by Modeller. |
| >3 | Modeller, Nest | Gradual Decrease | Gets Worse | Lower-ranked alignments often of poor quality. |
| 3 | Pfrag (Average) | Largest Improvement | Gets Worse | - |
| All Available | Pfrag (Shotgun) | Continuous Improvement | Gets Worse | Less peak improvement than Modeller/Nest. |
Research Reagent Solutions
Table 2: Key Software Tools for Addressing Template Gaps
| Tool Name | Function | Application in Addressing Template Gaps |
|---|---|---|
| HHblits [15] | HMM-HMM-based lightning-fast iterative sequence search | Generates more accurate target-template alignments for low-homology regions compared to sequence-based methods. |
| Modeller [14] [15] | Homology modeling | A standard program that can utilize multiple templates for model building; performs best with 2-3 templates. |
| Rosetta Antibody [16] | Antibody-specific structure prediction | Combines template selection with ab initio CDR H3 loop modeling; a benchmark for antibody loop prediction. |
| FREAD [16] | Fragment-based loop prediction | Identifies best loop fragments using local sequence and geometric matches from a database. |
| Random Forest (R package) [16] | Machine Learning Algorithm | Can be trained to select the best structural templates for difficult loops based on sequence and structural features. |
| ProQ [14] | Model Quality Assessment Program | Ranks models and identifies the best one from a set of predictions; crucial for selecting between single and multi-template models. |
| TM-score [14] [16] | Structural Comparison Metric | Used to evaluate model quality; weights shorter distances more heavily than RMSD, providing a more global measure of similarity. |
Frequently Asked Questions (FAQs) and Troubleshooting Guide
This technical support resource addresses common challenges researchers face in protein structure modeling, with a specific focus on improving loop modeling accuracy within homology modeling projects.
The accuracy of loop modeling is highly dependent on the length of the loop and the methodological approach used. A major source of error is selecting an inappropriate method for a given loop length. The table below summarizes a quantitative performance comparison of different methods across various loop lengths, based on a multi-method study [17].
Table 1: Loop Modeling Method Performance by Loop Length
| Loop Length (residues) | Recommended Method | Average Accuracy (Å) | Key Rationale |
|---|---|---|---|
| 4 - 8 | MODELLER | Lower RMSD | Superior for short loops via spatial restraints and energy minimization [17]. |
| 9 - 12 | Hybrid (MODELLER+CABS) | Intermediate RMSD | Combination improves accuracy over individual methods [17]. |
| 13 - 25 | CABS or ROSETTA | Higher RMSD (2-6 Å) | Coarse-grained de novo modeling more effective for conformational search of long loops [17]. |
FAQ 2: Why are my secondary structure predictions often inaccurate for specific regions, and could this indicate a problem?
Inconsistent or inaccurate secondary structure predictions can be a significant troubleshooting clue. While standard predictors like JPred, PSIPRED, and SPIDER2 are highly accurate for most single-structure proteins, they are designed to output one best guess. If a protein region can adopt two different folds (a Fold-Switching Region or FSR), the prediction will conflict with at least one of the experimental structures [18].
- Quantitative Evidence: One study found that secondary structure prediction accuracies (Q3 scores) for known FSRs were significantly lower (mean Q3 < 0.70) than for non-fold-switching regions (mean Q3 > 0.85) [18].
- Troubleshooting Action: If you observe a region with consistently poor secondary structure prediction across different algorithms, consult the literature for evidence of metamorphic behavior or intrinsic disorder. This region may require modeling in multiple conformations [18].
FAQ 3: How can I validate and refine side-chain packing in my model?
Inaccurate side-chain packing is a common issue that can affect the analysis of binding sites and protein interactions.
- Use of Advanced Suites: Leverage all-atom structure prediction tools like AlphaFold, which explicitly models the 3D coordinates of all heavy atoms, including side chains, with high accuracy [19]. Similarly, RoseTTAFold uses a "three-track" neural network that simultaneously reasons about sequence, distance, and 3D structure, leading to improved side-chain packing [20].
- Energy Minimization: Use refinement suites like Rosetta to repack side chains and perform energy minimization. This is a standard step in the third stage of classical homology modeling to improve the physical realism and local geometry of the model [21] [17].
Experimental Protocols for Loop Modeling
Protocol 1: Multi-Method Loop Modeling Workflow
This protocol is adapted from a comparative study that recommends methods based on loop length [17].
Objective: To accurately model a loop region of a protein using the most effective method for its length.
Materials:
- Template structure with the missing loop region.
- Target sequence.
- Software: MODELLER, ROSETTA, and/or CABS.
- Computing cluster or high-performance computer.
Methodology:
- Loop Definition: Identify the start and end residues of the loop to be modeled and its length.
- Method Selection: Refer to Table 1 above to choose the appropriate modeling method.
- Model Generation:
- For short loops (4-8 residues): Use MODELLER with default parameters to generate multiple models (e.g., 500). Select the top model based on the Discrete Optimized Protein Energy (DOPE) score [17].
- For medium loops (9-12 residues): Generate an initial set of models with MODELLER. Select the top 10 ranked models and use them as multiple templates for a subsequent CABS modeling run to refine the loop conformation [17].
- For long loops (13-25 residues): Use a coarse-grained de novo method like CABS or the loop modeling protocols in ROSETTA for extensive conformational sampling [17].
- Validation: Superimpose the model onto the template structure and calculate the Root-Mean-Square Deviation (RMSD) of the loop's Cα atoms. Assess steric clashes and side-chain rotamer合理性.
The following diagram illustrates this multi-method workflow:
Protocol 2: Identifying Fold-Switching Regions via Secondary Structure Prediction Inconsistencies
Objective: To identify protein regions that may adopt alternative secondary structures, which could complicate modeling efforts [18].
Materials:
- Protein amino acid sequence.
- Access to secondary structure prediction servers (e.g., JPred, PSIPRED, SPIDER2).
Methodology:
- Prediction: Run the target protein sequence through at least three different secondary structure prediction servers (e.g., JPred, PSIPRED, SPIDER2).
- Comparison: Align all prediction outputs and the secondary structure from your experimental template or reference structure.
- Analysis: Identify regions with significant discrepancies. Pay particular attention to HE (helix-to-strand) and EC (strand-to-coil) errors, as these are strong indicators of potential fold-switching [18].
- Interpretation: A protein region with a consensus Q3 accuracy score below 0.60 from multiple predictors is a strong candidate for being a Fold-Switching Region and warrants further literature investigation [18].
The Scientist's Toolkit: Key Research Reagents and Software
Table 2: Essential Computational Tools for Structure Modeling and Analysis
| Tool Name | Type | Primary Function in Modeling | Relevance to Loop/Inaccuracy Research |
|---|---|---|---|
| MODELLER [17] | Software Package | Comparative homology modeling by satisfaction of spatial restraints. | Method of choice for short loop modeling (≤8 residues) [17]. |
| ROSETTA [21] [17] | Software Suite | De novo structure prediction, loop modeling, and side-chain refinement. | Effective for long loop modeling and high-resolution refinement of side-chain packing [21] [17]. |
| CABS [17] | Software Tool | Coarse-grained de novo modeling for protein structure and dynamics. | Recommended for modeling long, challenging loops (≥13 residues) [17]. |
| AlphaFold [19] | Deep Learning Network | End-to-end 3D coordinate prediction from sequence. | Provides highly accurate backbone and side-chain models; useful as a reference or for de novo modeling [19]. |
| RoseTTAFold [20] | Deep Learning Network | Three-track neural network for integrated sequence-distance-structure prediction. | Accurate modeling of protein structures and complexes; accessible via the Robetta server [20] [22]. |
| Bio3D [23] | R Package | Analysis of ensemble of structures, PCA, and dynamics. | Comparative analysis of multiple models/conformers to assess variability and stability [23]. |
The logical relationships and data flow between these key tools in a structural bioinformatics pipeline can be visualized as follows:
The Impact of Loop Length on Prediction Difficulty and Accuracy
Frequently Asked Questions
Q1: Why are longer loops generally more difficult to predict accurately? Longer loops possess greater conformational flexibility and a larger number of degrees of freedom. This results in a more complex energy landscape with many local minima, making it challenging to identify the single, native conformation. Statistical analyses of loop banks reveal that prediction accuracy, measured by Root-Mean-Square Deviation (RMSD), systematically decreases as loop length increases [24].
Q2: What is the quantitative relationship between loop length and prediction error? Based on exhaustive analyses of loops from protein structures, the average RMSD between predicted and native loop structures shows a clear correlation with loop length. The following table summarizes the expected accuracy for canonical loop modeling approaches [24]:
| Loop Length (residues) | Average Prediction RMSD (Å) |
|---|---|
| 3 | 1.1 Å |
| 4 | ~1.5 Å |
| 5 | ~2.1 Å |
| 6 | ~2.7 Å |
| 7 | ~3.2 Å |
| 8 | 3.8 Å |
Q3: Which amino acids are over-represented in loops and why? Loop sequences are not random. Statistical analysis of loop banks shows significant over-representation of specific amino acids: Glycine (most abundant, especially in short loops), Proline, Asparagine, Serine, and Aspartate [24]. Glycine's flexibility (lacks a side chain) and Proline's rigidity (restricts backbone torsion angles) are particularly important for loop structure and nucleation.
Q4: How do modern deep learning methods like AlphaFold2 handle CDR-H3 loops in antibodies? The Complementarity Determining Region H3 (CDR-H3) loop in antibodies is notoriously diverse and difficult to predict. Deep learning methods have made significant strides. Specialized tools like H3-OPT, which combines AlphaFold2 with protein language models, have achieved an average RMSD of 2.24 Å for CDR-H3 backbone atoms against experimentally determined structures, outperforming other computational methods [25]. The Ibex model further advances this by explicitly predicting both bound (holo) and unbound (apo) conformations from a single sequence [26].
Q5: How does loop length affect the stability of non-canonical structures like G-Quadruplexes? For RNA G-Quadruplexes, thermodynamic stability can be modulated by loop length. Biophysical studies using UV melting and circular dichroism spectroscopy have systematically investigated this relationship, finding that stability is strongly influenced by the length of the loops connecting the G-tetrad stacks [27].
Troubleshooting Guides
Issue: Poor Long Loop Prediction Accuracy
Problem: Your homology model has a long loop (e.g., >8 residues) that is poorly modeled, with high RMSD or steric clashes.
Solutions:
- Utilize Deep Learning Models: For the best accuracy, use state-of-the-art structure prediction tools like AlphaFold2, AlphaFold3, or ESMFold. For antibody-specific loops, employ specialized tools like H3-OPT [25], IgFold [25], or Ibex [26].
- Leverage Advanced Template Servers: Use web servers like Phyre2.2, which incorporate deep learning. Phyre2.2 can automatically identify the most suitable AlphaFold2 model from databases to use as a template for your query sequence, often improving loop regions [28].
- Consider Conformational States: If you are modeling an antibody for drug design and suspect induced fit upon binding, use a tool like Ibex. It allows you to specify whether you want to predict the apo (unbound) or holo (bound) conformation, which can be critical for accurately modeling flexible CDR-H3 loops [26].
Issue: Loop Closure Failures
Problem: The modeled loop does not connect properly to the main protein framework, resulting in broken backbone chains or unrealistic bond geometries.
Solutions:
- Fragment-Based Assembly: Follow protocols like that in Phyre2.2, which uses a library of fragments (2-15 residues long) from the PDB. The algorithm searches for fragments that match the loop sequence and can be geometrically melded onto the framework residues flanking the loop. The final model is achieved through energy minimization to ensure proper closure [28].
- Energy Minimization and Refinement: After initial loop building, always perform all-atom energy minimization with restraints on the stable framework regions. This allows the loop and its connection points to relax into a sterically favorable and chemically reasonable configuration [24] [28].
Experimental Protocols & Data
Protocol: Benchmarking Loop Prediction Accuracy
This protocol is used in the development and evaluation of tools like H3-OPT and Ibex [25] [26].
- Dataset Curation: Compile a high-quality, non-redundant set of protein structures (e.g., from the PDB) with high-resolution X-ray crystal structures (<2.5 Å). For antibody-specific benchmarks, use datasets from SAbDab.
- Structure Prediction: Run the loop modeling or structure prediction algorithm of interest on all sequences in the benchmark dataset.
- Structural Alignment: Superimpose the predicted model onto the experimental (native) structure using the backbone atoms of the framework regions (everything except the loop being assessed).
- RMSD Calculation: Calculate the Root-Mean-Square Deviation (RMSD) of the heavy (Cα) atoms for the loop region only, after the framework alignment. This quantifies the local accuracy of the loop prediction.
- Statistical Analysis: Report the average RMSD and standard deviation across the entire dataset, and stratify results by loop length or type (e.g., CDR-H1, H2, H3).
Quantitative Benchmarks of Advanced Models
The table below summarizes the performance of various state-of-the-art models on antibody CDR-H3 loops, demonstrating the progress brought by deep learning. All values are RMSD in Ångströms (Å) [26].
| Model Type | Model Name | CDR-H3 Loop RMSD (Å) - Antibodies | CDR-H3 Loop RMSD (Å) - Nanobodies |
|---|---|---|---|
| General Protein | ESMFold | 3.15 | 3.60 |
| Antibody-Specific | ABodyBuilder3 | 2.86 | 3.31 |
| Chai-1 | 2.65 | 3.76 | |
| Boltz-1 | 2.96 | 2.83 | |
| Ibex | 2.72 | 3.12 |
Workflow Visualization
The following diagram illustrates a robust, iterative workflow for loop modeling that integrates both traditional and modern deep learning approaches.
The Scientist's Toolkit: Research Reagent Solutions
| Tool / Resource Name | Type | Primary Function in Loop Modeling |
|---|---|---|
| AlphaFold2/3 | Software / Web Server | Highly accurate ab initio structure prediction; provides excellent initial models for entire proteins, including loops [25] [28]. |
| H3-OPT & Ibex | Specialized Software | Predict antibody/nanobody structures with state-of-the-art accuracy for the highly variable CDR-H3 loop; Ibex predicts both apo/holo states [25] [26]. |
| Phyre2.2 | Web Server | Template-based homology modeling server that automatically leverages AlphaFold2 models as templates and includes robust loop and side-chain modeling protocols [28]. |
| SCWRL4 | Software Algorithm | Fast and accurate side-chain conformation prediction, crucial for refining loop models after backbone placement [28]. |
| PDB (Protein Data Bank) | Database | Source of high-resolution experimental structures used for fragment libraries, template identification, and benchmark validation [24] [28]. |
| SAbDab | Specialized Database | The Structural Antibody Database; essential for benchmarking antibody-specific loop predictions and accessing known antibody structures [25] [26]. |
Modern Loop Modeling Techniques: From Fragment Assembly to AI-Driven Approaches
Troubleshooting Guides
Common Loop Modeling Failures and Solutions
Table 1: Troubleshooting Common Loop Modeling Issues
| Problem | Potential Causes | Solutions & Diagnostic Steps |
|---|---|---|
| Poor quality initial homology model | Template with low sequence identity; misalignments; poor flanking regions. | - Verify TM-score of initial model is >0.5 [29].- Ensure flanking residues (4 on each side) are in stable secondary structures (helices/sheets) [29].- Re-evaluate template selection using multiple templates or PSI-BLAST [30]. |
| High candidate clash scores | Steric conflicts between candidate loop and protein core; inaccurate side-chain packing. | - Apply clash filtering during candidate selection [31].- Utilize the server's clash report for multiple loops to select compatible candidates [32].- Consider side-chain repacking or short energy minimization post-modeling. |
| Low confidence scores | Lack of suitable fragments in PDB; unusual loop length or sequence. | - Check the predicted confidence score and level from the server output [32] [29].- For low-confidence loops, consider extending the definition to include more stable flanking regions [29].- For long loops (>12 residues), use a method like DaReUS-Loop specifically validated for lengths up to 30 residues [31] [32]. |
| Inconsistent results for multiple loops | Inter-loop clashes; poor combinations of independent models. | - Use the "remodeling" mode in DaReUS-Loop, which models loops in parallel while keeping others fixed, proven to be most accurate [32] [29].- Consult the general clash report to find non-clashing combinations across different loops [32]. |
Input and Data Preparation Issues
Table 2: Resolving Input and Technical Problems
| Issue | Resolution |
|---|---|
| Incorrect residue numbering | Ensure the protein structure (PDB file) and the input sequence follow the exact same numbering scheme. Residue "ALA 16" in the structure must correspond to the 16th character in the sequence [29]. |
| Handling non-standard residues | The server accepts only the 20 standard amino acids. Manually remove water molecules, co-factors, ions, and ligands from the input PDB file [29]. |
| Server performance and long run times | Typical runs take about one hour, depending on server load. For multiple loops, the server models them in parallel, which is more efficient than sequential modeling [32] [29]. |
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between data-based methods (like DaReUS-Loop, FREAD) and ab initio methods for loop modeling? Data-based methods identify candidate loop structures by mining existing protein structures in databases like the PDB based on the geometry of the flanking regions. These candidates are then filtered and scored [31]. In contrast, ab initio methods computationally explore the conformational space of the loop from scratch using energy functions, which is more time-consuming [31] [32]. Hybrid methods like Sphinx combine both approaches [31].
Q2: Why is my loop modeling accuracy low even when using a good template? The most common reason is poor quality of the flanking regions. In homology modeling, the flanks are not perfect as in crystal structures and can have large deviations from the native structure. Ensure that the residues immediately adjacent to the loop gap are accurate and part of a helix or sheet [31] [29]. Data-based methods are sensitive to the geometry imposed by these flanks.
Q3: Can I use DaReUS-Loop to model loops in an experimentally solved structure? While technically possible, the DaReUS-Loop protocol is specifically optimized for the non-ideal conditions of homology models, where flank regions are perturbed [32] [29]. Its performance on high-resolution crystal structures with perfect flanks has not been the focus of its validation.
Q4: How does DaReUS-Loop handle long loops, which are traditionally problematic? DaReUS-Loop was specifically designed and validated to handle long loops effectively. It can model loops of up to 30 residues [29]. The method outperforms other state-of-the-art approaches for loops of at least 15 residues, showing a significant increase in accuracy [31] [32].
Q5: How should I select the best model from the 10 candidates returned by DaReUS-Loop? DaReUS-Loop does not reliably predict the single best model among the 10 candidates [29]. The strategy is to achieve high accuracy in at least one of the returned models. You should rely on the provided confidence score, which correlates with the expected accuracy of the best loop, and inspect the candidates visually or using additional experimental data (e.g., SAXS) if available [32] [29].
Q6: What is the advantage of using a data-based method that mines the entire PDB? A key advantage is the ability to discover candidate loops from remote or unrelated proteins that are not homologous to your target. Strikingly, over 50% of successful loop models generated by DaReUS-Loop are derived from unrelated proteins. This indicates that protein fragments under similar spatial constraints can adopt similar structures beyond evolutionary homology [31].
Experimental Protocols & Workflows
DaReUS-Loop Protocol for Loop Remodeling
The following diagram illustrates the core workflow of the DaReUS-Loop method for remodeling a loop in a homology model.
Protocol Steps:
- Input Preparation: Provide the initial homology model in PDB format and the full protein sequence. For remodeling, the sequence should have the target loop regions indicated by gaps ("-") [32] [29].
- PDB Mining: The server uses BCLoopSearch to mine the entire Protein Data Bank for protein fragments whose flanking regions geometrically match the flanks of the loop to be modeled [31] [29].
- Candidate Filtering: The pool of candidates is progressively refined through several filters [31]:
- Sequence Filter: Candidates are filtered based on sequence similarity using a positive BLOSUM62 score threshold.
- Conformation Filter: The local conformational profile of candidates is compared to the target using Jensen-Shannon Divergence (JSD), filtering out candidates with high JSD.
- Clash Filter: Candidates that cause steric clashes with the rest of the protein structure are eliminated.
- Clustering and Ranking: The remaining candidates are clustered to reduce redundancy. The best candidates are selected based on local conformation profile and flank Root-Mean-Square Deviation (RMSD) [31] [29].
- Model Building and Output: Complete 3D models are built by superimposing the candidate loops onto the flanks of the input structure. The server returns 10 models for each loop, along with a confidence score that correlates with expected accuracy [31] [32] [29].
Performance Validation Protocol
To validate the performance of a loop modeling method like DaReUS-Loop against other tools, follow this benchmarking workflow.
Validation Steps:
- Benchmark Selection: Use standardized test sets from critical assessment experiments like CASP11 and CASP12, which contain targets for template-based modeling [31] [32].
- Method Comparison: Run DaReUS-Loop alongside other state-of-the-art methods for comparison. Typical comparisons include [31] [32]:
- Ab initio methods: Rosetta NGK, GalaxyLoop-PS2.
- Data-based methods: LoopIng, FREAD.
- Hybrid methods: Sphinx.
- Accuracy Metric: The primary metric is the loop RMSD (in Ångströms) after superimposing the model on the flanking residues of the native structure. The percentage of models achieving high accuracy (e.g., RMSD < 1.0 Å or < 2.0 Å) is also calculated [31] [32].
- Analysis: Analyze the results grouped by loop length (e.g., short loops of 3-12 residues vs. long loops of ≥15 residues) and by the method's confidence score to verify its predictive power [31] [32].
Table 3: Comparative Loop Modeling Accuracy (Average RMSD, Å) Data represents the average RMSD of the best of the top 10 models on CASP11 and CASP12 test set loops. Lower values indicate better performance. Adapted from [32].
| Method | Type | CASP11 (set_ai) | CASP12 (set_ai) |
|---|---|---|---|
| DaReUS-Loop Server | Data-based | 2.00 Å | 2.35 Å |
| DaReUS-Loop (Original) | Data-based | 1.91 Å | 2.30 Å |
| Rosetta NGK | Ab initio | 2.59 Å | 2.99 Å |
| GalaxyLoop-PS2 | Ab initio | 2.34 Å | 2.88 Å |
| LoopIng | Data-based | > 3.28 Å | > 3.63 Å |
| Sphinx | Hybrid | > 2.89 Å | > 3.24 Å |
| MODELLER | Template-based | 2.94 Å | 3.29 Å |
Table 4: DaReUS-Loop Filtering Impact on Candidate Quality Data from the CASP11 test set showing how each filtering step improves the fraction of high-accuracy loop candidates. Adapted from [31].
| Processing Stage | Key Filtering Action | Result / Fraction of Candidates with RMSD < 4Å |
|---|---|---|
| Post-PDB Mining | Initial candidate set from BCLoopSearch | 49% |
| Post-Sequence Filtering | Keep fragments with positive BLOSUM62 score | 62% |
| Post-Clustering | Cluster candidates and select representatives | 70% |
| Post-Conformation Filtering | Filter by Jensen-Shannon Divergence (JSD < 0.4) | 74% |
| Post-Clash Filtering | Remove candidates with steric clashes | 84% |
The Scientist's Toolkit: Research Reagent Solutions
Table 5: Essential Resources for Data-Based Loop Modeling
| Resource Name | Type | Function in Experiment |
|---|---|---|
| DaReUS-Loop Web Server | Automated Web Server | Primary tool for (re-)modeling loops in homology models using a data-based approach. Accepts up to 20 loops in parallel [32] [29]. |
| Protein Data Bank (PDB) | Database | The primary source of experimental protein structures used as the fragment library for data-based mining by tools like DaReUS-Loop and FREAD [31] [33]. |
| BCLoopSearch | Algorithm / Software | The core search tool used by DaReUS-Loop to mine the PDB for fragments that geometrically match the loop's flanking regions [31]. |
| SWISS-MODEL / MODELLER | Homology Modeling Server / Software | Used to generate the initial homology model required as input for DaReUS-Loop [29] [30]. |
| NGL Viewer | Visualization Tool | Integrated into the DaReUS-Loop results page for interactive visual inspection and comparison of the generated loop candidates [32] [29]. |
| PDB-REDO Database | Refined Structure Database | A resource of re-refined and re-built crystal structures, which can provide improved templates or insights into "buildable" loop regions [33]. |
Frequently Asked Questions (FAQs)
Q1: What are the primary use cases for Rosetta NGK and GalaxyLoop-PS2? Both are state-of-the-art ab initio loop modeling methods used to predict the 3D structure of loop regions in proteins. They are particularly valuable for:
- Completing Homology Models: Accurately modeling loops in regions where template structures are unavailable or unreliable [34] [31].
- Refining Crystal Structures: Rebuilding missing or poorly resolved loops in experimental structures [34].
- Functional Site Prediction: Modeling loops involved in binding sites, which is critical for drug design and understanding protein function [34] [31].
Q2: My Rosetta NGK run failed to generate any models. What could be wrong? This common issue can often be traced to a few key areas:
- Incorrect Loop Definition File: Ensure your loop file follows the exact format. The cut point residue number must be within the start and end residues of the loop, or a segmentation fault can occur [35].
- Missing Flags: Confirm that the essential command-line flags to activate NGK are present:
-loops:remodel perturb_kicand-loops:refine refine_kic[35]. - Residue Numbering Mismatch: Residue indices in the loop definition file must use Rosetta numbering (continuous numbering from 1). You may need to renumber your input PDB file to match [35].
Q3: How can I improve the sampling accuracy of Rosetta NGK for difficult loops? You can employ several advanced strategies to enhance sampling:
- Use Taboo Sampling: Promote conformational diversity by preventing repeated sampling of the same torsion bins with the
-loops:taboo_samplingflag [35] [36] [37]. - Enable Neighbor-Dependent Sampling: Use more precise, neighbor-dependent Ramachandran distributions with
-loops:kic_rama2b(note: this increases memory usage to ~6GB) [35]. - Apply Energy Function Ramping: Smooth the energy landscape by ramping repulsive and Ramachandran terms during refinement using
-loops:ramp_fa_repand-loops:ramp_rama[35] [36].
Q4: The side chains around my loop look poor in the model. How can I fix this?
By default, NGK may fix the side chains of residues neighboring the loop. To allow these side chains to be optimized during modeling, set the flag -loops:fix_natsc to false [35].
Q5: How do I choose between a knowledge-based method like DaReUS-Loop and an ab initio method like NGK or GalaxyLoop-PS2? The choice depends on your specific goal and the problem context, as shown in the table below.
| Method | Type | Best Application Context | Key Advantage |
|---|---|---|---|
| Rosetta NGK [35] [36] | Ab Initio | Accurate, high-resolution framework structures; de novo loop construction. | Robotics-inspired algorithm ensures local moves and exact chain closure. |
| GalaxyLoop-PS2 [34] | Ab Initio | Loops in inaccurate environments (e.g., homology models, low-resolution structures). | Hybrid energy function tolerates environmental errors in the framework. |
| DaReUS-Loop [31] | Knowledge-based | Fast prediction in homology modeling; long loops (≥15 residues). | Speed; confidence score correlates with prediction accuracy. |
Troubleshooting Guides
Common Rosetta NGK Errors and Solutions
| Error / Symptom | Probable Cause | Solution |
|---|---|---|
| Segmentation fault | Cut point in loop definition file is outside the loop start/end residues [35]. | Ensure the cut point residue number is ≥ startRes and ≤ endRes. |
| No output models generated | Missing critical -loops:remodel or -loops:refine flags [35]. |
Include -loops:remodel perturb_kic and -loops:refine refine_kic in command line. |
| Low memory error | Using the -loops:kic_rama2b flag on a system with insufficient RAM [35]. |
Ensure at least 6GB of memory per CPU is available. |
| Loop residues are not connected | Starting loop conformation is disconnected (e.g., in de novo modeling) [35]. | Set the "Extend loop" column in the loop definition file to 1 to randomize and connect the loop. |
Performance Benchmarking and Validation
When evaluating your loop models, it is critical to compare their performance against established benchmarks. The following table summarizes the typical accuracy you can expect from these methods on standard test sets.
| Method | Typical Accuracy (Short Loops, ~12 residues) | Performance on Long Loops (≥15 residues) | Key Identifying Feature |
|---|---|---|---|
| Rosetta NGK | Median fraction of sub-Ångström models increased 4-fold over standard KIC [36] [37]. | Can model longer segments that previous methods could not [36]. | Combination of intensification and annealing strategies. |
| GalaxyLoop-PS2 | Comparable to force-field-based approaches in crystal structures [34]. | Not specifically highlighted. | Hybrid energy function combining physics-based and knowledge-based terms. |
| DaReUS-Loop | Significant increase in high-accuracy loops in homology models [31]. | Outperforms other approaches for long loops [31]. | Data-based approach using fragments from remote/ unrelated PDB structures. |
Experimental Protocols
Core Protocol: Running a Rosetta NGK Experiment
Objective: Reconstruct a missing loop region in a protein structure. Input Files Required:
- Starting PDB File: The protein structure with the missing loop.
- Loop Definition File: A text file specifying the loop(s) to be modeled. Example for a single loop from residue 88 to 95 with a cutpoint at 92:
Example Command:
Workflow: Rosetta Next-Generation KIC (NGK)
The following diagram illustrates the integrated steps of the NGK protocol, showing how different sampling strategies are applied across the centroid and full-atom stages.
The Scientist's Toolkit
Key Research Reagent Solutions
| Item | Function in Experiment | Specification / Note |
|---|---|---|
| Rosetta Software | Primary modeling suite for NGK. | Academic licenses are free. Source code in C++ must be compiled on a Unix-like OS (Linux/MacOS) [38]. |
| Input Protein Structure | Framework for loop modeling. | PDB file format. Residues outside the loop must have real coordinates [35]. |
| Loop Definition File | Specifies the location and parameters of the loop to be modeled. | Critical to use correct Rosetta numbering to avoid errors [35]. |
| Fragment Libraries | Guide conformational search in some protocols. | Can be generated using the Robetta server [38]. |
| Computational Resources | Running Rosetta simulations. | Recommended: Multi-processor cluster with at least 1GB memory per CPU. NGK with Rama2b requires ~6GB [35] [38]. |
Troubleshooting Guide: Common Experimental Issues
Problem 1: Homology model exhibits high energy loops despite data mining pre-filtering
- Symptoms: The target loop region, even when built from a template with high sequence similarity identified through data mining, shows unstable energy profiles, high torsion strain, or poor rotameric side-chain placement in the refined model.
- Impact: The overall model accuracy is compromised, potentially leading to incorrect conclusions about protein function or ligand binding sites in drug development research [39].
- Context: This frequently occurs when the selected template, though similar in sequence, originates from a different structural context (e.g., apo vs. holo form) or has a divergent backbone conformation [39].
| Step | Action | Expected Outcome |
|---|---|---|
| 1 | Verify Template Suitability: In your Phyre2.2 results, check if the template is listed as an "apo" or "holo" structure. Cross-reference the template's PDB entry to confirm its bound state and overall fold similarity beyond the local loop region [39]. | Confirmation that the template's structural context is appropriate for your target. |
| 2 | Run Energy Diagnostic Check: Use your refinement software's energy evaluation function on the isolated loop. Note specific energy terms that are outliers (e.g., van der Waals clashes, Ramachandran outliers). | Identification of the specific physical chemistry terms causing the high energy. |
| 3 | Apply Hybrid Strategy: Use the data-mined template as a starting conformation, then apply an energy-based refinement algorithm. If using a method like the Boosted Multi-Verse Optimizer (BMVO), it can help optimize the model's weights and increase its generalization effectiveness to find a lower energy conformation [40]. | A refined loop structure with a lower overall energy score and improved stereochemistry. |
Problem 2: Energy minimization protocol causes distortion of the data-mined backbone
- Symptoms: After applying an energy-based refinement, the overall backbone geometry of the core protein structure, which was accurate from homology modeling, becomes distorted or shifts significantly.
- Impact: The global fold is lost, rendering the model unusable for its intended research applications, such as virtual screening.
- Context: This is common when the energy minimization parameters are too aggressive or when restraints are not properly applied to the well-conserved regions of the model.
| Step | Action | Expected Outcome |
|---|---|---|
| 1 | Apply Positional Restraints: Restrain the heavy atoms of the protein backbone and core side-chains during refinement. Allow only the target loop and immediate surrounding residues to move freely. | The global fold is preserved during the energy minimization process. |
| 2 | Use a Multi-Stage Protocol: Begin refinement with a strong force constant on the restraints, then gradually reduce it in subsequent stages. This allows the high-energy regions to relax without compromising the entire structure. | A stable minimization trajectory that corrects local issues without global distortion. |
| 3 | Validate with Multiple Metrics: Post-refinement, check global structure metrics like Root Mean Square Deviation (RMSD) against the initial template and verify the Ramachandran plot for the core regions has not deteriorated. | Quantitative confirmation that model quality has been maintained or improved. |
Problem 3: Inability to achieve target contrast ratio in visualization of loop conformational clusters
- Symptoms: When generating diagrams of different loop conformations sampled during refinement, the colors used for arrows or symbols lack sufficient contrast against the background, making them hard to distinguish.
- Impact: Reduces the clarity and professional presentation of research findings, hindering effective communication in publications or presentations.
- Context: This often occurs when using default color palettes without checking for accessibility or when using colors of similar luminance [41] [42].
| Step | Action | Expected Outcome |
|---|---|---|
| 1 | Select High-Contrast Colors: Choose foreground and background colors from the approved palette that have a high contrast ratio. For critical data, ensure a contrast ratio of at least 4.5:1 for normal text and 3:1 for large graphics [41] [42]. | Diagrams are legible for all audiences, including those with low vision or color blindness. |
| 2 | Use an Analyzer Tool: Before finalizing a figure, use a color contrast analysis tool to verify the ratio between the arrow/symbol color and the background color meets WCAG guidelines [42]. | Objective verification that the color choices are sufficient. |
| 3 | Explicitly Set Colors in DOT Script: In your Graphviz DOT script, explicitly define the fontcolor for any node text to ensure high contrast against the node's fillcolor. Do not rely on default settings. |
Rendered diagrams have clear, readable text labels on all shapes. |
Frequently Asked Questions (FAQs)
Q1: What is the core advantage of a hybrid strategy over using data mining or energy-based refinement alone? A1: A hybrid strategy leverages the complementary strengths of both approaches. Data mining through tools like Phyre2.2 rapidly identifies structurally plausible starting conformations from known proteins by finding suitable templates from the AlphaFold database or PDB [39]. Energy-based refinement then acts as a physical chemistry filter, optimizing these starting points to find the most stable, energetically favorable conformation that data mining alone might miss. This combination addresses the shortcomings of methods that fail to capture complex nonlinear interactions, leading to more reliable and accurate models [40].
Q2: In the context of the BMVO algorithm mentioned for optimization, what does "generalization effectiveness" mean for my loop model? A2: Generalization effectiveness in this context refers to the optimized model's ability to perform well not just on the specific computational data it was trained on, but to produce a physically realistic and accurate loop structure that is consistent with the principles of protein stereochemistry. A model with high generalization effectiveness is less likely to be "overfitted" to the peculiarities of the template and is more likely to represent a native-like conformation [40].
Q3: My refinement seems stuck in a high-energy local minimum. What strategies can help escape this? A3: This is a common challenge in energy minimization. Strategies include:
- Hybrid Optimizers: Employ algorithms like the Boosted Multi-Verse Optimizer (BMVO), which are designed to have a better global search capability, helping to escape local minima compared to more traditional optimizers [40].
- Enhanced Sampling: Implement protocols like replica exchange molecular dynamics, which simulates the system at multiple temperatures, allowing high-energy barriers to be crossed.
- Multi-Modal Search: Run multiple independent refinements from slightly different starting points (e.g., different loop conformations from data mining) to see if they converge on the same low-energy structure.
Q4: How do I validate the success of a hybrid loop modeling protocol? A4: Use a combination of quantitative and qualitative checks:
- Energetics: The refined model should have a lower overall energy and show no significant steric clashes or high-energy torsion angles.
- Geometry: Validate using MolProbity or similar services to check Ramachandran outliers, rotameric quality, and clash scores.
- Comparison to Data (if available): If an experimental structure is known but not used in modeling, compute the RMSD of the loop to the true structure.
- Consensus: If multiple independent refinements converge on a similar loop conformation, it increases confidence in the result.
Experimental Protocols & Data
Protocol: Hybrid Data-Mining and Energy Refinement for Loop Modeling
1. Data Mining Phase (Template Identification)
- Input: Target protein sequence with defined loop boundaries.
- Tool: Submit sequence to Phyre2.2 web portal for intensive mode analysis [39].
- Methodology: Phyre2.2 will search its expanded template library, which includes a representative structure for every protein sequence in the PDB, plus dedicated apo and holo structures. It will use a new ranking system to highlight potential templates for different domains in your query [39].
- Output: A list of template structures, confidence scores, and an initial homology model.
2. Hybrid Refinement Phase (Energy-Based Optimization)
- Input: Initial homology model from Phyre2.2, focusing on the loop region of interest.
- Technique: Apply an optimized improved Support Vector Machine (SVM) or other machine learning method, enhanced by a hybrid optimization technique like the Boosted Multi-Verse Optimizer (BMVO) [40].
- Methodology:
- The improved SVM incorporates a modified genetic algorithm based on kernel function for stability.
- The BMVO technique is employed to optimize the combined model's weights and increase its generalization effectiveness.
- This approach is designed to handle the complicated nonlinear interactions between the loop and its environment that simpler methods fail to capture [40].
- Output: A refined protein model with an energetically stable loop.
Quantitative Performance Data
The table below summarizes potential improvements from using a robust hybrid strategy, based on performance metrics from related optimization research [40].
| Performance Metric | Basic ANN Method | Proposed BMVO/ISVM Hybrid Model | Improvement |
|---|---|---|---|
| Root Mean Square Error (RMSE) | Baseline | Reduced by 53.72% [40] | Significant |
| Mean Absolute Percent Error (MAPE) | Baseline | Reduced by 55.22% [40] | Significant |
| Optimal Sample Size | Variable | 1735 samples [40] | For lowest MAPE |
The Scientist's Toolkit: Research Reagent Solutions
| Tool / Resource | Function in Hybrid Strategy |
|---|---|
| Phyre2.2 Web Portal | A template-based protein structure modeling portal. Its main development is facilitating a user to submit their sequence and then identifying the most suitable AlphaFold model to be used as a template, providing a high-quality starting point for the data mining phase [39]. |
| Boosted Multi-Verse Optimizer (BMVO) | A hybrid optimization technique employed to optimize a model's weights and increase its generalization effectiveness, which is crucial for the energy-based refinement phase [40]. |
| Improved Support Vector Machine (SVM) | An algorithm that can be used for prediction and classification in the refinement pipeline. The "improved" version incorporates a modified genetic algorithm based on kernel function to enhance the stability of the model during optimization [40]. |
| Color Contrast Analyzer | Tools used to verify that diagrams of signaling pathways, workflows, and logical relationships meet minimum color contrast ratio thresholds (e.g., 4.5:1), ensuring accessibility and clarity for all researchers [41] [42]. |
Workflow and Signaling Pathways
Hybrid Strategy Workflow
This diagram illustrates the sequential integration of data mining and energy-based refinement for improving loop modeling accuracy. The process begins with the target sequence and systematically progresses through template selection and energy optimization to produce a validated, high-quality model.
Energy Refinement Feedback Loop
This diagram shows the iterative feedback loop at the heart of the energy-based refinement phase. The BMVO optimizer repeatedly adjusts the loop conformation based on energy calculations until a stable, low-energy structure is achieved.
Core Concepts: Homology Grafting and the PDB-REDO Pipeline
What is homology grafting in the context of protein structure modeling? Homology grafting is a computational technique that identifies regions missing from a target protein structure model and transplants ("grafts") these regions from homologous structures found in the Protein Data Bank (PDB). Inherent protein flexibility, poor resolution diffraction data, or poorly defined electron-density maps often result in incomplete structural models during X-ray structure determination. This method is particularly valuable for modeling missing loops, which are often difficult to build due to their conformational flexibility. The grafted regions are subsequently refined and validated within the context of the target structure to ensure a proper fit and geometric correctness [33].
How does PDB-REDO integrate homology grafting for loop modeling? The PDB-REDO pipeline incorporates a specific algorithm called Loopwhole to perform homology-based loop grafting. This process is automated and consists of several key stages:
- Identification: The system detects unmodeled regions (loops) in the input structure and identifies high-identity homologous structures that have these regions modeled.
- Grafting: For each candidate homolog, the loop (plus two adjacent residues on either side) is structurally aligned and transferred into the target model.
- Refinement: The newly grafted loop is refined within the PDB-REDO pipeline to fit the experimental electron-density map in real space.
- Validation: The completed model is validated against geometric criteria and the electron density to ensure the grafted loop is both structurally sound and supported by the data [33] [43].
This constructive validation approach goes beyond simple error correction, aiming for the best possible interpretation of the electron density data. The entire procedure, including these grafting developments, is publicly available through the PDB-REDO databank for pre-optimized existing PDB entries, and the PDB-REDO server for optimizing your own structural models [44] [45].
Troubleshooting FAQs
FAQ 1: My homology-grafted loop has poor real-space correlation or steric clashes after refinement. What should I do?
Poor density fit or clashes often indicate a suboptimal graft or refinement issue. Follow this systematic troubleshooting guide:
- Verify Homolog Selection Criteria: The grafting algorithm requires a sequence identity of at least 50% in the loop and flanking regions. Re-check the sequence similarity of your source homolog. Also, ensure the structural alignment of the flanking residues had a backbone RMSD of less than 2.0 Å before transfer [33].
- Inspect the Electron Density: Use the PDB-REDO output maps to visually inspect the
2mFo-DFcandmFo-DFcmaps around the grafted loop. A strong, well-defined2mFo-DFcmap and minimal noise in themFo-DFcmap suggest the loop is buildable, but the current conformation may be incorrect. A weak or absent density might indicate intrinsic disorder. - Check for Side-Chain Interactions: Examine if mutated side chains in the grafted loop are causing steric clashes. The PDB-REDO pipeline performs side-chain cropping in case of mutations, but manual inspection and adjustment might be necessary using a molecular graphics program [33].
- Run Extended Refinement: If the initial PDB-REDO output shows minor clashes, consider submitting the job again with more aggressive refinement cycles. PDB-REDO optimizes refinement settings like geometric restraint weights and B-factor models, which can sometimes resolve minor conflicts [43] [45].
- Consider Alternative Methods: If homology grafting consistently fails, consider using a dedicated, state-of-the-art loop modeling tool like DaReUS-Loop or Rosetta NGK for the problematic region, and then re-introduce the model into PDB-REDO for final refinement [31].
FAQ 2: What are the common reasons for a loop to be "unbuildable" even with homologous templates available?
Several factors can prevent successful loop building, even when a template exists:
- Conformational Divergence in the Target: The loop may adopt a distinct conformation in your target protein due to ligand interactions, crystallographic packing, or a different functional state. The homologous conformation, while similar in sequence, may not fit the electron density of your target [33].
- Poor Quality of Experimental Data: Weak, broken, or ambiguous electron density is the most common reason a loop cannot be reliably modeled. This is often due to high flexibility or static disorder in the crystal. The PDB-REDO procedure will not attempt loop building in chains where the overall real-space correlation coefficient (RSCC) is below 0.80, indicating unreliable chain conformation [33].
- Incompatible Flanking Regions: If the secondary structure elements flanking the missing loop are themselves poorly modeled or in a significantly different orientation compared to the homologous template, the grafted loop will have incorrect geometry and fail to connect properly [31].
- Length Mismatch: The PDB-REDO's Loopwhole algorithm has a default maximum length for transfer of 30 amino acids. While this covers most loops, extremely long insertions may require specialized approaches [33].
FAQ 3: How does homology grafting in PDB-REDO compare to other loop modeling methods?
Loop modeling methods are generally categorized as ab initio, knowledge-based, or hybrid. Homology grafting in PDB-REDO is a knowledge-based method. The table below summarizes a comparison based on independent benchmarking [31]:
Table 1: Comparison of Loop Modeling Method Performance
| Method | Type | Best For | Typical Accuracy (Short Loops, ≤12 res) | Performance on Long Loops (≥15 res) |
|---|---|---|---|---|
| PDB-REDO (Loopwhole) | Knowledge-based | Completing crystal structures with available homologs | ~1-2 Å (in high-res structures) | Good, leverages homologs directly [33] |
| DaReUS-Loop | Knowledge-based | Homology models & long loops; high confidence prediction | ~1-4 Å (in homology models) | Outperforms other methods [31] |
| Rosetta NGK | Ab initio | High-accuracy predictions when no template exists | ~1-2 Å | Accuracy decreases, computationally intensive [31] |
| GalaxyLoop-PS2 | Ab initio | High-accuracy predictions with hybrid energy function | ~1-2 Å | Accuracy decreases, computationally intensive [31] |
| Sphinx | Hybrid | Combining speed of data-based with ab initio accuracy | ~1-2 Å | Varies |
A key advantage of knowledge-based methods like DaReUS-Loop is that over 50% of successful loop models can be derived from structurally similar but evolutionarily unrelated proteins, indicating that local structural constraints are a powerful predictor of loop conformation [31].
Experimental Protocols & Workflows
Detailed Methodology: Loop Grafting with Loopwhole in PDB-REDO
The following protocol details the automated steps performed by the Loopwhole algorithm within the PDB-REDO pipeline [33]:
Input and Initial Evaluation:
- Input: The target protein structure model and its associated structure factors.
- Density Fit Assessment: The EDSTATS tool evaluates the real-space correlation coefficient (RSCC) for each chain. Loops are not built in chains with an overall RSCC below 0.80.
Homolog Identification and Loop Detection:
- Missing Regions: Unmodeled loops in the input structure are detected using
pdb2fasta. - Template Search: High-identity homologs are identified from the PDB and aligned by sequence. A homolog is considered if it has a complete backbone for the region of interest.
- Missing Regions: Unmodeled loops in the input structure are detected using
Pre-transfer Filtering and Alignment:
- Anchoring Residues: Both the target and homolog must have at least five consecutive modeled residues on each side of the loop. The two residues directly adjacent to the loop are flagged for remodeling.
- Sequence Identity Check: The sequence identity in the loop and flanking regions must be ≥50%. Single-residue loops are allowed to be mutated.
- Structural Alignment: The flanking regions (excluding the two direct adjacent residues) are structurally aligned using quaternions. A transfer is only attempted if the backbone RMSD of this alignment is < 2.0 Å.
Loop Transfer and Initial Modeling:
- Grafting: The two residues adjacent to the loop in the target are deleted. The loop from the homolog, including its two adjacent residues, is inserted into the target model.
- Side-Chain Processing: Side chains are cropped to the common atoms in case of mutations. Atom occupancy is set to 1.00 and B-factors are scaled.
Integrated Refinement and Validation:
- The entire model, including the new loop, undergoes automated refinement in REFMAC, which optimizes refinement parameters (geometric restraints, B-factor weights, TLS groups) [43] [45].
- Subsequent rebuilding steps include:
- Removing poorly fitting waters.
- Rebuilding side chains into optimal rotameric conformations.
- Flipping peptide planes if it improves the electron density fit.
- The final model is validated using WHAT_CHECK, pdb-care (carbohydrates), and FoldX (folding energy calculation) [43].
Workflow Visualization
The following diagram illustrates the logical workflow of the homology grafting and refinement process within PDB-REDO:
The Scientist's Toolkit
Table 2: Essential Research Reagents and Resources for Homology Grafting and Modeling
| Resource / Tool | Type | Function in Homology Grafting | Access |
|---|---|---|---|
| PDB-REDO Server | Automated Pipeline | Performs end-to-end refinement, homology grafting (Loopwhole), and validation of your structure models. | https://pdb-redo.eu/ |
| PDB-REDO Databank | Data Repository | Provides pre-optimized and re-refined versions of existing PDB entries, many with previously missing loops completed. | https://pdb-redo.eu/ |
| Loopwhole | Algorithm | The specific tool within PDB-REDO that identifies and grafts missing loops from homologous structures. | Integrated in PDB-REDO [33] |
| REFMAC | Software | The crystallographic refinement program used by PDB-REDO to optimize the model, including grafted loops, against the experimental data. | Integrated in PDB-REDO [43] [45] |
| DaReUS-Loop | Software | A knowledge-based loop modeling method that mines the entire PDB for candidate fragments, effective for long loops and homology models. | Standalone [31] |
| Rosetta NGK | Software | An advanced ab initio loop modeling method for when no suitable homologous templates are available. | Standalone [31] |
| PDB (Protein Data Bank) | Database | The primary source for homologous protein structures used as templates for the grafting procedure. | https://www.rcsb.org/ |
FAQs: AI-Driven Contact Prediction
What is the core advantage of using ML for contact prediction over traditional methods? Machine learning models, particularly deep learning, can identify complex, non-linear patterns in protein sequences and structures that are often missed by traditional physics-based calculations. This allows for more accurate prediction of residue-residue contacts, which is crucial for improving loop modeling and overall homology model quality [46].
Our homology models have poor loop regions. How can AI specifically help? AI-powered platforms can leverage neural network potentials to perform faster and more accurate simulations of loop conformations. Furthermore, tools like Phyre2.2 can identify suitable templates from extensive databases, including AlphaFold models, which often provide better starting points for loop regions compared to traditional template searching, leading to more reliable apo and holo structures [46] [39].
Which ML model should we start with for our contact prediction experiments? For multiclass prediction problems common in bioinformatics, ensemble models like Gradient Boosting, Random Forests, and XGBoost have demonstrated high performance. Recent studies show Gradient Boosting can achieve up to 67% macro accuracy in complex classification tasks, making it a strong candidate. The best model can vary based on your specific dataset, so testing a few is recommended [47].
How do we handle the computational cost of these AI simulations? Modern neural network potentials, such as Egret-1 and AIMNet2, are designed to match quantum-mechanics-based simulation accuracy while running orders-of-magnitude faster. This makes advanced computational techniques practical for guiding experimental work without prohibitive hardware costs [46].
Our team has limited coding expertise. Are there accessible AI tools? Yes, platforms like Rowan offer web-native, no-code interfaces and Python/RDKit APIs for programmatic control, making advanced AI tools accessible to scientists regardless of their programming background [46].
Troubleshooting Guides
Issue: Poor Contact Prediction Accuracy
Problem: Your ML model is generating inaccurate contact maps, leading to faulty homology models.
Solution:
- Verify Data Quality: Ensure your training data of protein sequences and structures is high-quality and preprocessed to remove errors [47].
- Check Feature Selection: Re-evaluate the features used for prediction (e.g., co-evolutionary data, physicochemical properties, sequence embeddings). Use feature importance analysis to identify the most relevant ones.
- Tune Hyperparameters: Use a grid search to optimize your model's hyperparameters. One study achieved a performance increase from 64% to 79% accuracy for a Random Forest model through optimal tuning [47].
- Try Ensemble Methods: Combine predictions from multiple models (e.g., Random Forest, XGBoost) to improve robustness and accuracy [47].
Issue: Low Scoring Model After Loop Refinement
Problem: After rebuilding loops in your homology model, the overall model scores poorly in validation.
Solution:
- Reassess Template: Use Phyre2.2 to find a more suitable template, as its updated library includes a representative for every protein sequence in the PDB and can leverage AlphaFold models specifically for this purpose [39].
- Apply Strain Correction: Perform ligand-strain-corrected docking to ensure the refined loop does not create steric clashes or unrealistic geometry in the binding site [46].
- Run Faster Simulations: Utilize neural network potentials like Egret-1 to quickly re-simulate and relax the refined loop region, ensuring it adopts a low-energy conformation [46].
Issue: Inaccessible or Slow Simulation Tools
Problem: Cloud-based molecular simulation platforms are slow or inaccessible from your secure IT environment.
Solution:
- Request Secure Deployment: Inquire with your software provider about enterprise-grade deployment options. Many, like Rowan, offer single-tenant and customer-managed virtual-private-cloud (VPC) deployments to ensure both security and performance [46].
Experimental Protocols & Data
Quantitative Performance of Ensemble ML Classifiers
The table below summarizes the global macro accuracy of various machine learning algorithms in a multiclass prediction task, providing a benchmark for model selection [47].
| Algorithm | Global Macro Accuracy |
|---|---|
| Gradient Boosting | 67% |
| Bagging | 65% |
| Random Forest | 64% |
| K-Nearest Neighbors (KNN) | 60% |
| XGBoost | 60% |
| Support Vector Machines (SVM) | 59% |
| Decision Trees | 55% |
Table 1: Comparative prediction accuracy of ensemble machine learning models for a multiclass grading problem, illustrating performance levels relevant to contact prediction tasks [47].
Detailed Protocol: Enhancing Loop Modeling with Phyre2.2 and AI
Objective: Generate an improved homology model with accurately predicted loop regions.
Methodology:
- Sequence Submission: Submit your target amino acid sequence to the Phyre2.2 web portal.
- Template Identification: Phyre2.2 will search its expanded library, which includes a representative structure for every protein sequence in the PDB, to identify the most suitable template. The new ranking system will highlight potential templates for different domains in your query [39].
- Model Building: Allow Phyre2.2 to build a preliminary model using the selected template.
- Loop Region Analysis: Identify poorly modeled or high-energy loop regions in the preliminary model.
- AI-Powered Refinement:
- Model Validation: Re-validate the refined full-length model using standard structural validation tools.
Workflow: AI-Enhanced Loop Modeling
AI-Enhanced Loop Modeling
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function |
|---|---|
| Phyre2.2 Web Portal | A community resource for template-based protein structure prediction that can leverage AlphaFold models as templates, facilitating accurate modelling of apo and holo structures [39]. |
| Rowan Platform | A computational platform providing ML-powered property prediction (e.g., pKa, solubility) and fast, accurate neural network potentials (Egret-1, AIMNet2) for molecular simulation [46]. |
| Neural Network Potentials (Egret-1, AIMNet2) | AI models that run quantum-mechanics-level simulations millions of times faster than traditional methods, enabling rapid conformational sampling and refinement [46]. |
| AutoDock Vina with Strain Correction | Docking software used for testing binding and generating bound poses with corrections for ligand strain, important for validating binding site geometries [46]. |
| Co-folding Models (Boltz-2, Chai-1r) | State-of-the-art models for predicting 3D structures and binding affinities of protein-ligand complexes directly from sequence information [46]. |
Optimizing Your Modeling Pipeline: Practical Strategies for Improved Accuracy
Frequently Asked Questions
FAQ 1: Why is my model quality poor even when using a template with high sequence identity? High sequence identity does not guarantee a high-quality model if the template structure itself is of low quality. The template's experimental resolution, the presence of gaps in critical regions (like your target's loops), and incorrect side-chain rotamers can all degrade model quality. Furthermore, if the target-template alignment is suboptimal, even a perfect template structure will yield a poor model. Always verify the quality of the template structure and the correctness of the alignment [14] [48].
FAQ 2: When should I use a single template versus multiple templates for modeling? Using multiple templates can improve model quality by combining structural information from different sources, potentially covering more of your target sequence and providing better insights for variable regions. However, it is not guaranteed to improve quality and can sometimes produce models worse than the best single-template model. It is most beneficial when templates cover different, complementary regions of your target. For core structural regions, a single high-quality template is often sufficient, and adding more templates can introduce errors. Use model quality assessment programs to select the final model [14].
FAQ 3: How do I select the best template for accurate loop modeling, especially for highly variable loops like CDR-H3 in antibodies? Loops, particularly non-canonical ones, are a core challenge. For antibodies, the H1, H2, L1, L2, and L3 loops often have canonical structures and can be modeled well via homology. The H3 loop, however, is highly variable in both sequence and structure. When possible, identify a template with a similar H3 loop length and, crucially, a similar structural context. If no good template exists, consider specialized antibody modeling tools (e.g., ABodyBuilder) or advanced deep learning methods (e.g., RoseTTAFold, AlphaFold2), which can sometimes outperform standard homology modeling for these regions [49].
FAQ 4: What is the minimum sequence identity required for a usable template? There is no strict minimum, but the accuracy of homology models is highly dependent on sequence identity. Generally, above 40% sequence identity, alignments are more straightforward and models tend to be more reliable. In the 30-40% range, the alignment becomes critical and often requires manual inspection and correction to avoid errors. Below 30% sequence identity, the relationship is considered remote, and template-based modeling becomes exceedingly difficult, making alternative methods like AlphaFold2 potentially more suitable [5] [48].
FAQ 5: How can I improve my target-template alignment for low-identity cases? For low sequence identity cases, move beyond simple pairwise sequence alignment. Use profile-based methods that leverage multiple sequence alignments (MSAs) for both the target and template sequences. Align the target sequence to a structure-based multiple alignment of your candidate templates. Manually inspect and edit the alignment in the context of the template's 3D structure, avoiding placing gaps in secondary structure elements or buried core regions [48].
Troubleshooting Guides
Issue: Model has severe steric clashes or unrealistic bond lengths. Possible Causes and Solutions:
- Cause: Incorrect side-chain packing during model building.
- Solution: Use a modeling program that employs a backbone-dependent rotamer library and energy minimization, like SWISS-MODEL's ProMod3, to optimize side-chain conformations and relieve clashes [50].
- Cause: Errors in the alignment, particularly in loop regions.
- Solution: Manually inspect the alignment in the problematic region against the template structure. Realign if necessary, ensuring gaps are placed in solvent-exposed, flexible regions rather than in rigid secondary structures [48].
Issue: Specific loop regions (e.g., CDR loops) are modeled inaccurately. Possible Causes and Solutions:
- Cause: The selected template has a different loop conformation or length.
- Cause: The loop is not covered by any template.
- Solution: Use a modeling server like SWISS-MODEL, which can perform a conformational space search using Monte Carlo techniques to model insertions and deletions not present in the template [50]. Alternatively, consider ab initio or deep learning-based loop modeling approaches.
Issue: The overall model quality is lower than expected based on template quality. Possible Causes and Solutions:
- Cause: The target-template alignment contains errors.
- Solution: This is the most common cause. Re-examine the alignment using more sophisticated profile-profile alignment methods. Check for consistent conservation of key functional residues [48].
- Cause: Over-reliance on a single, potentially flawed, template.
- Solution: Generate models using multiple templates and use a model quality assessment program (MQAP) like ProQ to select the best one. Studies show that while multi-template modeling can produce the best models, it can also produce worse ones, so careful selection is key [14].
Quantitative Data on Template Selection and Model Quality
The following table summarizes key findings from systematic studies on template-based modeling.
Table 1: Impact of Modeling Strategies on Model Quality
| Modeling Factor | Impact on Model Quality | Key Findings | Source |
|---|---|---|---|
| Multiple Templates | Can be positive or negative | - Using 2-3 templates in Modeller can improve the average TM-score by ~0.01 vs. a single template. - A significant number of multi-template models can be worse than the best single-template model. - Quality improvement is often due to model extension; improvement in core residues is smaller. | [14] |
| Sequence Identity | Directly correlated with accuracy | - >40% identity: alignments are generally trivial, models are accurate. - 30-40% identity: alignment is critical and often requires manual intervention. - <30% identity: alignment is highly nontrivial, models are less reliable. | [48] |
| AI vs. Homology Modeling | Context-dependent superiority | - AI methods (AlphaFold2) generally show superiority and can accurately model challenging loops (e.g., GPCR extracellular loops). - Homology modeling can still outperform AI for specific domains if a template with a highly relevant functional state (e.g., a G-protein complex) is used. | [5] |
| Antibody H3 Loop Prediction | Varies by method | - RoseTTAFold can achieve accuracy comparable to SWISS-MODEL for templates with GMQE < 0.8. - For H3 loops, RoseTTAFold's accuracy was better than ABodyBuilder but comparable to SWISS-MODEL. | [49] |
Experimental Protocols
Protocol 1: A Standard Workflow for Template Selection and Model Building
This protocol outlines the key steps for selecting templates and building a homology model, with a focus on obtaining the best possible loop regions.
- Input Target Sequence: Provide the full amino acid sequence of your target protein.
- Identify Candidate Templates: Use a protein structure prediction portal like Phyre2.2 or SWISS-MODEL. These servers search extensive template libraries (including the PDB and AlphaFold DB) using sequence profile methods (e.g., HHblits) to identify potential templates [39] [50].
- Evaluate and Select Templates: For each candidate, assess:
- Sequence Identity & Coverage: Prefer higher identity and greater coverage of your target sequence.
- Structure Quality: Check the experimental resolution of the template (lower is better, e.g., <2.5 Å).
- Biological Context: Prefer templates from the same protein family and, if relevant, with similar ligands or bound to similar proteins (e.g., for GPCR modeling, a template in the Gs-bound state is ideal) [5].
- Loop Presence: Check if the candidate template has resolved coordinates for the loop regions you are interested in.
- Generate and Refine Alignment: Use the server's default alignment. For difficult targets (<30% identity), manually inspect and edit the alignment in a viewer like DeepView, ensuring gaps are not placed in secondary structures or buried cores [50] [48].
- Build Model: Use the server's modeling engine (e.g., SWISS-MODEL's ProMod3, Modeller) to construct the 3D model. If multiple good templates exist, run builds using single and multiple templates [14] [50].
- Select and Validate the Final Model: Use the provided model quality estimates (GMQE, QSQE). Run your models through an MQAP like ProQ to select the best one. Visually inspect problematic loops and steric clashes [14].
Protocol 2: Benchmarking Modeling Methods for Antibody CDR Loops
This protocol is based on the comparative study of RoseTTAFold, SWISS-MODEL, and ABodyBuilder [49].
- Test Set Generation: Curate a non-redundant set of antibody sequences with known experimental structures. A common source is the Structural Antibody Database (SAbDab). Apply filters for sequence identity (e.g., <80%) and structure resolution (e.g., <3.2 Å).
- Model Generation with Multiple Methods:
- RoseTTAFold: Input the heavy and light chain sequences. Run the
make_msa.shscript to generate MSAs, then usepredict_complex.pyto model the Fv region. Refine with Rosetta FastRelax. - SWISS-MODEL: Submit the antibody sequences via the interactive web server in automated mode.
- ABodyBuilder: Use the web server or local installation.
- RoseTTAFold: Input the heavy and light chain sequences. Run the
- Structural Comparison: Superpose the modeled Fv region onto the experimental reference structure. Calculate the root-mean-square deviation (RMSD) for the backbone atoms of each CDR loop (H1, H2, H3, L1, L2, L3).
- Analysis: Compare the RMSD values across the different methods to determine which provides the most accurate prediction for each loop type, with special attention to the H3 loop.
Workflow Visualization
The following diagram illustrates the logical workflow and decision points for selecting templates with the goal of improving loop modeling accuracy.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Template-Based Modeling
| Item | Function in Experiment | Key Application Notes |
|---|---|---|
| SWISS-MODEL Workspace | A web-based service for automated protein structure homology modelling. | Provides automated template identification, model building, and quality evaluation. Excellent for standard homology modeling and offers oligomeric modeling [50]. |
| Phyre2.2 | A template-based protein structure modeling portal. | Searches a comprehensive template library, including AlphaFold DB models, and facilitates the modeling of apo/holo structures. Useful for identifying the most suitable template for a given query [39]. |
| Modeller | A program for comparative protein structure modeling. | Can use multiple templates and spatial restraints to build models. Shown to be effective in producing improved multi-template models, though care is needed [14]. |
| RoseTTAFold | A deep learning-based three-track neural network for protein structure prediction. | Can be used for antibody modeling and has shown promising results for predicting challenging H3 loops, especially when homology templates are weak [49]. |
| ProQ / Model Quality Assessment Programs (MQAPs) | Software to predict the quality of a protein model. | Essential for selecting the best model from a set of candidates, as visual inspection alone is often insufficient [14]. |
| DeepView (Swiss-PdbViewer) | An interactive molecular graphics tool. | Used for the manual manipulation and analysis of protein structures and alignments in the Project Mode of SWISS-MODEL, crucial for difficult modeling cases [50]. |
Troubleshooting Guides
PSI-BLAST Related Issues
Problem: PSI-BLAST fails to detect distant homologs
Question: My initial PSI-BLAST search does not find any distant homologs, even though I suspect they exist. What parameters should I adjust?
Solution:
- Lower the e-value threshold for inclusion: The e-value threshold determines which sequences are included in the Position-Specific Scoring Matrix (PSSM) for the next iteration. Beginners are advised to use a profile-inclusion threshold of E-value = 0.005. Users familiar with globular domains may use a threshold of 0.01 if the sequence lacks major compositionally biased segments [51].
- Increase the number of iterations: Distant relationships often require multiple iterations. For example, the relationship between human PCNA and E. coli DNA polymerase III β-subunit was only detected in the fifth iteration [51].
- Check the number of alignments and descriptions: When searching large databases like NCBI's non-redundant (nr) database, set the maximum number of alignments and descriptions to at least 1000 to retrieve all potentially significant hits [51].
- Verify sequence composition: Performance decreases with compositionally biased sequences. Adjust thresholds accordingly [51].
Preventative Steps:
- Use the "two-page formatting" option on the NCBI PSI-BLAST page for greater parameter control and save this configuration as a bookmark for future use [51].
- For standalone versions, utilize command-line flags to specify parameters precisely [51].
Problem: PSI-BLAST results contain false positives
Question: How can I distinguish true homologs from false positives in my PSI-BLAST results?
Solution:
- Evaluate statistical significance: As a heuristic, for compositionally unbiased queries encompassing a globular domain, a hit with an E-value ≤ 0.01 is likely indicative of a homologous relationship. However, always evaluate alignments on a case-by-case basis [51].
- Inspect alignment patterns: Examine the sequence alignment for biologically meaningful conservation. A true relationship, like that between PCNA and the β-subunit, will show patterns of conserved hydrophobic residues (critical for the hydrophobic core) and strategically placed polar/charged residues (often involved in solvent interaction or function) [51].
- Use taxonomy reports: Click the "taxonomy reports" link in the results to see the organisms of the hits. A scattered taxonomic distribution can support homology, while a very narrow distribution might warrant caution [51].
HMM and Multiple Template Related Issues
Problem: Integrating HMMs with PSI-BLAST for improved detection
Question: How can I combine HMMs with PSI-BLAST to find even more remote homologs?
Solution: This hybrid approach uses an HMM to create an initial, high-quality multiple alignment, which is then used to seed a more sensitive PSI-BLAST search [52].
Experimental Protocol:
- HMM Construction: Build an HMM for your protein family of interest, focusing on structurally conserved repeats. Incorporate predicted secondary structure to guide transition probabilities, favoring insertions or deletions in loop regions [52].
- Sequence Simulation: Use the optimized HMM to emit a large number of simulated sequences that embody the conserved characteristics of the family [52].
- Profile Generation: Use these simulated sequences as input for PSI-BLAST to generate a checkpoint file (PSSM) [52].
- Database Search: Perform a new PSI-BLAST search of your target database, initializing it with the checkpoint profile from the previous step. This leverages the statistical framework of PSI-BLAST while being guided by the structural constraints captured by the HMM [52].
Problem: Multi-template modeling produces worse models than single-template
Question: I used multiple templates for homology modeling, but the resulting model is worse than the one from the best single template. Why did this happen?
Solution: This is a known risk. While multi-template modeling can produce better models, the average quality does not always improve significantly because some models are worse [14].
- Cause 1: Low-quality input alignments. The most critical factor for success is the existence of high-quality target-template alignments [14].
- Cause 2: Incorrect combination of templates. Simply using all available templates can degrade model quality, especially in the core regions present in the best single-template model [14].
- Mitigation Strategy:
- Use model quality assessment programs (e.g., ProQ) to rank the generated models. This can improve the average selected model quality by about 5% [14].
- Prefer loop remodeling over loop modeling when dealing with multiple loops, as it typically gives more accurate results [29].
- Ensure the flanking regions (the four residues adjacent to each loop) are accurate and ideally located in a helix or sheet region of the initial homology model [29].
Frequently Asked Questions (FAQs)
General Workflow Questions
Q: What is the primary advantage of using PSI-BLAST over a standard BLASTp search? A: PSI-BLAST is significantly more sensitive for detecting distant evolutionary relationships. While the first iteration is identical to BLASTp, PSI-BLAST iteratively builds a Position-Specific Scoring Matrix (PSSM) from the significant hits. This PSSM captures the conservation patterns of a whole protein family, allowing it to find homologs that have diverged too much in sequence to be found by the single-sequence query used in BLASTp [51] [53].
Q: When should I consider using a multiple-template approach in homology modeling? A: Consider a multiple-template approach when no single template provides complete coverage of your target sequence or when different templates have high-quality alignments in different regions of the target. Systematic studies have shown that a multi-template combination algorithm can improve the average GDT-TS score of predicted models by 6.8% compared to using a single top template [54].
Q: Can these methods be applied to membrane proteins? A: Yes. Research indicates that profile-to-profile alignment methods (like those used in advanced PSI-BLAST and HMM techniques) perform well for membrane proteins. Furthermore, acceptable homology models (Cα-RMSD ≤ 2 Å in transmembrane regions) can be achieved with template sequence identities of 30% or higher, provided an accurate alignment is used [55].
Technical and Practical Questions
Q: My protein has long loops that are poorly modeled. What is a state-of-the-art approach for loop remodeling? A: For accurate loop modeling in homology models, use a dedicated data-based server like DaReUS-Loop. It mines the entire PDB for candidate loop fragments that fit the geometric and sequence constraints of your model's flanking regions [29] [31].
DaReUS-Loop Protocol:
- Input: Provide an initial homology model (in PDB format) and the full protein sequence.
- Define Loops: In "remodeling mode," indicate the loop regions to be remodeled by gapping the corresponding residues in the input sequence (using "-" or lower-case letters) [29].
- Execution: The server filters candidates based on sequence, local conformation profile, and structural fit, returning 10 models per loop [29] [31].
- Output Analysis: Use the provided confidence score, which correlates with expected accuracy, to select the best model. Strikingly, over 50% of successful loops identified by DaReUS-Loop come from unrelated proteins, demonstrating that structural constraints often transcend homology [31].
Q: How do I incorporate information from AlphaFold models into my template-based modeling workflow? A: Servers like Phyre2.2 now facilitate this. You can submit your sequence, and Phyre2.2 will automatically perform a BLASTp search to find the closest AlphaFold model in the EBI database and use it as a template for building your model. This provides an easy way to leverage the vast library of accurate AlphaFold predictions within a trusted homology modeling framework [28].
Performance of Multi-Template Modeling
Table 1: Improvement in Model Quality with Multi-Template Approaches
| Study Description | Performance Metric | Single-Template | Multi-Template | Improvement | Notes |
|---|---|---|---|---|---|
| CASP7 CM targets (45 proteins) [54] | Average GDT-TS Score | 66.59 | 71.15 | 6.8% (4.56 points) | Algorithm automatically selected and combined templates. |
| Modeller on CASP7 targets [14] | Average TM-Score (Core residues) | Baseline | ~0.01 increase | ~1% | Best improvement seen using 2-3 templates; more can degrade quality. |
Loop Modeling Accuracy
Table 2: Performance of DaReUS-Loop on Template-Based Test Sets [31]
| Loop Length | Modeling Context | Reported Accuracy (RMSD) | Key Advantage |
|---|---|---|---|
| Short to Long (4-30 residues) | Homology Models (CASP11, CASP12, HOMSTRAD) | High number of predictions with RMSD < 2 Å | Significant enhancement for long loops (≥15 residues). |
| All lengths | General | Confidence score provided | Score correlates well with expected accuracy. |
Workflow and Pathway Diagrams
PSI-BLAST and HMM Integrated Workflow
Multi-Template Combination Algorithm Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Advanced Alignment and Modeling
| Resource Name | Type | Primary Function | Access Link |
|---|---|---|---|
| NCBI PSI-BLAST | Web Server / Standalone Tool | Perform iterative protein sequence searches to detect distant homologs and build PSSMs. | https://www.ncbi.nlm.nih.gov/BLAST/ |
| DaReUS-Loop | Web Server | Accurately model loops in homology models by mining fragment candidates from the entire PDB. | https://bioserv.rpbs.univ-paris-diderot.fr/services/DaReUS-Loop/ |
| Phyre2.2 | Web Server | Template-based modeling server that can automatically leverage AlphaFold models as templates. | https://www.sbg.bio.ic.ac.uk/phyre2/ |
| Modeller | Software Package | Build homology models, including support for multiple templates and spatial restraints. | https://salilab.org/modeller/ |
Achieving atomic-level accuracy in comparative (homology) protein models is often limited by the challenge of refining loop regions closer to their native state. Unlike loop reconstruction in crystal structures, loop refinement in homology models is complicated by inaccuracies in the surrounding environment, including errors in side-chain conformations, backbone atoms flanking the loop, and other non-adjacent structural elements. This technical support center provides targeted guidance to address the specific issues researchers encounter when refining loops in such imperfect environments.
Why is loop refinement in homology models particularly difficult?
Loop refinement in homology models is more difficult than in crystal structures because side-chain, backbone, and other structural inaccuracies surrounding the loop create a complex sampling problem. The loop often cannot be refined successfully without simultaneously refining its adjacent portions [56].
Troubleshooting Guides and FAQs
FAQ: Core Concepts and Method Selection
Q1: What is the fundamental difference between loop prediction in crystal structures and loop refinement in homology models?
Loop prediction in crystal structures is performed in a nearly perfect, native-like environment. In contrast, loop refinement in homology models must contend with an inaccurate environment, including non-native side-chain conformations and backbone errors surrounding the loop. This makes the sampling problem much more challenging [56].
Q2: My loop refinement fails to improve the model. What is a common sampling-related cause?
A common cause is refining the loop in isolation without simultaneously optimizing its molecular environment. Errors in the conformations of side chains surrounding the loop can create steric clashes or disrupt favorable interactions, preventing the loop from adopting its correct, low-energy conformation [56].
Q3: How does simultaneous side-chain optimization improve loop refinement?
Methods that simultaneously optimize loop conformation and the rotamer states of surrounding side chains allow the local environment to adjust to accommodate the new loop. This can recover the native state in many cases where loop-only prediction fails [56].
Q4: When should I consider using Molecular Dynamics (MD) for refinement?
MD can be useful for exploring the conformational landscape and relaxing the model. A protocol often involves a quick geometry pre-optimization followed by an MD simulation in a specific ensemble (e.g., NVE). However, the effect on accuracy can be inconsistent, sometimes improving some domains while decreasing accuracy in others [5] [57].
Troubleshooting Guide: Common Experimental Issues
| Problem Area | Specific Issue | Potential Causes | Recommended Solutions |
|---|---|---|---|
| Sampling & Accuracy | Refined loop RMSD remains high. | Refining loop in isolation; inaccurate surrounding side-chains. | Use a method that samples loop and side-chains simultaneously (e.g., HLP-SS) [56]. |
| Refinement leads to steric clashes. | Insufficient sampling or poor initial model. | Run more sampling stages with constrained backbone atoms; use a hierarchical protocol [56]. | |
| Energy Evaluation | Low-energy model is far from native state. | Forcefield inaccuracies; insufficient sampling of near-native states. | Use an all-atom forcefield with implicit solvent; employ hierarchical sampling with knowledge-based dihedral libraries [56]. |
| MD Simulation | MD simulation becomes unstable. | Too long a timestep; bad initial contacts. | Pre-optimize the geometry with a forcefield; reduce timestep (e.g., to 1 fs); check initial velocities [57]. |
| Model Interpretation | Uncertainty in model quality. | Lack of experimental validation. | Compare multiple modeling approaches (Homology, AI, MD); focus on consistent regions [5]. |
Quantitative Data and Performance Metrics
Table 1: Loop Prediction Accuracy in Different Environments (Backbone RMSD in Ångströms)
This table summarizes the performance of loop prediction methods, highlighting the challenge of imperfect environments and the improvement offered by simultaneous side-chain optimization. Data is presented as median RMSD values [56].
| Loop Length | Native Environment (HLP) | Perturbed Surroundings (HLP) | Perturbed Surroundings (HLP-SS) |
|---|---|---|---|
| 6 residues | 0.3 Å | 1.1 Å | 0.4 Å |
| 8 residues | 0.6 Å | 2.2 Å | 0.8 Å |
| 10 residues | 0.4 Å | 1.5 Å | 1.1 Å |
| 12 residues | 0.6 Å | 2.3 Å | 1.2 Å |
Table 2: Comparison of Protein Structure Modeling Approaches
This table compares the relative strengths and weaknesses of different computational modeling methods, based on assessments of models for the GPR101 receptor [5].
| Modeling Method | Typical Use Case | Relative Strength | Key Limitation |
|---|---|---|---|
| Homology Modeling | Template-based structure prediction. | Can accurately predict specific domains (e.g., G protein binding mode) when using a closely related template [5]. | Overall accuracy generally lower than AI methods; quality highly template-dependent [5]. |
| AI Methods (AlphaFold2) | De novo structure prediction. | High overall accuracy; excels at predicting challenging loops (e.g., 2nd extracellular loop) [5]. | May be less accurate than specialized homology models for specific domains like TM6 [5]. |
| Molecular Dynamics | Refinement and conformational sampling. | Can relax models and explore dynamics. | Does not consistently improve accuracy; may increase RMSD in some regions [5]. |
Experimental Protocols and Workflows
Protocol 1: Hierarchical Loop Prediction with Surrounding Side-Chain Optimization (HLP-SS)
This protocol augments standard loop prediction by simultaneously optimizing the loop and its surrounding side chains, which is crucial for refining loops in homology models [56].
- Input Structure Preparation: Begin with your initial homology model. Identify the loop region to be refined.
- Initial Sampling Stages: Perform parallel initial loop predictions with varying degrees of allowed steric overlap (e.g., overlap factors of 0.7 and 0.6). This generates a diverse set of starting loop conformations.
- Selection and First Refinement: Select the five lowest-energy loops from each initial stage. Pass each of these structures to a refinement stage where the loop's Cα atoms are constrained to within 4 Å of their positions in the starting loop. This focuses the sampling.
- Second Refinement with Side-Chain Optimization: Select the five lowest-energy loops from all first refinement stages. Pass each to a final set of parallel refinement stages. In these stages, the Cα atoms are more tightly constrained (e.g., within 2 Å), and the rotamer states of side chains surrounding the loop are simultaneously sampled and optimized.
- Final Selection: The single loop conformation with the lowest all-atom forcefield energy, calculated with implicit solvent and correction terms, is selected as the final refined model.
Protocol 2: Molecular Dynamics for System Relaxation (Using AMS/PLOMS)
This protocol outlines a basic MD workflow for relaxing a system, which can be applied to a full protein or a refined loop model [57].
- System Setup: Create the molecular system. For a box of molecules, you can use tools like
packmolto set up the system at a desired density. - Pre-Optimization: Run a quick geometry optimization on the system using a forcefield (e.g., UFF) to remove any bad contacts and prepare a stable starting structure for MD. Use a setting like
maxiterations=50[57]. - MD Simulation Configuration:
- Set the
ForceField.Type(e.g., 'UFF'). - Define the ensemble (e.g., NVE for constant energy).
- Set the initial
temperature(e.g., 300 K) to initialize velocities from the Maxwell-Boltzmann distribution. - Choose a
timestep(e.g., 1.0 femtoseconds). - Set the total number of steps (
nsteps) and the sampling frequency (samplingfreq).
- Set the
- Run Simulation: Execute the MD job. Monitor the output for properties like potential energy, kinetic energy, and temperature to ensure stability.
- Analysis: After the run, analyze the trajectory file (e.g.,
ams.rkf) to plot energies, visualize the structural evolution, and extract relevant properties.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software and Computational Tools
| Item Name | Function / Role in Experiment |
|---|---|
| PLOP (Protein Local Optimization Program) | Software implementation of the Hierarchical Loop Prediction (HLP) and HLP-SS methods for high-accuracy loop construction and refinement [56]. |
| Knowledge-Based Dihedral Angle Libraries | Provides Ramachandran plot (backbone) and rotamer (side-chain) preferences to guide efficient and physically realistic conformational sampling [56]. |
| All-Atom Forcefield with Implicit Solvent (e.g., OPLS) | Provides the energy function for scoring and ranking sampled loop conformations. Includes terms for molecular mechanics, polar solvation (e.g., GBSA), and non-polar solvation [56]. |
| AMS with PLAMS | A computational environment that allows for running and scripting various molecular simulations, including Molecular Dynamics and geometry optimizations [57]. |
| PyDSSP / MDAnalysis.analysis.dssp | A Python module used for assigning protein secondary structure (helix, sheet, loop) from 3D coordinates, useful for analyzing simulation trajectories [58]. |
Frequently Asked Questions
1. What are the most common causes of failure in long loop modeling? Failure in long loop modeling is primarily caused by an insufficient number of decoys sampled, inaccurate energy functions that fail to identify native-like conformations, and weak spatial restraints from the flanking regions. Long loops have a much larger conformational space, making exhaustive sampling computationally expensive. Inaccurate scoring then compounds this problem by not guiding the search toward biologically relevant structures.
2. How do errors in flanking regions propagate into the loop model? The fixed flanking regions act as anchor points for the loop. If their relative orientation is incorrect due to errors in the template structure or the core model, the loop is forced to bridge an unnatural gap. This strain often results in modeled loops with high energy, poor stereochemistry, or clashes that would not occur with correct flanking geometry.
3. What are the key indicators of a poor-quality loop model? Key indicators include high energy terms (particularly for van der Waals clashes and torsional angles), poor rotameric states for side chains, deviation from ideal bond lengths and angles, and a high Root-Mean-Square Deviation (RMSD) from a known reference structure if available. Validation tools like MolProbity will typically flag these models.
4. When should I consider using an alternative template or method? Consider alternative approaches if the core homology model has large insertions/deletions (indels) in loop regions relative to your template, if the sequence identity of the template is very low (<20%), or if multiple independent loop modeling protocols consistently produce models that fail validation checks.
5. How can I improve the sampling efficiency for long loops? Combining multi-scale methods is effective. Start with coarse-grained sampling to explore large-scale conformational space cheaply, then refine a subset of promising low-energy decoys with all-atom molecular dynamics. Using enhanced sampling techniques like replica exchange can also improve the exploration of energy landscapes.
Table 1: Performance comparison of different loop modeling methods based on loop length. Accuracy is measured by RMSD (Å) from the native structure.
| Modeling Method | Loop Length (residues) | Average Accuracy (RMSD) | Computational Cost | Best Use Case |
|---|---|---|---|---|
| Knowledge-Based (e.g., FREAD) | 4-8 | < 1.0 Å | Low | Short loops with many homologs |
| Ab Initio (e.g., Rosetta) | 8-12 | 1.0 - 2.5 Å | High | Loops with no sequence homology |
| MD with Restraints | 12-20 | 2.0 - 4.0 Å | Very High | Refining models near native state |
| Hybrid (Knowledge + MD) | 8-14 | 1.5 - 3.0 Å | Medium | Balancing speed and accuracy |
Table 2: Impact of flanking region accuracy on loop modeling success rates.
| Flanking Region RMSD | Success Rate (Loop RMSD < 2.0 Å) | Observed Average Loop RMSD |
|---|---|---|
| < 0.5 Å | 75% | 1.2 Å |
| 0.5 - 1.0 Å | 45% | 2.5 Å |
| > 1.0 Å | 15% | 4.8 Å |
Experimental Protocol: Assessing Loop Quality and Flanking Region Stability
This protocol describes a workflow for evaluating and validating a modeled loop structure, with a focus on identifying issues stemming from the flanking regions.
1. Preparation of the Initial Model
- Begin with your homology model containing the loop of interest, generated by software like MODELLER, SWISS-MODEL, or a custom script.
- Ensure all hydrogen atoms are added and the protonation states of relevant residues (e.g., in the active site) are correct.
2. Energy Minimization of the System
- Use a molecular mechanics program like AMBER, GROMACS, or OpenMM.
- Employ a force field (e.g., ff19SB, CHARMM36).
- Solvate the protein model in a TIP3P water box, ensuring a minimum distance of 10 Å between the protein and the box edge.
- Neutralize the system's charge by adding ions (e.g., Na⁺, Cl⁻).
- Perform a two-step minimization:
- First, minimize only the hydrogen atoms while restraining the heavy atoms (backbone and side chains) with a force constant of 50 kcal/mol/Ų.
- Second, perform a brief, unrestrained minimization of the entire system (500-1000 steps) to relieve any minor steric clashes introduced during model building.
3. Restrained Molecular Dynamics for Relaxation
- Run a short MD simulation (50-100 ps) in explicit solvent.
- Apply strong positional restraints (e.g., 10-50 kcal/mol/Ų) on the Cα atoms of the core protein structure, but leave the loop and its immediate flanking residues (2-3 residues on each side) completely unrestrained.
- This allows the loop and its anchor points to relax and find a more natural conformation relative to the stable core.
4. Analysis of the Relaxed Model
- Loop Conformation: Calculate the RMSD of the relaxed loop against its initial model to see how much it has moved.
- Flanking Region Stability: Calculate the Cα Root-Mean-Square Fluctuation (RMSF) of the flanking residues during the simulation. High fluctuations can indicate inherent instability or strain.
- Energetic and Steric Quality: Run the final, relaxed model through a validation server like MolProbity. Pay close attention to:
- Clashscore (should be in the 100th percentile, i.e., low number of clashes).
- Ramachandran outliers (should be < 1% for a high-quality model).
- Rotamer outliers.
Workflow Diagram for Loop Modeling Troubleshooting
Troubleshooting workflow for loop modeling, guiding users to address issues with flanking regions or the loop itself.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential software tools and resources for loop modeling and validation.
| Tool / Resource | Function | Application in Loop Modeling |
|---|---|---|
| MODELER | Comparative protein structure modeling | Integrates spatial restraints from the template to model loops and side chains. |
| Rosetta | Ab initio structure prediction & design | Powerful for de novo loop modeling where no template is available. |
| MolProbity | Structure validation server | Provides clashscores, Ramachandran analysis, and rotamer statistics to identify poor regions. |
| Phenix | Software suite for macromolecular structure determination | Includes tools for real-space refinement and geometry minimization of loops. |
| GROMACS/AMBER | Molecular dynamics simulations | Used for energy minimization and restrained MD to relax and assess loop models. |
| PDB_REDO | Re-refined macromolecular structure database | Provides improved reference structures for benchmarking and analysis. |
Frequently Asked Questions
Q1: What are the primary advantages of integrating multiple modeling servers over a single-server approach? Integrating multiple modeling servers allows researchers to cross-validate results, leverage the unique strengths of different algorithms, and improve overall reliability. This is crucial for challenging targets in homology modeling, where no single method may be sufficient. The integrated approach mitigates the risk of method-specific biases and increases confidence in the final model.
Q2: A common error is "Job Submission Failed" when sending tasks to a modeling server. What are the initial diagnostic steps? First, verify your network connection and the server's operational status. Second, check that your input data format complies with the server's specific requirements (e.g., FASTA format, allowed characters). Third, confirm that your job parameters, such as sequence length or template selection, are within the server's accepted limits.
Q3: How should I handle conflicting results from different integrated servers? Conflicting results, such as different predicted loop conformations, should be analyzed systematically. First, check the quality and identity of the templates used by each server. Second, use statistical measures from each server's output (like Z-scores or model confidence scores) to assess reliability. Third, where possible, use a third, independent validation server or experimental data to arbitrate.
Q4: What is the recommended way to manage and track multiple modeling jobs across different servers? Implement a centralized job management system. The system should track:
- Job Identifier: A unique ID for your experiment.
- Server Name: The modeling server used.
- Submission Timestamp: When the job was sent.
- Status: (e.g., Queued, Running, Completed, Error).
- Result URL/Location: Where to retrieve the output.
Q5: Our automated pipeline occasionally fails to parse the output from a modeling server. What can be done? This is often due to changes in the server's output format. Implement a "contract testing" strategy where your pipeline periodically runs a known sequence and verifies it can find and parse the expected data fields. This alerts you to format changes before they disrupt major experiments. For critical servers, consider reaching out to the maintainers to inquire about a stable API.
Troubleshooting Guides
Issue 1: Server Integration and Data Flow Errors
Problem: Inability to connect to a remote modeling server or a failure in the data transfer process.
| Step | Action | Expected Outcome |
|---|---|---|
| 1 | Verify basic connectivity using tools like ping or traceroute. |
Confirmation of network reachability to the server. |
| 2 | Check for SSL/TLS certificate issues, especially with HTTPS connections. | Secure connection is established without certificate errors. |
| 3 | Validate the format and structure of the data being sent against the server's API documentation. | Data is accepted by the server without schema errors. |
| 4 | Check the server's status page or documentation for known outages or maintenance. | Confirmation that the server is operational. |
Issue 2: Parsing and Processing Server Outputs
Problem: The pipeline successfully retrieves results from a server but fails to correctly interpret or extract key data (e.g., model coordinates, confidence scores).
| Step | Action | Expected Outcome |
|---|---|---|
| 1 | Manually inspect the raw output file from the server. | Identification of the exact structure and delimiters used. |
| 2 | Compare the current output format with the format your parser was designed for. | Detection of any changes or inconsistencies in the data structure. |
| 3 | Update the parsing logic to handle the current format, adding robust error handling for missing data. | The parser successfully extracts all required data fields. |
| 4 | Run the updated parser on a set of historical outputs to validate it doesn't break previous functionality. | All test cases are processed successfully. |
Issue 3: Resolving Inconsistencies in Model Quality
Problem: Integrated servers return final models with significantly different quality assessment scores.
| Step | Action | Expected Outcome |
|---|---|---|
| 1 | Collect Scores: Gather all quantitative quality scores (e.g., QMEAN, MolProbity, DOPE) for each model. | A complete dataset for comparative analysis. |
| 2 | Normalize and Compare: Normalize scores where possible and tabulate them for easy comparison. | A clear overview of which model performs best on which metric. |
| 3 | Identify Strong Regions: Analyze per-residue or local quality scores to determine if one model is consistently better in your region of interest (e.g., the active site). | Identification of the most reliable model for your specific research question. |
| 4 | Consider a Meta-Prediction: Use the outputs as inputs to a consensus or model-averaging tool. | Generation of a single, refined model that incorporates the strengths of all inputs. |
Experimental Protocols for Server Integration
Protocol 1: Establishing a Basic Multi-Server Modeling Workflow
This protocol outlines the steps for a standard experiment that leverages multiple servers to generate a homology model.
1. Objective: To generate a high-confidence homology model for a target sequence by integrating results from three independent modeling servers.
2. Materials and Reagents:
- Target protein sequence in FASTA format.
- Access to a minimum of three modeling servers (e.g., SWISS-MODEL, Phyre2, I-TASSER, RaptorX).
- Centralized computing environment or job management script.
3. Methodology:
- Step 1 - Target Preparation: Prepare your target sequence, ensuring it is in the correct FASTA format and free of invalid characters.
- Step 2 - Parallel Job Submission: Submit the target sequence to each of the selected modeling servers simultaneously. Record the job IDs and server names.
- Step 3 - Result Retrieval: Periodically poll the servers to check job status. Upon completion, download the resulting model files (typically in PDB format) and any associated quality reports.
- Step 4 - Data Collation: Store all results in a structured directory, labeling each model with its source server and a timestamp.
Protocol 2: Quantitative Comparison and Consensus Model Building
This protocol details how to analyze the outputs from the basic workflow to select or create a final model.
1. Objective: To quantitatively compare models from multiple servers and build a consensus model for a challenging loop region.
2. Materials and Reagents:
- Output PDB files from Protocol 1.
- Model quality assessment software (e.g., ProSA-web, MolProbity, QMEANDisCo).
- Molecular visualization software (e.g., PyMOL, UCSF Chimera).
- Scripting environment (e.g., Python with Biopython).
3. Methodology:
- Step 1 - Quality Scoring: Run each generated model through a set of quality assessment tools. Record global scores (like ProSA-web Z-score) and local scores for the loop region of interest.
- Step 2 - Tabulate Results: Create a summary table of all quality metrics for easy comparison. Table: Example Model Quality Assessment Summary
| Model Source | ProSA-web Z-score | QMEANDisCo | MolProbity Clashscore | Best for Active Site? |
|---|---|---|---|---|
| Server A | -8.5 | 0.75 | 12 | Yes |
| Server B | -7.9 | 0.68 | 25 | No |
| Server C | -8.2 | 0.71 | 15 | Partial |
- Step 3 - Structural Alignment and Analysis: Superimpose all models onto a common frame of reference. Visually inspect regions of high divergence, particularly loops and termini.
- Step 4 - Generate Consensus: If no single model is superior overall, use the structural alignment to manually build a consensus model in molecular visualization software, or use the best parts from each model (e.g., the core from Server A, a loop from Server C).
Workflow Visualization
The following diagrams illustrate the logical flow of the integrated server protocols.
Integrated Server Modeling Workflow
Server Integration Error Resolution
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Computational Tools for Integrated Modeling
| Item | Function/Benefit |
|---|---|
| Modeling Servers (SWISS-MODEL, Phyre2) | Provide automated, rapid homology models based on established template structures, offering a strong starting point. |
| Ab Initio Server (I-TASSER, Rosetta) | Crucial for modeling regions with no detectable templates (e.g., long loops), using physical principles rather than homology. |
| Quality Assessment Tools (ProSA-web, MolProbity) | Evaluate the structural and energetic plausibility of generated models, helping to identify potential errors. |
| Molecular Visualization Software (PyMOL, Chimera) | Allows for visual inspection of models, manual model building, and analysis of specific regions like binding sites. |
| Job Management Script (Python/Bash) | Automates the submission, monitoring, and retrieval of jobs from multiple servers, ensuring reproducibility and saving time. |
Assessing Model Quality: Validation Metrics and Performance Benchmarking
Troubleshooting Guides
FAQ: Ramachandran Plot Outliers
Q: What does a Ramachandran plot outlier indicate in my model, and when should I be concerned? A: A Ramachandran plot outlier signifies a protein backbone conformation that is stereochemically rare and strained. While most outliers at lower resolution or in poor electron density indicate errors, a small number can be valid if supported by unambiguous electron density and a clear structural rationale [59]. Genuine outliers are occasionally observed in high-resolution protein and peptide structures where variations in bond length and angle relieve expected steric clashes [60]. You should be concerned if multiple outliers cluster in loop regions or near active sites, if they are accompanied by high clashscores, or if the electron density does not convincingly support the strained conformation.
Q: How can I correct a Ramachandran plot outlier? A: Follow this systematic protocol for correction:
- Inspect the Electron Density: First, visualize the outlier residue in your molecular graphics software (e.g., Coot, ChimeraX) alongside its
2F_o - F_candF_o - F_cmaps. Confirm whether the backbone conformation is truly supported by the density [59]. - Check for Supporting Evidence: Before moving the backbone, investigate if the outlier is stabilized by specific hydrogen bonds, ligand binding, or part of a functionally important strained motif. If evidence is strong, the outlier may be valid.
- Initiate Automated Correction: Use automated refinement suites like PHENIX or PDB-REDO, which can correct many outlier conformations during refinement cycles while maintaining the model-to-data fit [33].
- Manual Real-Space Refinement: If automation fails, manually refine the residue in real-time using a tool like Coot's "Regularize Zone" or "Real-Space Refine Zone" function. This allows you to adjust the φ and ψ angles while visually tracking the fit into the electron density.
- Validate the Correction: After adjustment, re-validate the structure. Ensure the correction has not introduced new steric clashes or poor rotamer states and that the real-space correlation coefficient (RSCC) for the residue has improved [33].
Q: What is the difference between a classical Ramachandran plot and a bond geometry-specific steric-map? A: Classical Ramachandran plots use idealized, standard bond lengths and angles to define allowed and disallowed regions. In contrast, bond geometry-specific steric-maps are highly sensitive to the precise bond length and angle values observed at each specific residue position in ultra-high-resolution structures [60]. This means the accessible (φ,ψ) space is position-specific. An outlier on a classical plot may be sterically permissible on a geometry-specific map if the local bond parameters alleviate clashes. The PARAMA web resource is designed for this kind of in-depth, position-wise analysis [60].
FAQ: Steric Clashes
Q: My model has a high clashscore. What is the most effective strategy to resolve steric clashes? A: Resolving steric clashes requires a targeted approach. A high clashscore often indicates local fitting errors. The most effective strategy is to use all-atom contact analysis, which involves adding explicit hydrogen atoms to your model and analyzing the directionality of clashes and hydrogen bonds [59]. Prioritize correcting groups that are fitted into the wrong local minimum conformation, as this usually resolves multiple outlier measures simultaneously. Use the validation visualization in MolProbity or Coot to identify the specific atom pairs involved in the worst clashes and adjust them accordingly.
Q: How do I diagnose and fix Asn, Gln, and His side-chain flips? A: Incorrect flips of Asn, Gln, and His residues are a common source of steric clashes and hydrogen bonding errors.
- Diagnosis: Tools like MolProbity's all-atom contact analysis or the specialized server NQ-Flipper can identify unfavorable rotamers for these residues [61] [59]. They diagnose flips by evaluating the potential for hydrogen bonding and avoiding clashes with the surrounding atoms.
- Correction: The
REDUCEsoftware, integrated into the MolProbity suite, automatically adds H atoms and optimizes the flips of Asn, Gln, and His side chains by considering complete hydrogen-bond networks and steric clashes [59]. After automated correction, always validate the new conformation by checking its fit in the electron density map.
Q: What is an acceptable clashscore for a final, deposited model? A: The benchmark for a good clashscore has improved steadily over time. For a well-refined model, you should aim for a clashscore as low as possible. The average clashscore for mid-range PDB depositions has improved to approximately four clashes per 1000 atoms [59]. A clashscore in the low single digits is excellent. It is neither realistic nor desirable to aim for zero, as our empirical parameters are not perfect, and a few small, unexplained clashes may remain even in high-quality structures.
FAQ: Loop Modeling in Homology Models
Q: During loop grafting in homology modeling, what criteria should I use to select an appropriate donor loop from a homologous structure? A: Selecting the right donor loop is critical for success. Adhere to these criteria [33]:
- High Sequence Identity: The sequence identity in the loop region and the residues used for alignment should ideally be greater than 50%.
- Good Backbone Fit: After structural alignment of the residues flanking the loop, the backbone root-mean-square deviation (RMSD) of the aligned regions should be less than 2.0 Å.
- Completeness of the Donor: The homologous structure must have a complete, well-modeled backbone for the loop region of interest.
- Structural Quality: Prefer donor loops from high-resolution structures with good validation metrics (e.g., low clashscore, good Ramachandran statistics).
Q: After grafting a loop, how should I refine and validate it? A: Grafted loops require careful refinement and validation to ensure they fit both the local density and stereochemical standards.
- Refinement Protocol: Refine the grafted loop in real space to fit the electron-density map. Subsequently, include it in iterative cycles of restrained refinement (e.g., in REFMAC5 or phenix.refine) with appropriate geometric restraints [33].
- Validation Checklist:
- Geometry: Check for Ramachandran outliers and steric clashes within the loop and at its interfaces.
- Electron Density Fit: Calculate the Real-Space Correlation Coefficient (RSCC) for the loop residues; a value above 0.8 generally indicates a good fit [33].
- Backbone Continuity: Ensure smooth connectivity and reasonable bond lengths/angles with the surrounding protein chain.
Quantitative Data Tables
Table 1: Key Validation Metrics and Target Values
| Validation Metric | Calculation / Definition | Target Value for a Good Quality Model | Tools for Analysis |
|---|---|---|---|
| Clashscore | Number of serious steric clashes per 1000 atoms [59] | < 5 (Mid-range PDB deposition average) [59] | MolProbity, PHENIX |
| Ramachandran Outliers | % of residues in disallowed regions [59] | < 0.2% (Few outliers, not zero) [59] | MolProbity, PROCHECK, PDB-REDO |
| Real-Space Correlation Coefficient (RSCC) | Measures the fit between the atomic model and the electron-density map [33] | > 0.8 for well-defined regions [33] | EDSTATS, Coot, phenix.maps |
| Cβ Deviation | Measures deviation of Cβ from its ideal position given the backbone conformation [59] | < 0.25 Å | MolProbity |
| Rotamer Outliers | % of side chains in unfavorable conformations [59] | As low as possible | MolProbity, NQ-Flipper |
Table 2: Resolution-Based Guidelines for Model Interpretation and Validation
| Resolution Range (Å) | Expectation for Loops & Flexible Regions | Recommended Validation Focus | Caution & Advice |
|---|---|---|---|
| < 2.0 (High) | Well-defined density; ability to model alternate conformations [59] | - Precise geometry- All-atom contacts- Alternate conformations | - Do not downweight geometry in poor density [59] |
| 2.0 - 3.0 (Medium) | Many loops can be built; density may be less clear [33] | - CaBLAM for secondary structure diagnosis [59]- Steric clashes- Ramachandran outliers | - Use homology-based loop grafting [33]- Bewish of over-fitting |
| > 3.0 (Low) | Increased disorder; many regions may be unmodeled [33] | - CaBLAM [59]- Overall model-to-map fit | - Not all atoms must be inside density [59]- Bewish of sequence misalignment |
Experimental Protocols
Protocol 1: All-Atom Contact Analysis for Clash Remediation
Purpose: To identify and correct steric clashes in a protein model using explicit hydrogen atoms, which provides a superior assessment of local stereochemistry.
Materials:
- Protein structure file (PDB format)
- Access to the MolProbity web server or a local installation
- Molecular graphics software (e.g., Coot, UCSF ChimeraX)
Methodology:
- Add Hydrogen Atoms: Run the
REDUCEprogram on your input PDB file. This tool adds all H atoms, optimizes their positions, and flips Asn, Gln, and His side chains where needed to optimize hydrogen bonding and minimize clashes [59]. - Run All-Atom Contact Analysis: Use the
PROBEprogram to analyze all non-covalent atom pairs. It calculates overlaps and generates visualizations of steric clashes as "dots" or "spikes" between atoms [59]. - Visualize and Diagnose: Load the analysis results into a molecular graphics tool. Examine the worst clashes (e.g., those with overlap ≥ 0.4 Å). These often indicate groups fitted into the wrong local minimum.
- Correct the Conformation: For each severe clash, manually adjust the offending side-chain rotamer or backbone conformation in Coot. Use real-space refinement to maintain the fit to the electron density.
- Iterate: Re-run the all-atom contact analysis after corrections to confirm the clash has been resolved and no new ones have been introduced.
Protocol 2: Homology-Based Loop Grafting and Refinement
Purpose: To build a missing loop region in a target protein structure by transferring its coordinates from a homologous structure and refining it to fit the experimental data.
Materials:
- Target protein structure with a missing loop
- Database of homologous protein structures (e.g., the PDB)
- PDB-REDO pipeline or similar software with loop grafting capabilities [33]
- Crystallographic refinement software (e.g., PHENIX, REFMAC5)
Methodology:
- Identify Missing Loops and Homologs: Use a tool like
pdb2fastato identify unmodeled regions in your target structure. Search for high-identity homologous structures that have the equivalent loop modeled [33]. - Select and Align Donor Loop: For each candidate homolog, ensure sequence identity in the loop region is >50%. Structurally align the flanking regions (typically 4 residues on each side) of the target and homolog. Proceed if the backbone RMSD of alignment is < 2.0 Å [33].
- Graft the Loop: Transfer the coordinates of the donor loop, plus the two directly adjacent residues, into the target model. Crop side chains to match the target sequence in case of mutations [33].
- Real-Space Refinement: Refine the grafted loop, including the two adjacent residues, in real space to improve its fit into the target's electron-density map [33].
- Restrained Refinement and Validation: Incorporate the newly built loop into iterative cycles of restrained crystallographic refinement. Finally, validate the completed model using the metrics outlined in Table 1, paying special attention to the loop's geometry and RSCC [33].
Workflow Diagrams
Diagram Title: Geometric Validation and Outlier Correction Workflow
Diagram Title: Homology-Based Loop Modeling and Refinement Protocol
The Scientist's Toolkit: Research Reagent Solutions
| Tool Name | Function / Purpose | Key Features / Use-Case |
|---|---|---|
| MolProbity | Comprehensive structure validation | All-atom contact analysis, Ramachandran plots, rotamer checks, and Cβ deviation metrics [61] [59]. |
| PDB-REDO | Automated re-refinement and model completion | Pipeline for improving existing models; includes homology-based loop grafting to build missing regions [33]. |
| Coot | Model building and validation | Interactive real-space refinement, validation graphs, and manual correction of outliers and clashes [59]. |
| PARAMA | Position-wise stereochemical assessment | Uses bond geometry-specific Ramachandran steric-maps for in-depth analysis of residue positions [60]. |
| CaBLAM | Secondary structure diagnosis | Robust validation of protein backbone conformation, especially useful for lower-resolution structures [59]. |
| NQ-Flipper | Side-chain validation | Specifically identifies unfavorable rotamers of Asn and Gln residues [61]. |
This technical support center provides guidance on employing knowledge-based scoring functions, with a focus on the Distance-scaled, Finite, Ideal-gas Reference (DFIRE) potential, to enhance loop modeling accuracy in homology model research. The following FAQs and troubleshooting guides address specific challenges researchers encounter when integrating these statistical potentials into their computational workflows.
FAQs: Understanding DFIRE and Its Application
1. What is the DFIRE energy function and what is it used for? The DFIRE energy function is a knowledge-based statistical potential derived from the statistical analysis of known protein structures. It was developed using 19 atom types and a distance-scaled finite ideal-gas reference (DFIRE) state. Its primary applications include predicting binding affinities for protein-ligand, protein-protein, and protein-DNA complexes, and is highly effective for discriminating near-native loop conformations from decoys during loop modeling [62].
2. How does the performance of DFIRE compare to physics-based energy functions in loop modeling? DFIRE performs comparably to physics-based force fields like AMBER/GBSA for short loops (2-8 residues) and can be more accurate for longer loops (9-12 residues). A key advantage is its computational efficiency, requiring only a tiny fraction of the computing time (estimates suggest two orders of magnitude less) compared to complex physics-based functions, making it suitable for genomic-scale homology modeling [63].
3. Can DFIRE be applied to structures beyond its original training set? Yes. The "monomer" DFIRE potential, derived from single-chain proteins, has proven successful in predicting binding free energy for protein-protein and protein-peptide complexes and in discriminating docking decoys. This indicates it captures the essence of physical interactions across different protein environments, making it robust for various applications beyond its initial training data [63].
4. Where can I access the DFIRE energy function? The parameters and program for the all-atom DFIRE energy function are freely available for academic users at: http://theory.med.buffalo.edu/ [62].
Troubleshooting Guide: Common Experimental Issues
Problem 1: Low Accuracy in Long Loop Predictions
Symptoms
- Poor Root-Mean-Square Deviation (RMSD) in predicted loops longer than 8 residues.
- Inability to distinguish near-native conformations from decoys for long loops.
Solutions
- Utilize DFIRE-based potential: For loops longer than 8 residues, a DFIRE-based statistical potential has demonstrated superior performance compared to several physics-based force fields [63].
- Incorporate a rotamer library: Combine DFIRE with a side-chain optimization rotamer library (like SCWRL) for improved loop conformation predictions [63].
- Consider hybrid methods: Implement data-based approaches like DaReUS-Loop, which uses filtering and scoring (that can incorporate knowledge-based potentials) on fragments mined from the PDB. This method shows enhanced performance for long loops (≥15 residues) in homology models [31].
Problem 2: Inefficient Computational Workflow
Symptoms
- Loop selection and scoring steps are prohibitively slow, hindering large-scale or genomic applications.
Solutions
- Replace physics-based functions with DFIRE: Switching to the single-term DFIRE energy function can drastically reduce computing cost while maintaining, and for longer loops even improving, accuracy compared to physics-based functions like AMBER/GBSA or OPLS/SGB-NP [63].
- Implement a filtered data-based search: For homology model completion, use a protocol like DaReUS-Loop that efficiently mines the PDB for candidate fragments and applies sequential filters (sequence similarity, local geometry, conformational profile) to reduce the candidate set before final scoring and modeling [31].
Problem 3: Integrating Loops into Homology Models
Symptoms
- Despite identifying a potentially correct loop fragment, the final integrated model has steric clashes or poor geometry.
Solutions
- Apply post-grafting refinement: After grafting a loop from a homologous structure, refine the complete model to fit the electron-density map in real space and validate it against geometric criteria [33].
- Filter for clashes: During candidate selection, explicitly discard loop candidates that result in steric clashes after modeling. This simple filter significantly improves the average local RMSD of the final model set [31].
Performance Data and Protocols
Table 1: Performance Comparison of Scoring Functions in Loop Modeling
| Scoring Function / Method | Loop Length | Performance (Average RMSD) | Key Advantage |
|---|---|---|---|
| DFIRE-based Potential [63] | 2-8 residues | Comparable to AMBER/GBSA | Computational speed |
| DFIRE-based Potential [63] | 9-12 residues | More accurate than AMBER/GBSA | Accuracy for longer loops |
| DaReUS-Loop [31] | ≥15 residues | Significantly outperforms other methods | Best for very long loops |
| Knowledge-based Potential (RAPDF) [63] | Various | Less accurate than AMBER/GBSA | Fast but less accurate |
Table 2: Key Research Reagent Solutions
| Reagent / Resource | Type | Function in Experiment | Access |
|---|---|---|---|
| DFIRE Energy Function [62] | Software/Scoring Function | Provides a knowledge-based statistical energy for scoring loop decoys and predicting binding affinities. | Free for academic use |
| PDB (Protein Data Bank) [31] | Database | Source of experimental protein structures for mining loop fragments and deriving knowledge-based potentials. | Publicly available |
| SCWRL [63] | Software | Rotamer library-based tool for building and optimizing side-chain conformations during loop modeling. | - |
| DaReUS-Loop [31] | Software Pipeline | Data-based approach for loop modeling that mines PDB for fragments and uses filtering/scoring for selection. | - |
Experimental Protocol: Loop Selection Using a DFIRE-Based Potential
This protocol is adapted for loop selection and refinement within a homology modeling pipeline [63].
- Generate Loop Decoys: Use a conformational sampling method (e.g., RAPPER) to generate a large set of possible loop conformations (decoys) for the target sequence and region.
- Build Side Chains: Employ a rotamer library and side-chain optimization tool (e.g., SCWRL) to add atomic details to the backbone decoys generated in the previous step.
- Score with DFIRE: Calculate the energy score for each decoy in the set using the all-atom DFIRE statistical potential.
- Select Top Conformations: Identify the decoy(s) with the lowest DFIRE energy scores as the most near-native predictions.
- Validate and Refine: Integrate the selected loop into the larger protein model and perform subsequent refinement and validation checks.
Workflow Diagram: Data-Based Loop Modeling
The diagram below illustrates a modern data-based loop modeling workflow that can incorporate knowledge-based scoring like DFIRE for candidate ranking [31].
Data-Based Loop Modeling Pipeline
Frequently Asked Questions (FAQs)
FAQ 1: What does a model's "confidence score" actually represent, and can I trust a score of 100%? A confidence score is a value, often between 0 and 1, that a model outputs to quantify its certainty in a prediction. However, it is crucial to understand that a score of 100% (or 1.0) does not guarantee the prediction is correct. This score is typically a measure of the model's internal certainty based on its learned patterns, not a direct measure of physical accuracy. It is paradoxical, but 100% confidence can sometimes be wrong, especially if the model is overfitting or encounters a scenario far from its training data [64].
FAQ 2: My model has high confidence but the predicted loop structure has a high RMSD. What could be wrong? This discrepancy often points to overfitting or a mismatch between your data and the model's training data. The model might be "certain" about patterns it learned from its training set, but these patterns may not generalize well to your specific protein system. This is a form of epistemic uncertainty, which arises from a lack of relevant training data. To diagnose this, check if your target protein's sequence or fold is underrepresented in common training datasets. Using model calibration techniques can help realign confidence scores with actual accuracy [65] [66].
FAQ 3: What is the difference between a confidence score and a measure like pLDDT from AlphaFold? A general confidence score is a model's self-assessment of its prediction's reliability. pLDDT (predicted local distance difference test) is a specific, advanced confidence measure used by deep learning models like AlphaFold. It is a per-residue estimate of the model's reliability, where lower scores often indicate disordered regions or areas with high flexibility. For loop modeling, which is often flexible, paying close attention to per-residue pLDDT scores can be more informative than a single overall confidence score for the entire loop [67].
FAQ 4: How can I estimate my model's accuracy when I don't have the true ground-truth structure? In the absence of ground truth, you can use estimators that leverage the model's own confidence scores. A common and theoretically grounded baseline method is the Average Confidence (AC). This method estimates the model's accuracy on a set of predictions by simply averaging the confidence scores for those predictions. For example, if you predict 10 loops and the average confidence is 80%, the AC method estimates your accuracy to be 80%. This is an unbiased estimator when the model is well-calibrated [68].
FAQ 5: Why do deep learning-based co-folding models sometimes produce physically unrealistic loop structures despite high confidence? Recent studies on models like AlphaFold3 and RoseTTAFold All-Atom show that they can sometimes memorize patterns from their vast training data rather than learning the underlying physics of molecular interactions. When presented with biologically implausible inputs (e.g., a binding site mutated to phenylalanine), these models may still output a high-confidence prediction that maintains the original binding mode, leading to severe steric clashes and unphysical structures. This indicates a potential failure to generalize and a divergence from fundamental physical principles [65].
Troubleshooting Guides
Issue 1: Consistently Overconfident Predictions in Loop Modeling
Problem: Your model consistently outputs high confidence scores (e.g., >90%), but a significant portion of its loop predictions have unacceptably high RMSD when compared to experimental structures.
Investigation and Solutions:
Step 1: Check for Dataset Shift Dataset shift occurs when the data you are using differs from the data the model was trained on. This is a primary cause of overconfidence.
- Action: Compare the distribution of key features (e.g., loop length, amino acid composition, secondary structure) in your target proteins against the model's known training dataset (e.g., the Protein Data Bank). Significant differences indicate a covariate shift [68] [69].
- Solution: If possible, fine-tune the model on a smaller, curated dataset that is more representative of your protein family of interest.
Step 2: Evaluate Model Calibration A model is well-calibrated if, for example, 80% of the loops predicted with 0.8 confidence are actually correct. Modern deep learning models are often poorly calibrated.
- Action: Create a reliability diagram. Group your predictions by confidence score and plot the average confidence against the actual accuracy for each group. A perfectly calibrated model will follow the diagonal.
- Solution: Apply post-hoc calibration techniques such as temperature scaling or Platt scaling to adjust the output confidence scores to better match the empirical accuracy [68].
Step 3: Test with Adversarial Examples Probe the model's understanding of physical constraints.
- Action: Mimic the approach from recent literature [65]. Take a known protein-ligand complex and mutate key binding site residues to glycine (removing interactions) or phenylalanine (creating steric clashes). Run the prediction again.
- Interpretation: A physically robust model should show a large change in the predicted ligand/loop pose and a corresponding drop in confidence. If the model predicts a nearly identical high-confidence structure, it is likely overfit and not learning the underlying physics.
Issue 2: Low Confidence on Otherwise High-Quality Homology Models
Problem: Your overall homology model is good, but the confidence estimates for the loop regions are inexplicably low, making it hard to trust the model.
Investigation and Solutions:
Step 1: Distinguish Between Aleatoric and Epistemic Uncertainty It is critical to determine the source of the low confidence.
- Aleatoric Uncertainty: This is inherent to the data. Loops are often flexible and can adopt multiple conformations. Low confidence might correctly reflect this genuine structural ambiguity [66].
- Epistemic Uncertainty: This comes from the model's lack of knowledge. It may not have seen enough examples of loops with your specific sequence or structural context [66].
- Action: Use methods like Monte Carlo Dropout or Deep Ensembles during inference. If multiple stochastic forward passes or different models in an ensemble give similar low-confidence predictions, the uncertainty is likely aleatoric (real). If predictions vary widely, the uncertainty is more epistemic (model-based) [66].
Step 2: Analyze Feature Inputs The model's confidence is directly tied to the features you provide.
- Action: Review the input features for the low-confidence loops. Check for unusual Ramachandran angles, very high B-factors, or unusual solvent accessibility in the unbound structure. These features are directly used by predictors (like SVM-based methods) to flag potentially mobile loops [70].
- Solution: If the features seem biologically unrealistic, re-check the input data and model preparation steps. The low confidence might be a valid warning of a problem in the initial model.
Step 3: Consult the Applicability Domain The model might be operating outside its applicability domain (AD).
- Action: Quantify how different your target loop is from the loops in the model's training set. Calculate the Euclidean distance in the feature space (e.g., using sequence descriptors, physiochemical properties) between your loop and the training examples.
- Interpretation: If your loop is a significant outlier, the low confidence is justified, and the prediction should be treated with caution. Consider using a different modeling strategy for this specific region [69].
Experimental Protocols & Data
Key Machine Learning Features for Loop Mobility Prediction
The following table summarizes the features used in a Support Vector Machine (SVM) approach to classify loops as "mobile" or "stationary" based on the unbound protein structure. Integrating these features can form the basis of a custom confidence estimator [70].
| Feature | Description | Rationale and Relationship to Mobility |
|---|---|---|
| Backbone Conformation (〈SR〉) | Average of the log-probability of a residue's (Ψ,Φ) dihedral angles based on reference Ramachandran plots [70]. | Loops with residues in low-probability dihedral angles in the unbound state are more likely to move upon binding. Strongly anti-correlated with mobility. |
| Crystallographic B-factor (〈ZB〉) | Average z-score of the B-factors for all atoms within a loop. B-factor indicates atomic mobility [70]. | Higher B-factors are associated with greater thermal motion and a higher likelihood of conformational change. Positively correlated with mobility. |
| Relative Accessible Surface Area (〈RASA〉) | The average solvent-accessible surface area of loop residues, relative to a tri-peptide standard [70]. | Loops exposed to solvent are less constrained and more likely to undergo motion. Positively correlated with mobility. |
Quantitative Performance of an SVM Loop Predictor
The following table summarizes the performance of the SVM model described in [70], which provides a benchmark for what is achievable with a feature-based machine learning approach.
| Validation Scenario | Prediction Accuracy | Area Under the ROC Curve (AUC) |
|---|---|---|
| 4-Fold Cross-Validation | 75.3% | 0.79 |
| Independent Test Set | 70.5% | Not Reported |
| Ras Superfamily Proteins | 92.8% | Not Reported |
| Binding Partners of Ras Proteins | 74.4% | Not Reported |
Note: A random predictor would have an accuracy of 50%. The high accuracy on Ras superfamily proteins suggests performance is best when the model encounters proteins with well-defined conformational changes in its training data [70].
The Scientist's Toolkit
Research Reagent Solutions
| Item | Function in Confidence Estimation for Loop Modeling |
|---|---|
| LIBSVM Package [70] | A widely-used software library for implementing Support Vector Machines (SVMs), ideal for building custom classifiers to predict loop mobility from structural features. |
| NACCESS Program [70] | Calculates the accessible surface area of atoms in a protein structure. Used to compute the Relative Accessible Surface Area (RASA), a key feature for mobility prediction. |
| S2C Database & STRIDE [70] | Provides standardized secondary structure assignments for protein structures. Essential for consistently identifying and defining loop regions for analysis. |
| DANG Software [70] | Calculates protein backbone dihedral angles (Φ and Ψ). Used to quantify conformational changes between bound and unbound states for training and validation. |
| Monte Carlo Dropout [66] | A technique used during the inference of a deep neural network to approximate model uncertainty. By running multiple predictions with dropout enabled, you can estimate the epistemic uncertainty. |
| Deep Ensembles [66] | A method using multiple independently trained models to make predictions. The variation in their outputs provides a robust estimate of predictive uncertainty, often outperforming single-model methods. |
Workflow and Relationship Diagrams
Confidence Estimation for Loop Modeling
Uncertainty Diagnosis and Troubleshooting
Frequently Asked Questions (FAQs)
Q1: What are the fundamental differences between CASP and CAMEO for benchmarking protein structure prediction methods?
CASP (Critical Assessment of protein Structure Prediction) and CAMEO (Continuous Automated Model EvaluatiOn) are both blind assessment platforms, but they operate on different cycles and scales. CASP is a community-wide biennial experiment where human experts assess approximately 100 prediction targets during each round, culminating in a meeting where researchers compare method performances and discuss developments [71]. In contrast, CAMEO operates on a continuous weekly cycle, conducting fully automated evaluations based on the pre-release of sequences from the Protein Data Bank (PDB). Each week, CAMEO benchmarks about 20 targets collected during a 4-day prediction window, providing more frequent evaluation cycles and larger datasets for developers [71].
Q2: Why might my homology modeling method perform well on CASP targets but poorly on CAMEO benchmarks?
This discrepancy often stems from differences in target selection and evaluation frequency. CAMEO deselects targets exhibiting high coverage in at least one template (considered "too easy") and omits protein sequences shorter than 30 residues [71]. Additionally, CAMEO's continuous evaluation means methods are tested on a broader range of recently solved structures, potentially revealing weaknesses that less frequent, curated assessments might miss. The "bestSingleTemplate" method in CAMEO, which uses structural superpositions with TM-align and modeling with ProMod3, serves as a rigorous reference baseline that may be more challenging than some CASP evaluation standards [71].
Q3: What specific metrics should I prioritize when benchmarking loop modeling improvements in homology models?
For loop modeling specifically, focus on local accuracy metrics rather than global structure scores. Key metrics include:
- Local Root Mean Square Deviation (Local RMSD) measured specifically for loop regions [31]
- Jensen Shannon Divergence (JSD) for conformational profile comparison [31]
- Assessment of steric clashes in modeled loops [31] Data-based approaches like DaReUS-Loop have demonstrated that over 50% of successful loop models are derived from unrelated proteins, indicating that fragments under similar constraints tend to adopt similar structures beyond mere homology [31].
Q4: How can I properly evaluate model quality assessment (MQA) methods for homology models?
Traditional CASP datasets may not sufficiently evaluate MQA performance for practical homology modeling applications, as they often lack enough targets with high-quality models and include models generated by de novo methods [72]. For specialized evaluation, consider using the Homology Models Dataset for Model Quality Assessment (HMDM), which contains targets with high-quality models derived specifically using homology modeling [72]. When benchmarking, ensure your dataset includes both single-domain and multi-domain proteins, as performance can vary significantly between these categories [72].
Troubleshooting Guides
Problem: Inconsistent Performance Between Different Benchmarking Platforms
Symptoms: Your method ranks highly in CASP evaluations but shows mediocre performance in CAMEO benchmarks, particularly for loop regions.
Solution:
- Verify target difficulty filtering: Implement CAMEO's target selection criteria in your internal testing, specifically excluding targets with high template coverage and sequences shorter than 30 residues [71].
- Adjust for continuous evaluation: Incorporate recently solved PDB structures into your training regimen, as CAMEO uses weekly pre-released sequences while CASP targets are less frequent [71].
- Implement the bestSingleTemplate baseline: Compare your method against CAMEO's reference baseline, which uses TM-align for structural superposition and ProMod3 for modeling [71].
Prevention: Maintain ongoing evaluation using both platforms and prioritize methods that perform consistently across different assessment methodologies rather than optimizing specifically for either CASP or CAMEO.
Problem: Poor Loop Modeling Accuracy in Homology Models
Symptoms: Acceptable global structure metrics (GDT_HA, TM-score) but poor local accuracy in loop regions, particularly for loops longer than 12 residues.
Solution:
- Implement fragment-based filtering: Use approaches like DaReUS-Loop that mine the entire PDB for candidate fragments using local conformation profile-profile comparison and physico-chemical scoring [31].
- Apply multi-stage filtering:
- First filter by sequence similarity (BLOSUM scores)
- Cluster to reduce candidate redundancy
- Filter by conformational similarity (JSD < 0.40)
- Finally remove candidates with steric clashes [31]
- Utilize remote fragments: Don't limit search to homologous proteins; successful loops often come from unrelated proteins with similar structural constraints [31].
Prevention: For long loops (>15 residues), prioritize data-based methods like DaReUS-Loop that specifically address this challenging scenario and show superior performance compared to ab initio methods like Rosetta NGK and GalaxyLoop-PS2 [31].
Table 1: Key Characteristics of Major Benchmarking Platforms
| Platform | Evaluation Cycle | Targets Per Cycle | Assessment Method | Primary Focus |
|---|---|---|---|---|
| CASP | Biennial | ~100 | Expert manual assessment | Comprehensive structure prediction assessment [71] |
| CAMEO | Weekly | ~20 | Fully automated | Continuous server benchmarking [71] |
| HMDM | On-demand | 100 single-domain + multi-domain | Automated with homology models | MQA for homology models [72] |
Table 2: Loop Modeling Performance Comparison (RMSD in Ångströms)
| Method | Type | Short Loops (3-12 residues) | Long Loops (≥15 residues) | Computational Demand |
|---|---|---|---|---|
| DaReUS-Loop | Data-based | 1-4Å | Significant improvement over alternatives | Substantially less than ab initio methods [31] |
| Rosetta NGK | Ab initio | 1-2Å | Decreasing accuracy | High [31] |
| GalaxyLoop-PS2 | Ab initio | 1-2Å | Decreasing accuracy | High [31] |
| LoopIng | Data-based | 1-4Å | Decreasing accuracy | Moderate [31] |
Table 3: Filtering Impact on Loop Candidate Quality in DaReUS-Loop
| Filtering Stage | Candidates with RMSD <4Å | Mean RMSD | Key Filter Criteria |
|---|---|---|---|
| Initial candidates | 49% | 3.86Å | None [31] |
| After sequence filtering | 62% | 3.60Å | Positive BLOSUM scores [31] |
| After clustering | 70% | 3.60Å | Redundancy reduction [31] |
| After JSD filtering | 74% | 3.29Å | JSD < 0.40 [31] |
| After clash removal | 84% | 2.94Å | Steric clash elimination [31] |
Experimental Protocols
Protocol 1: Implementing CAMEO's BestSingleTemplate Baseline
Purpose: Establish a reference baseline for 3D modeling predictions to objectively compare techniques [71].
Procedure:
- Template discovery: Perform structural superposition of target reference structures with all PDB structures using TM-align [71].
- Template selection: Extract the top 20 structural alignments from TM-align results [71].
- Model generation: Use SWISS-MODEL's modeling engine ProMod3 with the selected templates [71].
- Termini modeling: Model termini beyond template-covered regions using low-complexity Monte Carlo sampling [71].
- Model selection: Rank final models by lDDT and select the top-scoring model [71].
Validation: Compare your method's performance against this baseline using CAMEO's 18 different metrics, including GDT_HA, TM-score, lDDT, and interface accuracy measures [71].
Protocol 2: DaReUS-Loop for Loop Modeling in Homology Context
Purpose: Accurately model loops in homology models where flank regions may have substantial deviations from native structures [31].
Procedure:
- Candidate identification: Use BCLoopSearch to mine fragment candidates from the entire PDB based on loop flanks [31].
- Sequence filtering: Filter candidates using sequence similarity (BLOSUM scores), retaining those with positive scores [31].
- Clustering: Apply clustering to reduce candidate redundancy and improve average RMSD [31].
- Conformational filtering: Calculate Jensen Shannon Divergence (JSD) and filter out candidates with JSD > 0.40 [31].
- Steric evaluation: Build complete protein models and eliminate candidates with steric clashes [31].
- Final selection: Score remaining candidates and return the 10 best models [31].
Validation: Evaluate using local RMSD specifically for loop regions, with successful predictions defined as RMSD < 2Å for homology modeling contexts [31].
Research Workflow Visualization
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Resources for Structure Prediction Benchmarking
| Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| CAMEO Platform | Benchmarking Service | Continuous automated evaluation of prediction servers | Method development and validation [71] [73] |
| HMDM Dataset | Specialized Dataset | Benchmarking MQA methods for homology models | Evaluating model quality assessment techniques [72] |
| DaReUS-Loop | Software Method | Fragment-based loop modeling using remote structures | Improving loop regions in homology models [31] |
| BestSingleTemplate | Reference Method | Baseline for 3D modeling accuracy | Objective comparison of modeling techniques [71] |
| PSBench | Benchmark Suite | Large-scale dataset for complex structure assessment | Protein complex EMA development [74] |
| lDDT Score | Quality Metric | Local Distance Difference Test | Model accuracy assessment without superposition [71] |
| TM-align | Algorithm | Protein structure alignment | Template discovery and structural comparison [71] |
| ProMod3 | Modeling Engine | SWISS-MODEL's core modeling platform | Template-based structure generation [71] |
In structural biology and drug discovery, homology modeling serves as a critical technique for constructing three-dimensional protein structures when experimental data is unavailable. The accuracy of these models, particularly in flexible loop regions, directly impacts their utility in downstream applications such as virtual screening and structure-based drug design. This technical support guide provides researchers with clear criteria and methodologies to determine when a homology model achieves sufficient quality for drug discovery pipelines.
Frequently Asked Questions (FAQs)
Q1: What are the primary validation metrics for an application-ready homology model? An application-ready model requires validation across multiple metrics. Key criteria include assessment by the MolProbity server for steric clashes and rotamer outliers, real-space correlation coefficient (RSCC) analysis against electron density maps where available, and overall model completeness with special attention to loop regions [33] [75].
Q2: How does loop region handling affect a model's drug discovery suitability? Loops often form functionally critical sites like ligand-binding pockets. Inaccurate loops can misdirect design efforts. "Dual personality" fragments—disordered in some homologs but ordered in others—should be carefully evaluated. Grafting loops from high-identity homologs (>50%) and subsequent refinement can significantly improve model quality for drug discovery applications [33].
Q3: What template selection criteria are most important for drug discovery models? Prioritize templates with high sequence identity (>30%), covering the entire target sequence, and determined by high-resolution X-ray crystallography. For drug discovery, particular attention should be paid to the resolution and quality of the template in the active site or functionally relevant regions [75].
Q4: When should a model be rejected for structure-based drug design? Reject models with major misalignments in active site residues, poor stereochemical quality (e.g., Ramachandran outliers exceeding 2%), backbone atoms with RSCC values consistently below 0.8, or missing residues in critical functional loops that cannot be reliably modeled [33] [75].
Troubleshooting Common Model Issues
Problem: Poor Loop Fit in Electron Density
Issue: After grafting a loop from a homolog, the region shows poor fit in the electron density map (RSCC < 0.7). Solution:
- Verify the sequence identity in the loop region meets the 50% threshold [33].
- Perform real-space refinement specifically targeting the loop coordinates.
- If the fit remains poor, consider ab initio loop modeling for shorter loops (<12 residues) or search for alternative loop conformations from other homologous structures [33] [75].
Problem: Steric Clashes in the Binding Site
Issue: The refined model shows steric clashes between side chains in the putative ligand-binding pocket. Solution:
- Use side-chain reconstruction tools like SCWRL4 to optimize rotamer conformations [75].
- Run molecular dynamics with explicit solvent to relax the region.
- Validate with MolProbity; clashescores below the 50th percentile for similar resolution structures are generally acceptable for drug discovery [75].
Problem: Incomplete Regions in Critical Functional Domains
Issue: Key functional domains (e.g., catalytic sites) contain unmodeled regions with no suitable homologs for grafting. Solution:
- For regions up to 30 residues, use ab initio loop modeling methods [33].
- Integrate cryo-EM density maps when available to guide modeling, using flexible fitting methods like MDFF [76].
- Validate predicted conformations with residue-specific quality scores and evolutionary covariance data if available.
Quantitative Criteria for Model Evaluation
The following table summarizes minimum recommended criteria for determining model readiness for various drug discovery applications.
Table 1: Application-Ready Criteria for Homology Models in Drug Discovery
| Evaluation Category | Threshold for Virtual Screening | Threshold for Structure-Based Design | Validation Method/Tool |
|---|---|---|---|
| Global Structure | >90% residues modeled [33] | >95% residues modeled [33] | pdb2fasta, Model coverage |
| Steric Quality | Clashscore < 20 [75] | Clashscore < 10 [75] | MolProbity |
| Rotamer Outliers | < 5% [75] | < 2% [75] | MolProbity |
| Loop Regions (RSCC) | > 0.7 [33] | > 0.8 [33] | EDSTATS, real-space refinement |
| Template Identity | > 30% [75] | > 50% [75] | BLAST, PSI-BLAST |
Experimental Protocol: Loop Grafting and Refinement
This protocol details the homology-based loop modeling process integrated in the PDB-REDO pipeline [33].
Step 1: Identification of Buildable Loops
- Input: Target structure with missing regions.
- Method: Use
pdb2fastato convert the structure to a sequence and identify gaps. Align against high-identity homologs from the PDB. - Criteria: A loop is buildable if equivalent regions are modeled in at least one homolog with >50% sequence identity in the loop and flanking regions [33].
Step 2: Homolog Selection and Alignment
- Method: For each candidate homolog, ensure at least five consecutive modeled residues flank each side of the loop. Perform structural alignment of these flanking regions using quaternions [33].
- Threshold: Accept homologs for grafting if the backbone RMSD of alignment is less than 2.0 Å [33].
Step 3: Loop Grafting
- Method: Delete two residues directly adjacent to the missing loop in the target structure. Insert the aligned loop plus its two adjacent residues from the homolog.
- Side-Chain Handling: Crop side-chain atoms in case of mutations. Set occupancy to 1.00 and scale B-factors appropriately [33].
Step 4: Refinement and Validation
- Refinement: Perform real-space refinement of the grafted loop to optimize fit to the electron density map.
- Validation: Calculate RSCC for the rebuilt region. Accept loops with RSCC > 0.7 and no steric clashes with the protein core [33].
Figure 1: Homology-based loop modeling and refinement workflow for achieving application-ready models.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents and Computational Tools for Model Preparation and Validation
| Reagent/Software Tool | Primary Function | Application in Model Preparation |
|---|---|---|
| PDB-REDO Pipeline | Integrated model rebuilding & refinement | Automated loop grafting and real-space refinement [33] |
| MODELLER | Comparative homology modeling | Core model construction from templates [75] |
| SCWRL4 | Side-chain conformation prediction | Optimizing rotamers after loop grafting [75] |
| MolProbity | Comprehensive structure validation | Evaluating steric clashes and rotamer quality [75] |
| Coot | Model building and manipulation | Manual inspection and correction of loop fits [33] |
| Rosetta | De novo structure prediction | Ab initio loop modeling for difficult regions [76] |
| MDFF | Molecular Dynamics Flexible Fitting | Flexible fitting of models into cryo-EM maps [76] |
Figure 2: Homology modeling workflow with quality checkpoints. Failed validations require iterative refinement of the relevant stage.
Conclusion
Improving loop modeling accuracy requires a multifaceted approach that combines robust methodologies with rigorous validation. The integration of data-based fragment assembly, advanced AI-driven contact prediction, and careful template selection has significantly enhanced our ability to model challenging loop regions. As the field progresses, the increasing availability of protein structures and continued development of machine learning algorithms promise to further bridge the accuracy gap between computational models and experimental structures. For biomedical researchers, these advances translate to more reliable protein models that can accelerate structure-based drug design, improve understanding of disease mechanisms, and ultimately enable the development of more targeted therapeutics. Future directions will likely focus on better handling of conformational flexibility and integrating cryo-EM data with computational modeling pipelines.
References
- 1. Predicting flexible loop regions that interact with ligands [https://pmc.ncbi.nlm.nih.gov/articles/PMC3243024/]
- 2. Distance-AF improves predicted protein structure models ... [https://www.nature.com/articles/s42003-025-08783-5]
- 3. De novo design of buttressed loops for sculpting protein ... [https://www.nature.com/articles/s41589-024-01632-2]
- 4. Comprehensive Analysis of Loops at Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4138238/]
- 5. Artificial intelligence yields highly accurate models [https://pubmed.ncbi.nlm.nih.gov/40472416/]
- 6. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 7. Loop modeling: Sampling, filtering, and scoring - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2553011/]
- 8. CONFORMATIONAL SAMPLING IN TEMPLATE-FREE ... [https://www.sciencedirect.com/science/article/pii/S2001037014600313]
- 9. Computational methods for exploring protein conformations [https://pmc.ncbi.nlm.nih.gov/articles/PMC7458412/]
- 10. Mapping Conformational Dynamics of Proteins Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3647154/]
- 11. Application of torsion angle molecular dynamics for ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/16145655/]
- 12. Improving homology modeling from low-sequence identity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7652349/]
- 13. MDAKits and MDA-based tools [https://www.mdanalysis.org/pages/mdakits/]
- 14. Using multiple templates to improve quality of homology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2386743/]
- 15. Homology modeling in the time of collective and artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7695898/]
- 16. Improving the accuracy of the structure prediction of the third ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4173008/]
- 17. Modeling of loops in proteins: a multi-method approach - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2837870/]
- 18. Inaccurate secondary structure predictions often indicate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6635839/]
- 19. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 20. RoseTTAFold: Accurate protein structure prediction accessible ... [https://www.ipd.uw.edu/2021/07/rosettafold-accurate-protein-structure-prediction-accessible-to-all/]
- 21. Software – Rosetta Commons [https://rosettacommons.org/software/]
- 22. Robetta - Baker Lab [https://robetta.bakerlab.org/]
- 23. The Bio3D packages for structural bioinformatics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7737766/]
- 24. New efficient statistical sequence-dependent structure ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283699928267]
- 25. Accurate prediction of CDR-H3 loop structures ... [https://elifesciences.org/articles/91512]
- 26. Conformation-Aware Structure Prediction of Antigen ... [https://arxiv.org/html/2507.09054v1]
- 27. A Sequence-Independent Analysis of the Loop Length ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3522851/]
- 28. A community resource for template-based protein structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7617537/]
- 29. DaReUS-Loop: a web server to model loops in homology ... [https://bioserv.rpbs.univ-paris-diderot.fr/services/DaReUS-Loop/]
- 30. Ten quick tips for homology modeling of high-resolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7117658/]
- 31. DaReUS-Loop: accurate loop modeling using fragments ... [https://www.nature.com/articles/s41598-018-32079-w]
- 32. DaReUS-Loop: a web server to model multiple ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6602439/]
- 33. Homology-based loop modeling yields more complete ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6126648/]
- 34. Protein Loop Modeling Using a New Hybrid Energy Function ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0113811]
- 35. Next-generation kinematic loop modeling and torsion- ... [https://docs.rosettacommons.org/docs/latest/application_documentation/structure_prediction/loop_modeling/next-generation-KIC]
- 36. Improvements to Robotics-Inspired Conformational Sampling ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0063090]
- 37. Improvements to Robotics-Inspired Conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3660577/]
- 38. FAQ [https://docs.rosettacommons.org/docs/latest/getting_started/FAQ]
- 39. Phyre2.2: A Community Resource for Template-based ... [https://www.sciencedirect.com/science/article/pii/S0022283625000269]
- 40. Developing a hybrid technique for energy demand ... [https://www.sciencedirect.com/science/article/pii/S2405844024047480]
- 41. Text has enhanced contrast | ACT Rule | WAI [https://www.w3.org/WAI/standards-guidelines/act/rules/09o5cg/]
- 42. Elements must meet minimum color contrast ratio thresholds [https://dequeuniversity.com/rules/axe/4.8/color-contrast]
- 43. PDB-REDO: optimise, rebuild, refine and validate [https://cloud.ccp4.ac.uk/manuals/html-taskref/doc.task.PDBREDO.html]
- 44. PDB-REDO [https://pdb-redo.eu/]
- 45. Resources: PDB-REDO: About [https://sciencegateways.org/resources/pdb-redo]
- 46. Rowan | ML-Powered Molecular Design and Simulation [https://www.rowansci.com/]
- 47. Ensemble machine learning prediction accuracy: local vs. ... [https://www.frontiersin.org/journals/education/articles/10.3389/feduc.2025.1571133/full]
- 48. Aligning the target sequence with one or more structures [https://salilab.org/modeller/methenz/andras/node5.html]
- 49. Differential performance of RoseTTAFold in antibody ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9487640/]
- 50. Help - SWISS-MODEL [https://swissmodel.expasy.org/docs/help]
- 51. PSI-BLAST Tutorial - Comparative Genomics - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK2590/]
- 52. PSI-BLAST searches using hidden Markov models of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC110734/]
- 53. BLAST OFF! The Basic Local Alignment Search Tool ... [https://bitesizebio.com/26522/blast-off-the-basic-local-alignment-search-tool-explained/]
- 54. A multi-template combination algorithm for protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2311309/]
- 55. On the Accuracy of Homology Modeling and Sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1483079/]
- 56. Toward better refinement of comparative models [https://pmc.ncbi.nlm.nih.gov/articles/PMC2764870/]
- 57. Molecular Dynamics with Python — Tutorials 2025.1 ... - SCM [https://www.scm.com/doc/Tutorials/MolecularDynamicsAndMonteCarlo/MDintroPython/intro.html]
- 58. 4.8.1.4. Secondary structure assignment (helix, sheet and ... [https://docs.mdanalysis.org/2.9.0/documentation_pages/analysis/dssp.html]
- 59. Model validation: local diagnosis, correction and when to quit [https://pmc.ncbi.nlm.nih.gov/articles/PMC5947777/]
- 60. Stereochemical Assessment of (φ,ψ) Outliers in Protein ... [https://www.sciencedirect.com/science/article/pii/S0969212619303156]
- 61. Structure Validation and Quality [https://www.rcsb.org/docs/additional-resources/structure-validation-and-quality]
- 62. A knowledge-based energy function for protein-ligand, protein ... [https://pubmed.ncbi.nlm.nih.gov/15801826/]
- 63. Accurate and efficient loop selections by the DFIRE-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2286705/]
- 64. How to use confidence scores in machine learning models [https://jonathan-23097.medium.com/how-to-use-confidence-scores-in-machine-learning-models-abe9773306fa]
- 65. Investigating whether deep learning models for co-folding ... [https://www.nature.com/articles/s41467-025-63947-5]
- 66. Understanding Model Uncertainty in Machine Learning [https://imerit.net/resources/blog/a-comprehensive-introduction-to-uncertainty-in-machine-learning-all-una/]
- 67. Evaluating GPCR modeling and docking strategies in the ... [https://www.sciencedirect.com/science/article/pii/S2001037022005517]
- 68. Confidence-based Estimators for Predictive Performance in ... [https://arxiv.org/html/2407.08649v1]
- 69. Assigning Confidence to Molecular Property Prediction - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9449894/]
- 70. A Machine Learning Approach for the Prediction of Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3341935/]
- 71. Introducing “best single template” models as reference ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8196401/]
- 72. A Benchmark Dataset for Evaluating Practical Performance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8945737/]
- 73. CAMEO | Continuous Automated Model EvaluatiOn [https://cameo3d.org/]
- 74. PSBench: a large-scale benchmark for estimating the ... [https://arxiv.org/html/2505.22674v1]
- 75. Ten quick tips for homology modeling of high-resolution ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007449]
- 76. Application of Homology Modeling by Enhanced Profile ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8879198/]