Pathway Enrichment Analysis: A Comprehensive Guide from Basics to Advanced Applications in Biomedicine
This article provides a complete guide to pathway enrichment analysis (PEA), a foundational bioinformatics method for interpreting gene lists from omics experiments.
Pathway Enrichment Analysis: A Comprehensive Guide from Basics to Advanced Applications in Biomedicine
Abstract
This article provides a complete guide to pathway enrichment analysis (PEA), a foundational bioinformatics method for interpreting gene lists from omics experiments. Tailored for researchers, scientists, and drug development professionals, it covers core concepts, statistical methods, and practical workflows. Readers will learn to define gene lists, select appropriate enrichment tools like g:Profiler and GSEA, and interpret results using visualization platforms such as Cytoscape and EnrichmentMap. The guide also addresses common pitfalls, optimization strategies for robust results, and advanced applications in drug repositioning and biomarker discovery, empowering users to confidently apply PEA in their research.
Understanding Pathway Enrichment Analysis: Core Concepts and Definitions
What is Pathway Enrichment Analysis? Defining the Method and Its Purpose
Pathway Enrichment Analysis (PEA) is a core bioinformatic technique used to interpret lists of genes derived from genome-scale experiments. It identifies biological pathways—structured series of molecular interactions that lead to a cellular product or change—that are statistically overrepresented in a gene list, thereby transforming large, complex omics datasets into mechanistically interpretable biological insights [1] [2]. Its primary purpose is to help researchers move beyond a gene-by-gene interpretation of their data and instead understand the coordinated activity of genes within established biological systems, which is crucial for uncovering disease mechanisms and identifying potential therapeutic targets [1] [3].
Core Principles and Definitions
At its heart, pathway enrichment analysis addresses a fundamental challenge in modern biology: how to extract meaningful biological understanding from long lists of genes, often comprising thousands of entries, generated by technologies like RNA sequencing or genome sequencing [1] [3].
- Pathway: A pathway is a model describing a series of interactions among molecules in a cell that leads to a certain product or a change. It is not a simple list but a structured network that captures knowledge about mechanisms, interactions, and dependencies, such as those found in KEGG or Reactome databases [3].
- Gene Set: In contrast, a gene set is an unordered and unstructured collection of genes, defined by a shared biological property, such as involvement in a specific biological process (e.g., cell cycle) or location on a chromosome. A pathway can be represented as a gene set, but this conversion loses all the topological and interaction information [3] [2].
- Gene List of Interest: This is the input for the analysis—a set of genes derived from an omics experiment, such as differentially expressed genes from an RNA-seq study or somatically mutated genes from cancer sequencing [1].
- Pathway Enrichment Analysis: This method identifies pathways that are statistically enriched in the gene list more than would be expected by chance alone. For example, if an experimental dataset contains 40% cell cycle genes, this would be surprisingly enriched given that only about 8% of human protein-coding genes are involved in this process [1].
The following diagram illustrates the foundational concept of how a structured pathway is often simplified into a gene set for enrichment analysis, a process that discards valuable topological information.
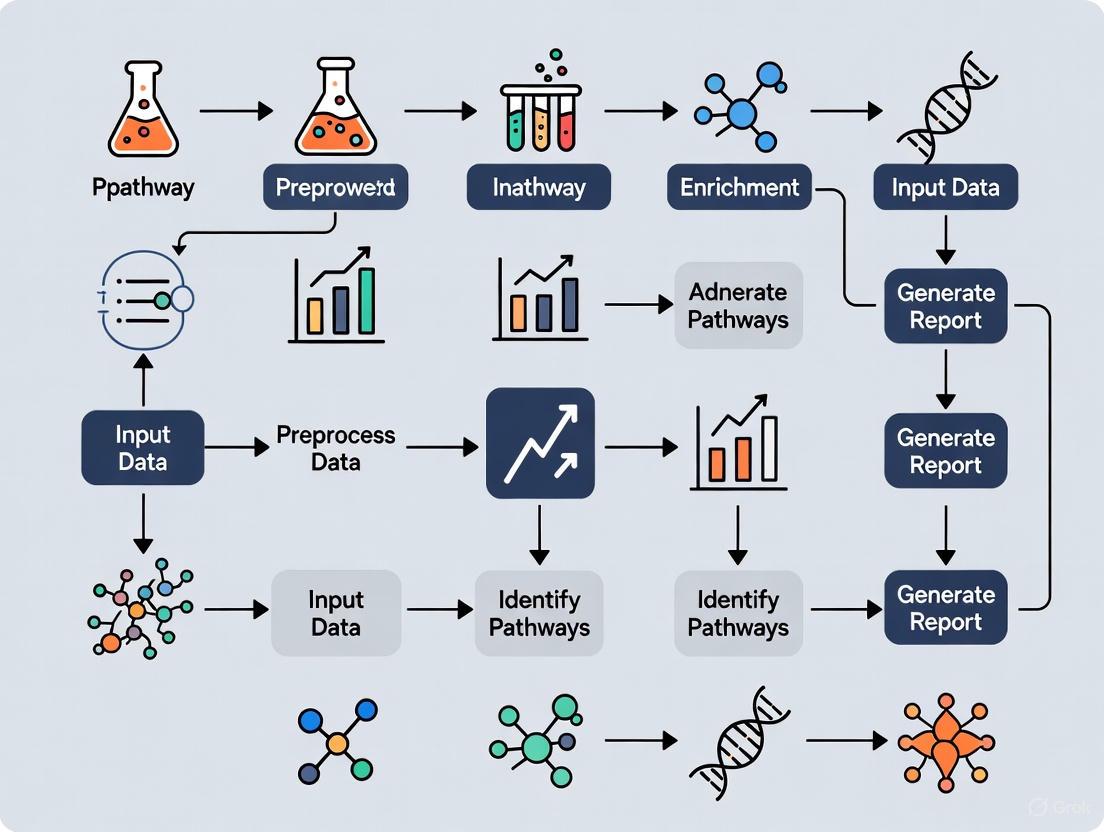
The Analytical Workflow: From Data to Discovery
A standard protocol for pathway enrichment analysis comprises three major stages, which can be performed in approximately 4.5 hours using freely available software [1].
Stage 1: Definition of a Gene List from Omics Data
The first step involves processing raw omics data to create a gene list suitable for analysis. The input can take one of two primary forms:
- Gene List: A simple set of genes, such as all somatically mutated genes in a tumor identified by exome sequencing. This is suitable for direct input into tools like g:Profiler [1].
- Ranked Gene List: A list of all genes measured in an experiment, ranked by a score such as the level of differential expression or a p-value. This format preserves more information and is the required input for methods like Gene Set Enrichment Analysis (GSEA) [1].
Stage 2: Determination of Statistically Enriched Pathways
A statistical method is applied to identify pathways that are significantly overrepresented in the gene list. There are three general methodological approaches, each with its own strengths.
Stage 3: Visualization and Interpretation
The final stage involves making sense of the list of enriched pathways, which often includes many related terms. Visualization tools like Cytoscape and EnrichmentMap help identify the main biological themes and their relationships for in-depth study and experimental validation [1].
The complete workflow, integrating these stages, is visualized below.
Key Methodological Approaches
Researchers can choose from several methodological approaches for enrichment analysis, each with distinct underlying principles and data requirements.
| Method | Description | Input Required | Key Advantage |
|---|---|---|---|
| Over-Representation Analysis (ORA) [2] | Statistically tests if a pathway contains more genes from the input list than expected by chance. | A list of genes (e.g., differentially expressed genes). | Simple, intuitive, and requires only gene identifiers. |
| Functional Class Scoring (FCS) [2] | Considers the full ranked list of genes to identify pathways where members are clustered at the top or bottom. | A ranked list of all genes from the experiment. | More sensitive; does not require an arbitrary significance cutoff for individual genes. |
| Pathway Topology (PT) [3] | Incorporates the pathway structure (interactions, positions, and roles of genes) into the analysis. | A gene list or ranked list, plus pathway topology data. | Uses more biological knowledge; can predict downstream effects and pathway activity. |
Over-Representation Analysis (ORA) is often the simplest starting point. It uses statistical tests like the hypergeometric test to ask whether the number of genes from a particular pathway found in the experimental list is larger than what would be expected if genes were selected at random from the background genome [2]. Its main limitation is its dependence on an often-arbitrary threshold to define the input gene list [3].
Functional Class Scoring (FCS) methods, such as the widely used Gene Set Enrichment Analysis (GSEA), address this limitation. GSEA uses a ranked list of all genes and a Kolmogorov-Smirnov-like running sum statistic to determine if members of a predefined gene set are randomly distributed throughout the list or found primarily at the top or bottom [1] [2]. A positively enriched pathway has its genes clustered at the top of the ranked list (e.g., highly upregulated), while a negatively enriched pathway has its genes clustered at the bottom [1].
Pathway Topology (PT) methods represent a more advanced approach. They leverage the detailed knowledge embedded in pathway diagrams, such as activation/inhibition relationships and signal flow. For example, if a pathway is triggered by a single receptor and that gene is not expressed, the entire pathway may be shut off. Conversely, changes in downstream genes might have less impact. Methods like Impact Analysis use this information to calculate a pathway perturbation score, producing more biologically accurate results [3].
Essential Databases and Research Toolkit
The utility of any enrichment analysis is directly tied to the quality and comprehensiveness of the pathway databases used. The table below summarizes key resources.
| Database | Type | Description & Key Features |
|---|---|---|
| Gene Ontology (GO) [1] | Gene Set | A hierarchically organized set of thousands of standardized terms for biological processes, molecular functions, and cellular components. Biological Process terms are most commonly used. |
| MSigDB [1] | Gene Set | A large, curated database of gene sets from various sources, including GO, pathways, and published studies. Its "Hallmark" gene set collection is a relatively non-redundant, useful resource. |
| Reactome [1] | Detailed Pathway | An actively updated, general-purpose public database of human pathways with detailed biochemical reactions and regulatory events. |
| KEGG [1] | Detailed Pathway | Provides intuitive pathway diagrams for metabolism, signaling, and disease. Licensing restrictions can affect free access to up-to-date files. |
| WikiPathways [1] | Meta-Database | A community-driven, open-source platform that collects and creates pathways from various sources. |
| PFOCR [4] | Novel Database | Uses machine learning to extract pathway information and gene sets directly from published pathway figures in the literature, offering exceptional breadth and direct literature support. |
Beyond databases, a successful analysis relies on a toolkit of software and platforms.
| Tool / Resource | Function | Key Characteristics |
|---|---|---|
| g:Profiler [1] | Enrichment Analysis Tool | A free web tool for ORA, known for ease of use, extensive documentation, and up-to-date databases. |
| GSEA [1] | Enrichment Analysis Software | The original software for FCS, widely used for analyzing ranked gene lists against gene sets, notably from MSigDB. |
| Cytoscape & EnrichmentMap [1] | Visualization | Free, open-source platforms for visualizing molecular interaction networks and enrichment results, helping to identify overarching themes. |
| STAGEs [5] | Integrated Web Tool | A web-based platform that integrates data visualization (e.g., volcano plots) with pathway enrichment analysis using Enrichr and GSEA, simplifying the workflow. |
| PEANUT [6] | Network-Based Tool | A newer tool that enhances traditional analysis by integrating protein-protein interaction networks to amplify signals from connected gene sets. |
| QIAGEN IPA [7] | Commercial Platform | A comprehensive, commercial software built on an expert-curated knowledge base, offering causal reasoning and upstream regulator analysis. |
Purpose and Impact in Biomedical Research
The core purpose of pathway enrichment analysis is to add mechanistic insight and biological context to observational gene lists. It is a critical step in translating data into discovery.
- Gaining Mechanistic Insight: PEA helps answer the question, "What biological processes are most relevant to my experimental condition?" It shifts the focus from individual genes to systems-level biology, revealing the orchestrated activity that underlies phenotypes [1] [3].
- Prioritizing Findings: By providing a statistical framework, PEA allows researchers to prioritize pathways, rather than individual genes, for further experimental investigation. This is especially valuable in drug repurposing efforts, where analyzing shared mechanisms of action across multiple drugs can increase on-target signal and reduce false leads [8].
- Generating Testable Hypotheses: The list of enriched pathways serves as a source of new, testable hypotheses about disease mechanisms or treatment effects. For instance, identifying histone and DNA methylation as an enriched pathway in childhood ependymoma led to the rational therapeutic use of 5-azacytidine, which stopped rapid metastatic tumor growth in a terminally ill patient [1].
Pathway Enrichment Analysis is an indispensable bioinformatic method for interpreting high-throughput biological data. By statistically evaluating the collective behavior of genes within the context of predefined biological pathways, it provides a powerful lens through which researchers can discern meaningful patterns and mechanisms in complex datasets. The field continues to evolve with the integration of network biology, more sophisticated topological analyses, and the development of expansive new resources like PFOCR. For researchers in basic science, translational medicine, and drug development, a firm grasp of PEA's principles, methods, and tools is fundamental to transforming genomic data into actionable biological knowledge and therapeutic advances.
Pathway enrichment analysis is a cornerstone of functional genomics, enabling researchers to move beyond a simple list of differentially expressed genes to a mechanistic understanding of the biological processes underlying their experimental data. This analytical approach statistically evaluates whether pre-defined sets of genes (pathways or gene sets) are over-represented in an experimentally derived gene list more than would be expected by chance [2] [9]. By harnessing prior biological knowledge, pathway analysis increases statistical power, eases interpretation, and helps predict new roles for genes, making it particularly valuable for studying complex diseases where individual genetic effects may be modest but concerted pathway-level effects are substantial [2] [9].
The fundamental motivation for pathway analysis stems from observations that multiple disease-associated genetic variants often impinge on a limited number of common pathways or interacting networks. Notable examples include synaptic biology in schizophrenia, cytokine pathways in immune diseases, and complement pathways in age-related macular degeneration [9]. This approach stands in contrast to single-locus analysis, as it takes a multilocus strategy that capitalizes on biological knowledge, thereby increasing discovery power while facilitating biological interpretation of statistical associations [9].
Core Statistical Frameworks in Pathway Analysis
Over-Representation Analysis (ORA)
Over-Representation Analysis represents the first generation of pathway analysis methods. ORA statistically evaluates whether the fraction of genes in a particular pathway found among a set of differentially expressed genes is greater than what would be expected by random chance [2]. The method begins with a list of differentially expressed genes, typically identified using an arbitrary threshold (e.g., p-value < 0.05, fold change > 2), and then identifies pathways that are over- or under-represented in this gene list [2] [3].
The statistical foundation of ORA typically relies on the hypergeometric distribution, Fisher's exact test, chi-square test, or binomial distribution. These tests determine the probability that the number of genes from a particular pathway observed in the differentially expressed gene list would occur by random chance [2]. The hypergeometric test is conceptually equivalent to the "urn problem": if you have a total of N genes in the genome, with K genes belonging to a pathway of interest, and you draw n genes (your list of differentially expressed genes), what is the probability that k or more of these drawn genes belong to the pathway of interest?
Key assumptions and limitations of ORA include:
- It requires an appropriate background gene set for comparison, which could be all genes in the organism, all protein-coding genes, or only genes measured/expressed in the experiment [2]
- It depends heavily on the arbitrary threshold used to select differentially expressed genes [3]
- It assumes independence between genes [2]
- It discards quantitative information about the magnitude of expression changes [3]
Functional Class Scoring (FCS) Methods
Functional Class Scoring methods represent a second generation of pathway analysis approaches designed to overcome some limitations of ORA. Rather than relying on an arbitrary threshold to select differentially expressed genes, FCS methods consider all measured genes and their expression values [2] [3]. The fundamental hypothesis behind these methods is that small but coordinated changes in sets of functionally related genes may be biologically important, even if individual genes do not show large expression changes [3].
Gene Set Enrichment Analysis (GSEA) is arguably the most prominent FCS method. Instead of pre-selecting genes based on significance thresholds, GSEA uses all genes ranked by the magnitude of their expression change between conditions [2]. The ranking is typically based on a combination of fold change and statistical significance, with the most strongly upregulated and significant genes at the top and the most strongly downregulated and significant genes at the bottom [2]. GSEA then determines whether members of a predefined gene set are randomly distributed throughout this ranked list or primarily found at the top or bottom, suggesting coordinated differential expression [2].
The method creates a running sum statistic (enrichment score) that increases when a gene in the set is encountered and decreases when genes not in the set are encountered. The enrichment score is then normalized and assessed for statistical significance using a permutation-based approach [2]. The Molecular Signatures Database (MSigDB) is a curated resource of thousands of gene sets specifically designed for use with GSEA and similar methods [2].
Pathway Topology-Based Methods
Pathway Topology methods represent the third generation of pathway analysis approaches that aim to incorporate the rich biological knowledge embedded in pathway structures. While both ORA and FCS methods treat pathways as simple gene sets (unordered collections of genes), topology-based methods recognize that pathways are complex models describing biological processes, mechanisms, and interactions [3].
These methods utilize prior knowledge about pathway topology - including the positions and roles of genes, types of interactions (activation, repression, phosphorylation), direction of signal propagation, and other relational information - to derive more biologically meaningful assessments of pathway perturbation [2] [3]. Impact Analysis, for example, constructs a mathematical model that captures the entire topology of a pathway and uses it to calculate perturbations for each gene, which are then combined into a total perturbation for the entire pathway [2].
Key advantages of topology-based methods include:
- They account for the type and direction of interactions within pathways [3]
- They consider the positions and roles of genes within pathways [3]
- They can predict or explain downstream or pathway-level effects [3]
- They help identify specifically affected mechanisms in an experiment [3]
Table 1: Comparison of Pathway Analysis Methodologies
| Feature | Over-Representation Analysis (ORA) | Functional Class Scoring (FCS) | Pathway Topology (PT) |
|---|---|---|---|
| Input | List of differentially expressed genes | All genes with expression values | All genes with expression values plus pathway structure |
| Statistical Basis | Hypergeometric, Fisher's exact test | Kolmogorov-Smirnov, permutation tests | Network perturbation models |
| Handles Subtle Effects | No | Yes | Yes |
| Uses Pathway Structure | No | No | Yes |
| Key Advantage | Simple, intuitive | No arbitrary threshold needed | Biologically realistic |
| Key Limitation | Depends on arbitrary threshold | Ignores pathway structure | Requires curated pathway data |
The Multiple Testing Problem in Pathway Analysis
Understanding the Multiple Comparisons Problem
In pathway analysis, researchers typically test hundreds or thousands of gene sets simultaneously, which creates a substantial multiple testing problem. When conducting multiple independent statistical tests, the probability of obtaining at least one false positive result increases dramatically with the number of tests performed [10]. For example, if 20 independent tests are conducted with a significance level (α) of 0.05, the probability of observing at least one false positive is approximately 64% [10].
The multiple comparisons problem arises because the significance level α represents the probability of rejecting the null hypothesis when it is actually true (Type I error). When conducting m independent tests, the probability of making at least one Type I error (called the family-wise error rate or FWER) is given by:
1 - (1 - α)^m
For m = 20 tests with α = 0.05, this becomes 1 - (0.95)^20 ≈ 0.64, meaning there's a 64% chance of at least one false positive [10] [11]. In pathway analysis, where the number of tests can be much larger, this problem becomes even more pronounced, making multiple testing correction an essential step in the analytical workflow [9].
Correction Methods
Bonferroni Correction
The Bonferroni correction is the simplest and most conservative method for multiple testing correction. It controls the family-wise error rate (FWER), which is the probability of making at least one Type I error across all tests [10] [11]. The method works by dividing the desired significance level (α) by the number of tests performed (m):
Adjusted significance threshold = α/m
For example, with an original α of 0.05 and 20 tests, the Bonferroni-corrected significance threshold would be 0.05/20 = 0.0025 [10]. Any p-value below this adjusted threshold would be considered statistically significant after correction.
The Bonferroni correction is based on the union bound, which states that the probability of at least one false positive is less than or equal to the sum of the individual false positive probabilities [10]. While this method provides strong control over false positives, it can be overly conservative, especially when dealing with many tests or correlated hypotheses, leading to increased Type II errors (false negatives) and reduced statistical power [10] [11].
False Discovery Rate (FDR) Control
As an alternative to the conservative Bonferroni approach, methods controlling the False Discovery Rate (FDR) have gained popularity in genomic applications. The FDR is the expected proportion of false positives among all significant tests [10]. Unlike the FWER, which controls the probability of at least one false positive, FDR methods allow a small proportion of false positives while maintaining higher statistical power [10].
The Benjamini-Hochberg procedure is the most widely used FDR-controlling method. It works by:
- Sorting all p-values from smallest to largest: p(1) ≤ p(2) ≤ ... ≤ p(m)
- Finding the largest k such that p(k) ≤ (k/m) × α
- Declaring the tests corresponding to p(1), p(2), ..., p(k) as significant
This approach is less conservative than Bonferroni correction and is particularly useful in high-throughput genomic studies where researchers are willing to tolerate some false positives in exchange for greater power to detect true effects [10].
Table 2: Multiple Testing Correction Methods
| Method | Controls | Approach | Best Use Cases |
|---|---|---|---|
| Bonferroni | Family-Wise Error Rate (FWER) | Divide α by number of tests (α/m) | When false positives are very costly; small number of tests |
| Holm-Bonferroni | FWER | Step-down procedure: order p-values and compare to α/(m+1-i) | Less conservative than Bonferroni; general FWER control |
| Benjamini-Hochberg | False Discovery Rate (FDR) | Step-up procedure controlling expected proportion of false discoveries | Genomic studies; large number of tests; balance of power and precision |
Experimental Design and Workflow
Pathway Analysis Workflow
A comprehensive pathway analysis involves multiple critical steps, each requiring careful consideration to ensure biologically meaningful and statistically valid results. The major analytical procedures include hypothesis selection, SNP-to-gene mapping (for genetic data), enrichment testing, and multiple testing correction [9].
Pathway Analysis Workflow
Critical Experimental Considerations
Several key decisions throughout the pathway analysis workflow can significantly impact results and interpretation:
Gene Set Selection: The choice of gene set database fundamentally shapes analytical outcomes. Major categories include functional annotation-based sets (Gene Ontology, KEGG, Reactome), disorder-based sets, and high-throughput data-derived sets [9]. Each database has different coverage, curation standards, and biological emphasis, making database selection a critical consideration.
Background Set Definition: The appropriate background set for comparison must reflect the experimental context. Options include all genes in the genome, all protein-coding genes, only genes measured on the specific platform used, or only genes expressed in the experimental system [2]. An improperly specified background can introduce substantial bias.
SNP-to-Gene Mapping (for GWAS): When analyzing genetic variation data, the strategy for connecting genetic variants to genes significantly influences results. Approaches include mapping to the nearest gene, using a specific window size, incorporating regulatory information, or employing chromatin interaction data [9].
Handling Gene Length and GC Content Bias: Certain analysis methods may be susceptible to biases related to gene length or GC content, particularly for RNA-seq data. These technical artifacts can disproportionately influence results if not properly addressed [9].
Key Databases and Knowledgebases
Successful pathway analysis relies heavily on high-quality, curated biological knowledge resources. These databases provide the gene sets and pathway information that form the foundation of enrichment analysis.
Table 3: Essential Pathway Analysis Resources
| Resource | Type | Key Features | Common Applications |
|---|---|---|---|
| Gene Ontology (GO) | Functional Annotation | Three domains: Molecular Function, Cellular Component, Biological Process; species-agnostic | General functional enrichment; ORA analysis |
| KEGG | Pathway Database | Curated biological pathways; molecular interaction networks; pathway maps | Metabolic and signaling pathway analysis |
| Reactome | Pathway Database | Human-specific; curated signaling, metabolic processes; disease pathways | Detailed pathway analysis; visualization |
| MSigDB | Gene Set Collection | 34,837+ gene sets; curated for GSEA; hallmark collections with reduced redundancy | GSEA analysis; immunological research |
| PANTHER | Classification System | Protein families and phylogenetic trees; evolutionary relationships | Evolutionarily informed analysis |
| WikiPathways | Pathway Database | Community-curated; continuously updated; diverse pathways | Novel pathway discovery; less established mechanisms |
Analytical Tools and Software
The pathway analysis landscape includes numerous software tools and packages implementing various statistical approaches:
Web-Based Tools: DAVID, Qiagen IPA, and WebGestalt provide user-friendly interfaces for ORA and basic enrichment analysis, making them accessible to wet-lab researchers without programming expertise [2].
R/Bioconductor Packages: Tools like clusterProfiler, fgsea, and SPIA offer programmatic access to advanced analysis methods, enabling customized workflows and integration with other bioinformatics analyses [2].
Specialized Software: GSEA from the Broad Institute provides a standalone desktop application specifically optimized for gene set enrichment analysis, with tight integration to MSigDB [2].
Advanced Applications and Future Directions
Pathway analysis continues to evolve with methodological advancements and expanding applications. Integrative approaches that combine multiple data types (genetic variation, gene expression, epigenetic modifications) represent the cutting edge of pathway analysis methodology [9]. These methods leverage complementary information to provide more comprehensive biological insights than single-data-type analyses.
Emerging applications include:
- Multi-omics pathway analysis integrating genomic, transcriptomic, and proteomic data
- Cell-type-specific pathway analysis using single-cell sequencing data
- Cross-disorder pathway analysis identifying shared biological mechanisms
- Pharmacogenomic pathway analysis for drug target identification
As pathway analysis methodologies mature, considerations of power, sample size, and analytical validity become increasingly important. Future developments will likely focus on improving statistical power for detecting pathway-level signals, enhancing methods for multi-omics integration, and developing more sophisticated approaches for modeling pathway dynamics and interactions [9].
Integrative Multi-omics Pathway Analysis
Pathway Enrichment Analysis (PEA) is a cornerstone bioinformatics method for interpreting the results of genome-scale experiments. It helps researchers move from seemingly impenetrable lists of genes to a mechanistic understanding of the underlying biology by identifying predefined sets of biologically related genes that are statistically overrepresented [12] [1]. In modern research, technologies like RNA-seq, proteomics, and genome sequencing comprehensively measure cellular molecules but often produce lists of hundreds or thousands of significant genes. Manually sifting through these lists is impractical [1]. PEA addresses this challenge by summarizing large gene lists into a smaller, more interpretable set of biological pathways or processes, effectively translating data into biological insight [1]. For instance, it has been used to identify histone and DNA methylation as a therapeutic target in a childhood brain cancer, leading to a compassionate treatment that stopped tumor growth [1]. This protocol is essential for researchers and drug development professionals aiming to understand complex disease mechanisms, identify novel therapeutic targets, and generate testable hypotheses from high-throughput omics data.
Core Terminology and Definitions
A precise understanding of the key terms is fundamental to correctly applying and interpreting pathway enrichment analysis. The following table structures and defines the essential vocabulary in this field.
Table 1: Essential Terminology in Pathway Enrichment Analysis
| Term | Definition | Key Characteristics |
|---|---|---|
| Gene Set | An unordered, unstructured collection of genes grouped by a shared biological property, location, or involvement in a pathway [3] [13]. | Lacks internal structure; a simple list. Examples: genes on chromosome 1, genes from a KEGG pathway. |
| Pathway | A series of interactions among molecules in a cell that leads to a product or change, describing specific mechanisms, phenomena, and dependencies [3]. | A model with structure, interactions, and directionality (e.g., KEGG, Reactome pathways). |
| Pathway Enrichment Analysis (PEA) | A statistical technique to identify pathways significantly overrepresented in a gene list more than expected by chance [12] [1]. | An umbrella term; sometimes used interchangeably with Functional Enrichment Analysis. |
| Gene Set Enrichment Analysis (GSEA) | A specific computational method determining if a predefined gene set shows significant, concordant differences between two biological states [14]. | Both an analysis type (see below) and a specific software tool from Broad Institute [14]. |
| Enrichment Score (ES) | A statistic quantifying the degree to which a gene set is overrepresented at the extremes (top or bottom) of a ranked gene list [15]. | A Kolmogorov-Smirnov-like statistic; core to the GSEA method. |
| Leading Edge Genes | A subset of genes in an enriched set that appear at the start of the enrichment peak and are considered the primary drivers of the enrichment signal [1]. | Often account for a pathway being defined as enriched. |
Pathways vs. Gene Sets: A Critical Distinction
While the terms "pathway" and "gene set" are sometimes used interchangeably, they represent fundamentally different concepts. A pathway is a detailed model that describes a biological process, such as a signaling cascade or a metabolic reaction. It contains crucial information about the roles, interactions, and directionality between genes and gene products. For example, the KEGG MAPK signaling pathway shows which genes activate others, the location of interactions, and the flow of information [3].
In contrast, a gene set is simply the list of genes involved in that pathway, stripped of all its structural and relational context [3]. Treating a pathway as a mere gene set discards valuable biological knowledge about how genes interact. This distinction is critical because topology-based analysis methods that use full pathway information can produce more accurate and biologically meaningful results than those that use gene sets alone [3] [13].
Key Methodological Approaches
There are three primary methodological approaches to functional enrichment analysis, each with its own strengths, limitations, and statistical foundations.
Over-Representation Analysis (ORA)
Concept: Over-Representation Analysis (ORA) is the simplest and most straightforward approach. It tests whether genes from a pre-defined gene set are present in a submitted list of interesting genes more than would be expected by chance [13].
Workflow and Statistical Foundation:
- Input: A list of significant genes derived from an omics experiment, typically created by applying a cutoff (e.g., adjusted p-value < 0.05 and fold-change > 2) [1] [13].
- Statistical Test: A Fisher's exact test or a hypergeometric test is commonly used to calculate the probability (p-value) of observing the overlap between the submitted list and the gene set by random chance [12] [16] [13].
- Output: A list of gene sets that are statistically overrepresented in the submitted gene list.
Limitations: ORA is sensitive to the arbitrary cutoff used to create the input gene list and assumes gene independence, which is often biologically unrealistic [13]. It also ignores the magnitude of gene expression changes [3].
Functional Class Scoring (FCS) / Gene Set Enrichment Analysis (GSEA)
Concept: Functional Class Scoring (FCS) methods, most famously Gene Set Enrichment Analysis (GSEA), were designed to overcome the cutoff dependency of ORA. Instead of a simple list, these methods use a ranked list of all genes from an experiment (e.g., ranked by differential expression statistic) to identify gene sets enriched at the top or bottom of the list [15] [13].
Workflow and Statistical Foundation (GSEA):
- Input: A ranked list of all genes from an omics dataset [1].
- Enrichment Score (ES) Calculation: The ES is the primary statistic. It is calculated by walking down the ranked list, increasing a running sum when a gene is in the set (S) and decreasing it when it is not. The increment is based on the gene's correlation with the phenotype. The ES is the maximum deviation from zero encountered [15].
- Phit(S,i) = ∑ (|rj|^p / NR) for genes gj in S, j ≤ i
- Pmiss(S,i) = ∑ (1/(N-NH)) for genes gj not in S, j ≤ i
- ES = max|Phit(S,i) - Pmiss(S,i)|, where rj is the gene's correlation, p is a weighting exponent, NR is a normalization factor, N is the total genes, and NH is genes in the set [15].
- Significance Estimation: The significance of the ES is estimated by comparing it to a null distribution generated by permuting the phenotype labels [15].
- Multiple Testing Correction: The final step adjusts for testing multiple gene sets simultaneously, typically by controlling the False Discovery Rate (FDR) [15].
Advantages: GSEA is more sensitive than ORA because it can detect subtle but coordinated changes in a group of genes, where individual genes may not be significant on their own [15] [13].
Pathway Topology (PT) Methods
Concept: Pathway Topology (PT) methods, also known as topology-based (TB) or "pathway analysis," represent a more advanced approach that moves beyond simple gene sets. They incorporate the detailed structure of pathways, including the positions of genes, the types of interactions (e.g., activation, inhibition), and the direction of signal flow [3] [13].
Workflow and Statistical Foundation:
- Input: Gene expression data and a structured pathway model from databases like KEGG or Reactome.
- Analysis: The method considers how measured gene expression changes propagate through the pathway's network structure. For example, the absence of an upstream receptor (e.g., INSR in the insulin pathway) would have a much greater impact than a change in a downstream component [3].
- Output: A ranked list of pathways whose overall activity is deemed significantly perturbed, considering the network topology.
Advantages: PT methods can more accurately model biological reality and predict the functional impact of expression changes, potentially leading to more relevant and robust results [3]. Limitations: They require high-quality, detailed pathway models, which are not available for all organisms or processes [13].
Diagram: Three primary methodological approaches for enrichment analysis, highlighting their distinct inputs and key characteristics.
Quantitative Foundations: Enrichment Scores and Statistics
The core of any enrichment method is its statistical engine. The following table compares the quantitative foundations of the major approaches.
Table 2: Statistical Foundations of Enrichment Methods
| Method | Core Statistical Test | Key Metrics & Scores | Data Input Requirement |
|---|---|---|---|
| ORA | Fisher's Exact Test / Hypergeometric Test [12] [16] | P-value, Odds Ratio, Enrichment Score (Observed/Expected) [16] | List of significant genes (uses a cutoff) [13] |
| GSEA | Kolmogorov-Smirnov-like statistic with permutation testing [15] | Enrichment Score (ES), Normalized ES (NES), False Discovery Rate (FDR), Leading-edge genes [15] [1] | Ranked list of all genes (no cutoff) [1] |
| Topology-Based | Varies by method (e.g., Impact Analysis) [3] | Pathway Impact P-value, Perturbation Statistic | Gene expression data and a structured pathway model [3] |
The GSEA Enrichment Score in Detail
The GSEA Enrichment Score (ES) is a pivotal statistic in modern enrichment analysis. It is calculated by walking down a ranked list of genes (e.g., ranked by correlation with a phenotype) and evaluating the distribution of genes in a set S [15].
- Enrichment Score (ES): The maximum deviation from zero of a running sum statistic, Phit - Pmiss [15].
- Phit(S,i): Increases when a gene in the set S is encountered. The increment is proportional to |rj|^p, where rj is the gene's correlation with the phenotype, allowing genes more strongly correlated with the phenotype to contribute more to the score [15].
- Pmiss(S,i): Increases when a gene not in the set S is encountered, ensuring sets that are randomly distributed receive a low score [15].
- Normalized ES (NES): The ES is normalized for the size of the gene set, allowing for comparison across gene sets of different sizes [15].
- Significance: The NES is compared to a null distribution generated by permuting phenotype labels, yielding a nominal p-value. This is then adjusted for multiple hypothesis testing across all gene sets, resulting in an FDR q-value [15].
Diagram: The workflow for calculating and evaluating the Gene Set Enrichment Analysis (GSEA) Enrichment Score.
The Researcher's Toolkit: Databases and Software
Successful pathway enrichment analysis relies on using curated knowledge bases and robust software tools.
Essential Pathway and Gene Set Databases
Table 3: Key Databases for Pathway and Gene Set Information
| Database | Type | Scope and Key Features |
|---|---|---|
| Gene Ontology (GO) [1] | Ontology | A hierarchically structured, standardized vocabulary of terms for Biological Processes, Molecular Functions, and Cellular Components [1]. |
| Molecular Signatures Database (MSigDB) [14] [1] | Gene Set Database | A large, comprehensive collection of over 10,000 annotated gene sets, including those from GO, pathways, and literature signatures [14] [1]. |
| Reactome [1] | Pathway Database | An open-access, peer-reviewed database of detailed human biological pathways, actively curated and updated [1]. |
| KEGG [1] [3] | Pathway Database | Known for intuitive pathway diagrams; includes metabolic, signaling, and disease-related pathways [1] [3]. |
| WikiPathways [1] | Pathway Meta-Database | A community-driven, collaborative platform for pathway curation and collection [1]. |
Software Tools for Analysis
A wide array of tools exists, from web-based platforms to command-line packages.
- g:Profiler: A widely used tool for ORA, available as both a web server and an R package. It supports multiple organisms and statistical corrections [12] [1].
- Enrichr: A popular, user-friendly web tool that provides ORA against a vast and frequently updated collection of gene set libraries [15] [17].
- GSEA Software: The original desktop implementation of the GSEA algorithm from the Broad Institute, tightly integrated with the MSigDB [14].
- clusterProfiler: An R/Bioconductor package that is highly versatile and powerful, supporting both ORA and GSEA methods for comparative functional analysis [15] [13].
- Cytoscape & EnrichmentMap: A powerful visualization platform. The EnrichmentMap app can create network-based visualizations of enrichment results, helping to identify overarching biological themes [1].
- Topology-Based Tools: R packages like ROntoTools and TPEA implement various topology-based algorithms for a more in-depth pathway impact analysis [3] [13].
Experimental Protocol: A Step-by-Step Guide
This protocol, adapted from a Nature Protocols article, outlines a standard workflow for performing and visualizing a pathway enrichment analysis, suitable for data from RNA-seq or genome-sequencing experiments [1].
Stage 1: Define a Gene List of Interest
The first step is to process your omics data to create a gene list for analysis. The type of list depends on your data and chosen method [1].
- For ORA (e.g., using g:Profiler): Generate a list of significant genes. This typically involves applying a threshold to your data, such as an adjusted p-value (FDR) < 0.05 and an absolute fold-change > 2, to define a set of differentially expressed genes [1] [13].
- For GSEA (e.g., using GSEA software): Create a ranked list of all genes. Genes are typically ranked by a metric that reflects their association with the phenotype, such as the signed -log10(p-value) (where the sign is taken from the fold-change) or the signal-to-noise ratio [1].
Stage 2: Perform Pathway Enrichment Analysis
Using g:Profiler (ORA):
- Access the g:Profiler web tool (g:GOSt) or R package.
- Input your list of significant genes.
- Select the organism and relevant data sources (e.g., GO:BP, KEGG, Reactome).
- Set the significance threshold (e.g., FDR < 0.05) and run the analysis [1].
Using GSEA Software:
- Prepare your input files: a normalized gene expression dataset (.gct) and a phenotype labels file (.cls).
- Load your data and the desired gene set database (e.g., from MSigDB).
- Set the permutation type (usually phenotype) and the number of permutations (e.g., 1000).
- Run the GSEA analysis. The output will include the enriched gene sets, their ES, NES, FDR, and leading-edge genes [1].
Stage 3: Visualize and Interpret Results
Visualization is key to interpreting the often long list of enriched pathways.
- EnrichmentMap for Cytoscape: This is a highly effective visualization technique.
- Import your GSEA or g:Profiler results into Cytoscape via the EnrichmentMap app.
- The app automatically generates a network where nodes represent enriched gene sets and edges represent the overlap of genes between sets. This clusters related pathways (e.g., all immune-related processes), allowing you to see broad biological themes rather than isolated terms [1].
- Voronoi Maps (Reactome): Tools like Reactome provide alternative visualizations like Voronoi maps, which offer a tiled overview of analysis results, with tile size and color often representing the statistical significance of the enrichment [18].
Advanced Concepts and Future Directions
As the field evolves, several advanced concepts are becoming critical for robust and cutting-edge research.
- Multi-Omics Integration: A major frontier is the integration of multiple omics datasets (e.g., transcriptomics, proteomics, epigenomics) to gain a holistic understanding. Methods like ActivePathways and its extension Directional P-value Merging (DPM) allow for the fusion of p-values and directional changes (e.g., fold-changes) across datasets, prioritizing genes and pathways with consistent signals [19].
- Best Practices and Pitfalls: To ensure meaningful results:
- Clarify Your Analysis Type: Before starting, decide whether ORA, GSEA, or PT is most appropriate for your data and question [12].
- Ensure Input Data Quality: The principle of "garbage in, garbage out" applies. Use high-quality, well-processed input gene lists [12].
- Apply Multiple Testing Correction: Always use FDR or another correction method to account for the thousands of pathways tested, reducing false positives [1].
- Understand the Limitations: No single method is perfect. Be aware that gene set analysis does not inherently indicate if a pathway is activated or inhibited, and it relies on the completeness and accuracy of the underlying databases [12] [13].
Pathway Enrichment Analysis is an indispensable technique for translating high-throughput genomic data into biological insight. A firm grasp of the essential terminology—distinguishing a gene set from a pathway, and understanding what an enrichment score represents—is the foundation. By selecting the appropriate methodological approach (ORA, FCS/GSEA, or PT) and leveraging the powerful databases and software tools available, researchers can systematically uncover the functional themes and mechanistic underpinnings of their experiments. As the field moves towards multi-omics integration and more sophisticated topology-based models, these core concepts will continue to be vital for driving discovery in biology and drug development.
Pathway Enrichment Analysis (PEA) is a foundational computational biology method used to interpret lists of genes or proteins derived from high-throughput omics experiments. It identifies biological pathways—predefined sets of genes that collectively perform a specific function—that are overrepresented in a gene list more than would be expected by chance [12]. This process helps researchers move from a simple list of differentially expressed genes to a functional understanding of the underlying biology, revealing the processes most affected in a given condition, such as disease states or drug treatments.
Two primary computational approaches are used: Overrepresentation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA). ORA uses statistical tests like the hypergeometric test or Fisher's exact test to determine if certain pathways contain a disproportionately high number of genes from an input list, typically a set of differentially expressed genes identified using a significance cutoff [20] [12]. In contrast, GSEA considers the entire ranked list of genes (e.g., by expression fold-change or p-value) without requiring an arbitrary cutoff. It identifies pathways where genes are concentrated at the extreme ends (top or bottom) of the ranked list, detecting subtle but coordinated changes in expression that might be missed by ORA [14] [20] [12]. The choice between these methods depends on the research question and data type, a critical decision point for generating robust results [12].
Core Pathway Databases
Several curated databases provide the biological pathway and gene set definitions essential for enrichment analysis. The table below summarizes the key features of five major resources.
Table 1: Core Features of Major Pathway Databases
| Database | Primary Focus | Key Features & Content | Species Coverage | Update Status |
|---|---|---|---|---|
| Gene Ontology (GO) [12] | Structured, hierarchical vocabulary (ontologies) for gene function. | Three independent aspects: Biological Process, Molecular Function, and Cellular Component. | Extensive, many species | Continuously updated |
| KEGG (Kyoto Encyclopedia of Genes and Genomes) [12] | Reference knowledge on biological pathways and systems. | Well-known pathway maps for metabolism, genetic information processing, and human diseases. | Extensive, many species | Updated regularly (e.g., Nov 2023) [21] |
| Reactome [22] [23] | Expert-authored, detailed molecular pathways. | ~2,825 human pathways with 16,002 reactions; includes detailed pathway topology and expression overlay. | Extensive, but projects other species to human by default [22] | Version 94 (Sept 2025) [23] |
| MSigDB (Molecular Signatures Database) [14] | Broad collection of annotated gene sets for GSEA. | Includes Hallmark sets, curated pathways, GO terms, and computational signatures from published studies. | Human, mouse, rat [24] | MSigDB 2025.1 (Jun 2025) [14] |
| WikiPathways [12] | Collaborative, community-curated pathway resource. | Diverse pathway content curated by researchers; pathways are editable and versioned. | Extensive, many species | Continuously updated [25] |
Detailed Database Profiles
Gene Ontology (GO): GO is not a pathway database in the traditional sense but a comprehensive, structured vocabulary that describes the roles of genes and gene products. Its value in enrichment analysis lies in its ability to provide a deep functional context. An enrichment result for a term like "positive regulation of cell migration" (a Biological Process) can offer a more granular understanding of phenotype than a broader pathway might [12].
KEGG (Kyoto Encyclopedia of Genes and Genomes): KEGG provides manually drawn reference pathway maps that are widely recognized in the scientific community. It is particularly strong in metabolic pathways and disease-related pathways. The KEGG Mapper Color tool allows users to visualize their own data (e.g., gene identifiers with color specifications) directly onto these pathway maps for intuitive interpretation [21].
Reactome: Reactome is an open-access, peer-reviewed database known for its highly detailed and accurate molecular pathways. A key strength is its powerful analysis toolkit, which supports not only standard over-representation analysis but also pathway topology analysis, which considers the connectivity between molecules in a pathway. Furthermore, Reactome allows the overlay of expression data or other numerical values onto its pathway diagrams, enabling powerful visualization of experimental results [22] [23].
MSigDB (Molecular Signatures Database): MSigDB is a massive, diverse collection of gene sets designed specifically for use with GSEA software. Its collections extend beyond canonical pathways to include gene sets derived from perturbation studies, genetic signatures, and immunologic signatures. A notable feature is the "Hallmark" gene sets, which summarize and represent specific well-defined biological states or processes, reducing redundancy and simplifying interpretation [14].
WikiPathways: As a wiki-based platform, WikiPathways leverages the power of community curation to keep pathways current with the latest research. This model allows for rapid updates and the creation of highly specialized pathways that might not be available in other databases. The platform provides detailed curation guidelines to ensure the quality and consistency of its content [25] [12].
Experimental Protocols for Pathway Enrichment Analysis
This section provides detailed methodologies for performing enrichment analysis using both ORA and GSEA approaches, which represent the two primary paradigms in the field.
Protocol A: Overrepresentation Analysis (ORA) with g:Profiler
ORA is used when the input is a flat, unordered list of genes, typically a set of significantly differentially expressed genes.
Table 2: Research Reagent Solutions for ORA with g:Profiler
| Item Name | Function/Description | Example/Format |
|---|---|---|
| Input Gene List | A list of significant genes (e.g., DEGs). | A single-column text file with gene identifiers (HGNC symbols, Ensembl IDs, etc.). |
| Background Gene Set | The set of all genes considered in the experiment. | Often implied by the tool; can be set to all genes in the genome. |
| Pathway Gene Sets (GMT File) | The database of pathways used for the enrichment test. | A GMT file containing pathways from GO, Reactome, etc. [20]. |
| g:Profiler Web Tool | The ORA software tool for performing the analysis. | Accessible at http://biit.cs.ut.ee/gprofiler/ [20]. |
Step-by-Step Procedure:
Prepare Input Data: Compile your list of significant genes (e.g., differentially expressed genes with p-value < 0.05) into a single-column plain text file. Ensure gene identifiers are consistent with the database being used (e.g., HGNC symbols) [20].
Access g:Profiler: Open a web browser and navigate to the g:Profiler website (http://biit.cs.ut.ee/gprofiler/) [20].
Input Data and Set Parameters:
- Paste the gene list into the "Query" field.
- Check the box for "Ordered query" if your list is ranked.
- Check "No electronic GO annotations" to increase result reliability.
- Click "Show Advanced Options" [20].
Configure Advanced Options:
- Data Sources: In the legend, select the pathway databases for the analysis. For an initial analysis, it is recommended to select Biological Processes (BP) from GO and pathways from Reactome [20].
- Pathway Size Filtering: Set the minimum size of a functional category to
5and the maximum to350. This filters out overly broad pathways and those that are too small for meaningful statistical testing [20]. - Statistical Threshold: Set the "Size of query/term intersection" to
3, meaning a pathway will only be considered if it shares at least 3 genes with your input list [20].
Execute and Retrieve Results:
- Click "g:Profile!" to run the analysis. The results will be displayed as a heatmap.
- To prepare results for visualization in tools like Cytoscape, change the "Output type" to "Generic Enrichment Map (TAB)" and run the analysis again.
- Download the results file in this format for the next steps [20].
Protocol B: Gene Set Enrichment Analysis (GSEA) with the GSEA Software
GSEA is applied when the input is a ranked list of all genes from an experiment, as it does not require a pre-defined significance cutoff.
Table 3: Research Reagent Solutions for GSEA
| Item Name | Function/Description | Example/Format |
|---|---|---|
| Ranked Gene List (RNK File) | A genome-wide list of genes ranked by a metric of differential expression. | A two-column text file: Gene ID and ranking metric (e.g., log2 fold-change). |
| Gene Set Database (GMT File) | A collection of gene sets (e.g., from MSigDB) against which the RNK file is tested. | A GMT file from MSigDB or Baderlab [20]. |
| GSEA Desktop Application | The Java-based software used to perform the GSEA algorithm. | Downloaded from the GSEA-MSigDB website [14] [20]. |
| Java Runtime Environment | Required to run the GSEA application. | Version 8 or higher must be installed [20]. |
Step-by-Step Procedure:
Software and Data Setup:
- Install the latest version of Java if not already present.
- Download and launch the GSEA desktop application from the official GSEA-MSigDB website (registration is free) [14] [20].
- Prepare your ranked gene list (.rnk file), a two-column text file where the first column contains gene identifiers and the second contains the ranking metric (e.g., signal-to-noise ratio, fold-change). The file should have a header row starting with
#[20].
Load Data into GSEA:
- In the GSEA application, click "Load Data" in the "Steps in GSEA Analysis" section.
- Use the file browser to select your
.rnkfile and your pathway gene set (.gmt) file. Click "Choose" to load them. A success message will appear once the files are processed [20].
Run GSEA Preranked:
- In the left-hand sidebar, under "Tools," click "Run GSEAPreranked".
- In the form that appears:
- Select your
.rnkfile for the "Gene expression dataset" parameter. - Select your
.gmtfile for the "Gene sets database" parameter. - Leave other parameters at their defaults for an initial run.
- Select your
- Click "Run" to start the analysis [20].
Interpret Results:
- GSEA generates a detailed HTML report. The key result is the Enrichment Score (ES), which reflects the degree to which a gene set is overrepresented at the top or bottom of your ranked list.
- The report includes Normalized Enrichment Score (NES), which allows for comparison across gene sets, and the False Discovery Rate (FDR),- which indicates statistical significance. An FDR < 0.25 is often considered significant in GSEA [20].
Visualization and Interpretation of Results
Effective visualization is critical for interpreting the complex results of an enrichment analysis. The following diagram illustrates the logical workflow and decision points involved in a typical PEA.
Diagram 1: PEA Workflow and Method Selection
The relationships between core databases, analysis tools, and the biological concepts they represent can be visualized as a network.
Diagram 2: Database and Concept Relationships
Advanced Visualization with Cytoscape and EnrichmentMap
For complex analyses involving many enriched pathways, tools like the EnrichmentMap app for Cytoscape are invaluable. EnrichmentMap creates a network visualization of enrichment results where each node represents a significantly enriched pathway, and edges connect pathways that share a significant number of genes. This helps researchers see functional clusters and themes, such as a large cluster of related immune response pathways, rather than interpreting a long, flat list of results [20].
The major pathway databases—GO, KEGG, Reactome, MSigDB, and WikiPathways—each offer unique content and perspectives, making them collectively indispensable for modern biological research. The choice of database and analytical method (ORA vs. GSEA) should be guided by the specific biological question and the nature of the available data. A well-executed pathway enrichment analysis, following established protocols and leveraging robust visualization, transforms raw gene lists into coherent biological narratives, directly fueling hypothesis generation and accelerating discovery in biomedical research and drug development.
Pathway Enrichment Analysis (PEA) is a cornerstone bioinformatics method for interpreting the complex data generated by modern omics technologies. By contextualizing long lists of genes, proteins, or metabolites within predefined biological pathways, PEA transforms statistical results into mechanistically meaningful insights [26] [1]. In practice, omics experiments such as RNA sequencing or mass spectrometry-based proteomics generate extensive datasets that quantify molecular abundance across different experimental conditions. The initial analysis typically identifies differentially expressed genes or significantly altered proteins—a molecular signature of the phenomenon under study [1] [27]. PEA then examines these signature molecules against knowledge bases of established biological pathways to determine which cellular processes are statistically overrepresented compared to chance occurrence [26]. This process effectively distills thousands of molecular measurements into a focused set of biologically relevant pathways, enabling researchers to formulate testable hypotheses about underlying mechanisms driving observed phenotypes, from disease progression to treatment response [1].
The fundamental value of PEA lies in its systems-level perspective. Rather than considering individual genes or proteins in isolation, PEA recognizes that cellular functions emerge from complex networks of molecular interactions [27]. For example, while a single differentially expressed gene might hint at relevant biology, the coordinated enrichment of multiple genes operating within the same pathway provides compelling evidence for the pathway's activation or repression [1]. This approach has proven instrumental across diverse applications, from identifying histone and DNA methylation by the polycomb repressive complex as a therapeutic target in childhood brain cancer to unraveling pathway dysregulation in autism spectrum disorder through copy-number variant analysis [1]. As omics technologies continue to evolve, generating increasingly complex and multidimensional data, PEA remains an essential tool for extracting biological insight from molecular signatures.
Core Methodologies and Statistical Foundations
Pathway enrichment analysis encompasses several distinct methodological approaches, each with specific statistical foundations and optimal use cases. Understanding these methodologies is crucial for selecting appropriate analytical strategies for different omics data types and research questions. The three primary categories of PEA methods include over-representation analysis, functional class scoring, and pathway topology-based approaches, each with particular strengths and considerations for implementation [26].
Over-representation analysis (ORA), the most established approach, tests whether genes of interest are overrepresented in predefined pathway gene sets more than would be expected by chance [26] [1]. ORA typically uses hypergeometric, chi-square, or Fisher's exact tests to calculate statistical significance, comparing the proportion of pathway genes in the target list against their proportion in a appropriate background set [26]. While straightforward to implement and interpret, ORA has limitations including dependence on arbitrary significance thresholds for creating gene lists and disregard for gene interactions and pathway topology [26]. Functional class scoring (FCS) methods, such as Gene Set Enrichment Analysis (GSEA), address some limitations by considering all measured genes ranked by their association with a phenotype rather than relying on arbitrary thresholds [1]. FCS methods evaluate whether members of a gene set tend to appear toward the top or bottom of the ranked list, identifying pathways where coordinated but potentially subtle changes might be biologically important [1]. Topology-based methods incorporate information about pathway structure, including the positions of genes within pathways and their relationships, to provide more biologically contextualized enrichment scores [26].
Table 1: Comparison of Major Pathway Enrichment Analysis Methodologies
| Method Type | Statistical Approach | Key Advantages | Common Tools |
|---|---|---|---|
| Over-representation Analysis (ORA) | Hypergeometric, Fisher's exact tests | Simple implementation, intuitive interpretation, works with simple gene lists | g:Profiler, DAVID |
| Functional Class Scoring (FCS) | Kolmogorov-Smirnov-like running sum statistics | Uses full gene rankings, no arbitrary thresholds, detects subtle coordinated changes | GSEA, GSVA |
| Pathway Topology-Based | Impact analysis incorporating pathway structure | Accounts for gene interactions and positions, more biological context | PathwayMapper, SPIA |
The statistical rigor of PEA requires careful consideration of multiple testing correction, as thousands of pathways are typically tested simultaneously [1]. Without appropriate correction, false positive results become likely. Common correction methods include Bonferroni (stringent) and Benjamini-Hochberg (false discovery rate) approaches [1] [27]. Additionally, proper definition of the background gene set is essential, as it represents the universe of possible genes from which the target list was drawn [26]. For RNA-seq experiments, this should typically include all genes detected above a minimum expression threshold, rather than the entire genome [26].
Experimental Design and Workflow Implementation
Implementing a robust pathway enrichment analysis requires careful attention to experimental design and computational workflow. The following diagram illustrates the standard PEA workflow, from raw omics data processing through biological interpretation:
Figure 1. Standard PEA workflow from data to interpretation
Stage 1: Input Preparation from Omics Data
The initial stage involves processing raw omics data into a gene list suitable for enrichment analysis. For RNA-seq data, this typically includes quality control, read alignment, quantification, and differential expression analysis to identify genes associated with the experimental condition [1]. The resulting output can be either a simple list of significant genes (e.g., FDR < 0.05 and fold-change > 2) or a ranked list of all genes sorted by a measure of association with the phenotype (e.g., t-statistic or fold-change) [1]. Proper definition of the background set is crucial—it should reflect all genes detectable in the experimental system, as this represents the statistical universe from which the gene list was drawn [26]. For example, in an RNA-seq experiment, the background should include all genes expressed above a minimum threshold across samples, rather than the entire genome [26].
Stage 2: Database Selection and Enrichment Calculation
Selecting appropriate pathway databases significantly influences PEA results. Commonly used resources include Gene Ontology (GO) for biological processes, molecular functions, and cellular components; Reactome for detailed human biological pathways; Molecular Signatures Database (MSigDB) for curated gene sets; and KEGG for metabolic and signaling pathways [1]. The emerging Pathway Figure OCR (PFOCR) database offers exceptional breadth, covering 77% of human genes through automated extraction of pathway figures from literature, providing unique depth with multiple pathway instances for biological processes typically represented by single canonical pathways in other databases [4]. Each database has distinct strengths—GO offers extensive coverage, Reactome provides mechanistic detail, while PFOCR excels in disease-specific pathway diversity [4].
Stage 3: Results Interpretation and Visualization
Effective visualization is essential for interpreting PEA results, which often include dozens of significantly enriched pathways. Enrichment maps create network visualizations where nodes represent enriched pathways and edges indicate gene overlap, helping identify broader biological themes [1] [27]. The Hyperpathway tool introduces innovative hyperbolic embedding of pathway-molecule bipartite networks, where radial positions indicate hierarchical importance and angular positions reflect functional similarity, effectively revealing modular organization of biological processes [27]. Directional enrichment methods like DPM (Directional P-value Merging) incorporate directional relationships between omics layers, such as the expected negative correlation between DNA methylation and gene expression, to prioritize genes with consistent directional changes across datasets [19].
Advanced Applications and Integrative Approaches
Multi-omics Integration
Pathway enrichment analysis has evolved to address the challenges and opportunities presented by multi-omics experiments. The DPM (Directional P-value Merging) method enables directional integration of multiple omics datasets by incorporating user-defined constraints based on biological relationships between data types [19]. For example, when integrating transcriptomic and proteomic data, researchers can specify an expected positive correlation between mRNA and protein expression, prioritizing genes with consistent up- or down-regulation across both molecular layers while penalizing those with discordant directions [19]. This approach increases power to detect coordinated biological events while filtering out inconsistent signals. The method can be represented as:
Figure 2. Directional multi-omics data integration
For complex longitudinal omics studies, linear mixed models (LMM) and generalized linear mixed models (GLMM) account for within-subject correlations and time-varying covariates, enabling identification of pathways with dynamic activity patterns over time [28]. These approaches are particularly valuable in clinical development for tracking pathway modulation in response to therapeutic interventions.
Machine Learning Enhancement
Machine learning (ML) approaches are increasingly integrated with pathway analysis to handle the high-dimensional, nonlinear relationships prevalent in multi-omics data [29]. ML models can capture complex interactions between pathway components that traditional statistical methods might miss, potentially improving prediction accuracy for clinical endpoints [29]. In vaccine development, multi-omics data integration using ML frameworks like MOFA has identified key pathway signatures correlated with immunogenicity, informing vaccine design and efficacy assessment [30]. The combination of ML and pathway enrichment creates a powerful framework for biomarker discovery and mechanism elucidation, particularly for complex traits like disease resistance in crops or drug response in oncology [29] [31].
Table 2: Pathway-Centric Machine Learning Applications in Biology
| Application Domain | ML Approach | Pathway Integration | Key Outcome |
|---|---|---|---|
| Crop Improvement | Random Forest, Multilayer Perceptron | Multi-trait performance pathways | Identification of climate-resilient genotypes [31] |
| Vaccine Development | MOFA, Stabl Algorithms | Immune response pathways | Prediction of immunogenicity and vaccine response [30] |
| Disease Resistance | Multi-omics ML integration | Plant-pathogen interaction pathways | Accurate prediction of resistance mechanisms [29] |
| Cancer Subtyping | Classification algorithms | Cancer hallmark pathways | Improved subtype prediction and biomarker discovery [4] |
Single-Cell and Spatial Resolution
The emergence of single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics technologies has necessitated adaptation of PEA methods to address data sparsity and cellular heterogeneity [32]. GSDensity represents a novel pathway-centric approach that bypasses conventional clustering by directly evaluating pathway activity heterogeneity across cells using graph-based modeling [32]. This method projects both cells and genes into a shared latent space using multiple correspondence analysis, then quantifies pathway coordination through kernel density estimation of pathway genes in this space [32]. For spatial transcriptomics, GSDensity calculates spatially weighted pathway activity, identifying pathways with non-random spatial expression patterns that may reflect tissue organization and cell-cell communication networks [32].
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of pathway enrichment analysis requires both computational tools and conceptual understanding of key resources. The following table summarizes essential components of the PEA research toolkit:
Table 3: Essential Research Reagents and Resources for Pathway Enrichment Analysis
| Resource Category | Specific Examples | Primary Function | Key Considerations |
|---|---|---|---|
| Pathway Databases | Gene Ontology, Reactome, KEGG, WikiPathways, PFOCR | Provide curated gene-pathway associations | Coverage, curation quality, update frequency [1] [4] |
| Enrichment Tools | g:Profiler, GSEA, Enrichr, clusterProfiler | Perform statistical enrichment calculations | Method suitability, visualization options [1] |
| Visualization Platforms | Cytoscape, EnrichmentMap, Hyperpathway | Visualize results and pathway relationships | Interpretability, customization options [1] [27] |
| Multi-omics Integrators | DPM, ActivePathways, MOFA | Combine multiple omics datasets | Directional constraints, data compatibility [19] |
| Specialized Environments | GSDensity (single-cell), Longitudinal LMM | Handle specific data types and designs | Accounting for data structure and dependencies [32] [28] |
Pathway Enrichment Analysis has evolved from a simple over-representation method to a sophisticated framework for biological mechanism elucidation. By contextualizing omics signatures within known biological processes, PEA bridges the gap between statistical associations and testable hypotheses about underlying biology. The continuing development of directional integration methods for multi-omics data, ML-enhanced pathway analysis, and specialized approaches for emerging technologies like single-cell and spatial transcriptomics ensures that PEA will remain an indispensable tool for researchers and drug development professionals seeking mechanistic insights from complex biological data. As pathway resources continue to expand in both breadth and depth, and analytical methods become increasingly refined, the power of PEA to reveal meaningful biological insights from high-dimensional data will only continue to grow.
Executing PEA: A Step-by-Step Guide to Methods and Real-World Applications
Pathway enrichment analysis is a cornerstone of modern computational biology, providing researchers with a powerful method to extract mechanistic insight from large-scale omics data. By interpreting gene lists generated from genome-scale experiments (e.g., RNA-seq, proteomics) in the context of existing biological knowledge, this approach helps identify underlying biological processes, pathways, and molecular functions that are systematically altered in a given condition [1]. The core premise is to determine whether defined sets of genes, representing specific pathways or biological themes, are over-represented in an experimental gene list more than would be expected by chance [1] [2]. This technique has proven invaluable in diverse applications, from identifying rational therapeutic targets in childhood brain cancers to unraveling the complex genetics of neurodevelopmental disorders [1]. Over time, the methodologies have evolved from simple over-representation tests to more sophisticated frameworks that incorporate gene expression magnitudes and, most recently, pathway topology, each paradigm offering distinct advantages and addressing specific analytical challenges [33] [34].
Foundational Concepts and Definitions
- Pathway: A series of interactions among molecules in a cell that leads to a certain product or a change in the cell. Pathways are models describing the interactions of genes, proteins, or metabolites within cells, tissues, or organisms, not simple lists of genes [3].
- Gene Set: An unordered and unstructured collection of genes formed on the basis of shared biological or functional properties as defined by a reference knowledge base [2] [13].
- Gene List of Interest: The list of genes derived from an omics experiment that serves as input to pathway enrichment analysis [1].
- Ranked Gene List: In many omics datasets, genes can be ranked according to a score (e.g., level of differential expression) to provide more information for pathway enrichment analysis [1].
- Multiple Testing Correction: A statistical technique to correct the P values from individual enrichment tests to reduce the chance of false-positive enrichment, necessary when thousands of pathways may be individually tested [1].
The Three Analytical Paradigms
Over-Representation Analysis (ORA)
Core Principle and Workflow Over-representation Analysis represents the first generation of pathway analysis methods. ORA statistically evaluates the fraction of genes in a particular pathway found among a set of genes showing significant changes in expression, typically determined by an arbitrary threshold [2] [13]. The method operates by asking a straightforward question: "Are there more annotations in the gene list than expected by chance?" [2]
The standard ORA workflow involves three key steps:
- Identify Differentially Expressed Genes (DEGs): From the full dataset, select genes that meet specific significance thresholds (e.g., adjusted p-value < 0.05 and fold-change > 2) [35].
- Check for Pathway Over-Representation: For each predefined gene set (pathway), examine whether the DEGs are disproportionately represented compared to a background set [35].
- Perform Statistical Testing: Calculate the probability (p-value) that the observed overlap between DEGs and pathway genes occurred by chance using statistical tests like Fisher's exact test or hypergeometric distribution [35].
Mathematical Foundation and Key Assumptions ORA methods typically employ tests based on hypergeometric, Fisher's exact, chi-square, or binomial distributions [2]. These tests determine the probability that the number of genes in a experimental gene list found in a given gene set would be observed by chance, considering the size of the pathway and the background gene set [2]. A crucial requirement for ORA is defining an appropriate background gene set for comparison, which could include all genes in the organism, all protein-coding genes, only genes measured on a specific platform, or only genes expressed in the experiment [2]. The method assumes independence between genes, a condition that rarely holds true in biological systems where genes often function in coordinated networks [13].
Table 1: Characteristics of Over-Representation Analysis (ORA)
| Aspect | Description |
|---|---|
| Statistical Test | Hypergeometric test, Fisher's exact test [2] |
| Input Requirements | List of differentially expressed genes (DEGs) based on arbitrary threshold [13] |
| Key Assumptions | Gene independence; appropriate background definition [2] |
| Strengths | Conceptually easy to understand; fast computation; requires only gene identifiers, not full dataset [2] [35] |
| Limitations | Sensitive to arbitrary thresholds; ignores expression magnitude; assumes gene independence; performs poorly with small gene lists (<50 genes) [13] |
| Example Tools | DAVID, g:Profiler, Enrichr, Qiagen IPA [1] [13] |
Rank-Based Methods (GSEA)
Core Principle and Workflow Gene Set Enrichment Analysis (GSEA) represents the second generation of pathway analysis methods, known as Functional Class Scoring (FCS) approaches [34]. Unlike ORA, GSEA does not require pre-selection of genes based on arbitrary thresholds. Instead, it considers all genes measured in an experiment, ranked by their degree of differential expression, and examines whether genes in a predefined set are randomly distributed throughout this ranked list or clustered at the top or bottom [35].
The GSEA methodology involves four key computational steps:
- Rank the Genes: Genes are ranked based on the magnitude of their differential expression between experimental conditions. The most upregulated genes appear at the top of the list, while the most downregulated appear at the bottom [35].
- Calculate Running Enrichment Score: For each gene set, GSEA walks down the ranked list, increasing a running sum when encountering a gene in the set and decreasing it when encountering genes not in the set. The amount of increment is determined by the gene's correlation with the phenotype [1].
- Determine Enrichment Score (ES): The enrichment score is the maximum deviation from zero encountered in the running sum, representing the degree to which the gene set is overrepresented at the extremes of the ranked list [35].
- Normalize and Assess Significance: The ES is normalized for gene set size, and statistical significance is estimated through permutation testing, generating a Normalized Enrichment Score (NES) that allows comparison across different experiments [35].
Interpretation of Results A high positive NES indicates that the pathway is strongly upregulated (genes clustered at the top of the ranked list), while a high negative NES indicates strong downregulation (genes clustered at the bottom) [35]. The "leading-edge" subset of genes - those appearing at or just before the maximal ES - often accounts for a pathway being defined as enriched and provides biological insights into which specific genes drive the enrichment [1].
Table 2: Characteristics of Gene Set Enrichment Analysis (GSEA)
| Aspect | Description |
|---|---|
| Statistical Approach | Kolmogorov-Smirnov like running sum statistic; permutation testing [2] |
| Input Requirements | Full ranked list of genes (all genes measured); requires expression data [2] [35] |
| Key Features | No arbitrary threshold; considers coordinated small changes; identifies direction of regulation [35] [3] |
| Strengths | More sensitive than ORA; detects subtle coordinated changes; utilizes full expression dataset [13] |
| Limitations | Computationally intensive; ignores gene position and interactions within pathways [2] [3] |
| Example Tools | GSEA, ssGSEA, GSVA, GSA, CAMERA [1] [34] [36] |
Topology-Based Methods
Core Principle and Workflow Topology-Based (TB) methods represent the third generation of pathway analysis, addressing a fundamental limitation of both ORA and GSEA: their treatment of pathways as simple gene sets while ignoring the biological knowledge embedded in pathway structures [3]. These methods incorporate information about the positions of genes within pathways, the types of interactions between them (activation, inhibition, phosphorylation), and the direction of signal flow [33] [3].
The core innovation of TB methods lies in their ability to leverage pathway topology to understand how measured expression changes propagate through biological networks. Instead of treating all genes in a pathway equally, these approaches recognize that the position and role of a gene within a pathway determines its importance [3]. For instance, if a pathway is triggered by a single receptor and that protein is not produced, the entire pathway may be shut off, whereas changes in downstream genes may have less impact [3].
The analytical framework of TB methods typically involves:
- Pathway Modeling: Representing pathways as graphs G = (V, E), where V is a set of vertices/nodes (gene products) and E is a set of edges (interactions between them) [33].
- Gene-Level Statistic Calculation: Utilizing prior knowledge of pathway topology to derive gene-level statistics that account for network position and interaction types [34].
- Pathway-Level Statistic Computation: Combining gene-level statistics into an overall pathway-level statistic used to rank pathways by their differential activity [34].
- Perturbation Assessment: Evaluating how experimental perturbations affect the entire pathway system, often considering the type and direction of interactions [3].
Advanced Implementation: SEMgsa Example A recently developed TB method called SEMgsa implements topology-based analysis within the framework of structural equation models (SEM) [34]. This approach combines p-values regarding node-specific group effect estimates in terms of activation or inhibition, after statistically controlling for biological relations among genes within pathways. The method adds a binary group (treatment or disease class) node to the pathway graph and models its effect on gene expressions while accounting for the pathway topology through linear structural equations [34].
Table 3: Characteristics of Topology-Based Analysis Methods
| Aspect | Description |
|---|---|
| Statistical Approach | Varied: Impact Analysis, Structural Equation Modeling, Network-based statistics [3] [34] |
| Input Requirements | Gene expression data + pathway topology information [33] |
| Key Features | Incorporates gene position, interaction types, and directionality; models signal propagation [3] |
| Strengths | Biologically more realistic; higher accuracy; predicts downstream effects; explains mechanisms [3] [34] |
| Limitations | Requires detailed pathway topologies; computationally complex; limited for organisms with poorly annotated pathways [13] |
| Example Tools | SPIA, Impact Analysis, DEGraph, NetGSA, Pathway-Express, SEMgsa [33] [3] [34] |
Comparative Performance Analysis
Methodological Comparison
The three paradigms offer complementary strengths and address different research needs. A systematic comparison of seven topology-based methods (SPIA, PRS, CePa, TAPPA, TopologyGSA, Clipper, and DEGraph) revealed wide variability in their performance, sensitivity to sample and pathway size, and ability to detect target pathways [33]. This underscores the importance of selecting methods appropriate for specific experimental conditions and research questions.
Table 4: Comparative Analysis of the Three Methodological Paradigms
| Characteristic | ORA | GSEA | Topology-Based |
|---|---|---|---|
| Generation | First | Second | Third |
| Information Utilization | Gene membership only | Gene membership + expression ranks | Full topology + interactions + expression |
| Threshold Dependency | High (requires DEG selection) | Low (uses all genes) | Variable |
| Biological Realism | Low | Medium | High |
| Statistical Power | Lower, especially for small gene sets | Higher, detects coordinated subtle changes | Highest in simulated benchmarks [34] |
| Computational Complexity | Low | Medium | High |
| Ideal Use Case | Quick initial screening; small studies | Comprehensive analysis without arbitrary thresholds; subtle coordinated changes | Mechanistic insights; understanding pathway deregulation |
Practical Applications and Validation
Drug Response Prediction In a comprehensive study comparing method performance for predicting response to anti-cancer drugs, a topology-based approach called NEAmarker demonstrated superior performance in correlating pathway-level features with drug sensitivity [37]. The method transformed the original space of altered genes into a lower-dimensional space of pathways using network enrichment analysis scores, which proved more robust than single-gene features or alternative enrichment methods across independent drug screens [37]. This approach successfully identified predictors of both in vitro response and patient survival following administration of the same drug, a challenging task that highlights the practical value of advanced pathway analysis methods in translational research [37].
Neurodevelopmental and Neurodegenerative Disorders In neurodevelopmental disorders, topology-based approaches have enabled the identification of key pathways from personalized protein-protein interaction networks generated from genomic alterations [38]. Similarly, in neurodegenerative diseases, centrality-based GSEA applied to interaction networks revealed enriched pathways like "Metabolism of amino acids and derivatives" and "Cellular response to stress or external stimuli" as top-ranked pathways, providing insights into disease mechanisms beyond what traditional methods could identify [38].
Experimental Protocols and Implementation
Standard Experimental Workflow
Protocol for Topology-Based Analysis Using SEMgsa
Materials and Reagent Solutions
Table 5: Essential Research Reagents and Computational Tools for Pathway Analysis
| Item | Function/Purpose |
|---|---|
| RNA-seq or Microarray Data | Raw gene expression measurements from experimental conditions |
| Pathway Databases | Source of curated pathway information (KEGG, Reactome, WikiPathways) |
| R Statistical Environment | Platform for implementing analysis algorithms [33] [34] |
| SEMgraph R Package | Implements SEMgsa method for topology-based analysis [34] |
| Graphite R Package | Provides pathway topologies for analysis [33] |
| High-Performance Computing Resources | For computationally intensive permutations and large-scale analyses |
Step-by-Step Methodology
Data Preprocessing and Quality Control
- Obtain normalized, log2-transformed gene expression profiles from high-throughput technology after standard pre-processing [33].
- Ensure appropriate sample size (typically > 5 per group for reasonable power).
- Verify data quality through principal component analysis and sample clustering.
Pathway Topology Acquisition and Pre-processing
- Download pathway topologies from databases such as KEGG, Reactome, or WikiPathways.
- Convert pathways into simple interaction networks represented as graphs G = (V, E), where V is a set of vertices/nodes (gene products) and E is a set of edges (interactions) [33].
- Resolve any inconsistencies in gene identifiers across data and pathway databases.
Implementation of SEMgsa Algorithm
- Install and load the SEMgraph package in R (available at https://CRAN.R-project.org/package=SEMgraph) [34].
- Fit a structural equation model for each pathway, adding a binary group node to represent experimental conditions.
- Estimate parameters using maximum likelihood, with the model defined by:
- For exogenous genes: Yj = βjX + Uj
- For endogenous genes: Yj = ΣβjkYk + βjX + Uj where Yj represents gene expression, X is the group variable, and Uj is the error term [34].
- Extract node-specific group effects (β_j coefficients) representing activation or inhibition.
Pathway Enrichment Scoring
- Combine p-values for node-specific group effects using Fisher's method or similar approaches.
- Calculate an overall pathway perturbation statistic that considers both node perturbation and direction of regulation.
- Adjust for multiple testing using Benjamini-Hochberg false discovery rate (FDR) control.
Results Interpretation and Visualization
- Identify significantly enriched pathways based on adjusted p-values (typically FDR < 0.05).
- Examine the direction of pathway perturbation (activation or inhibition).
- Identify key driver genes within significant pathways that contribute most to the enrichment signal.
- Generate publication-quality visualizations of perturbed pathways highlighting key alterations.
The evolution of pathway enrichment analysis from simple over-representation tests to sophisticated topology-based methods represents a paradigm shift in how researchers extract biological meaning from high-throughput data. Each methodological paradigm - ORA, GSEA, and topology-based analysis - offers distinct advantages and is suited to different research scenarios. ORA provides a straightforward, accessible entry point for initial hypothesis generation. GSEA offers a more nuanced approach that leverages complete expression datasets without arbitrary thresholds. Topology-based methods represent the current state-of-the-art, incorporating biological context to provide mechanistic insights into pathway dysregulation.
Future developments in pathway analysis will likely focus on better integration of multi-omics data, improved scalability for single-cell applications, and more sophisticated modeling of dynamic pathway alterations across time and conditions. As these methods continue to evolve, they will further empower researchers and drug development professionals to unravel the complexity of biological systems and translate these insights into improved human health.
Pathway enrichment analysis is a foundational computational biology method that helps researchers interpret genome-scale (omics) data by identifying biological pathways that are statistically overrepresented in a gene list more than would be expected by chance [1]. This method transforms large, complex molecular datasets into biologically meaningful insights about underlying mechanisms, disease processes, and potential therapeutic targets [1] [39]. The quality and appropriate formatting of input data fundamentally determine the validity and biological relevance of enrichment results [12]. Properly prepared inputs allow researchers to gain mechanistic insights into cellular organization in both health and disease states through systematic interpretation of multiple molecular datasets [39].
The first critical step in this process involves deriving appropriate gene lists from raw omics data, which varies by experimental type and technology [1]. This guide provides a comprehensive technical framework for preparing these essential inputs, covering both fundamental concepts and advanced multi-omics integration strategies, with particular attention to the needs of researchers and drug development professionals.
Fundamental Concepts: Gene Lists Versus Ranked Gene Lists
Omics experiments generate raw data that require computational processing to produce gene-level information suitable for pathway enrichment analysis [1]. The two primary formats for input data are simple gene lists and ranked gene lists, each with distinct characteristics and applications.
Table 1: Comparison of Gene List Types for Pathway Enrichment Analysis
| Feature | Simple Gene List | Ranked Gene List |
|---|---|---|
| Data Structure | Unordered set of genes | Genes ordered by a quantitative score |
| Typical Sources | Mutated genes, protein interactors, CRISPR hits | Differential expression, correlation statistics, drug sensitivity |
| Information Captured | Presence/absence in condition | Magnitude and direction of effect |
| Preferred Methods | Overrepresentation Analysis (ORA) | Gene Set Enrichment Analysis (GSEA) |
| Statistical Approach | Fisher's exact test, hypergeometric test | Rank-based permutation tests |
| Key Advantage | Simplicity, intuitive interpretation | Utilizes full dataset, no arbitrary thresholds |
Simple gene lists consist of unordered sets of genes identified through omics experiments, such as somatically mutated genes from exome sequencing or proteins interacting with a bait in proteomics experiments [1]. These lists are suitable for direct input into tools like g:Profiler using Overrepresentation Analysis (ORA) methods [1] [40].
Ranked gene lists contain genes ordered by a quantitative score that reflects the magnitude and direction of biological effect [1]. Examples include genes ranked by differential expression scores from RNA-seq experiments, correlation coefficients with a phenotype, or drug sensitivity measures from CRISPR screens [1] [40]. Ranked lists preserve continuous biological information and are analyzed using specialized methods like Gene Set Enrichment Analysis (GSEA) that detect pathways enriched at the top or bottom of the ranking [1] [12].
Figure 1: Workflow for preparing gene lists from omics experiments, showing the divergence point for simple versus ranked lists based on data type and analysis goals.
Experimental Protocols: Generating Gene Lists from Omics Data
Protocol 1: Preparing Ranked Gene Lists from RNA-seq Data
RNA sequencing (RNA-seq) provides comprehensive transcriptome profiling that naturally generates data suitable for ranked gene lists [1] [40]. The standard protocol involves multiple computational steps implemented through specialized tools:
- Quality Control of Raw Reads: Assess sequence quality using FastQC to identify potential issues with base quality, adapter contamination, or GC content [41].
- Read Alignment to Reference Genome: Map sequencing reads to a reference genome using aligners such as STAR, which accounts for splice junctions in eukaryotic transcripts [41].
- Read Quantification: Generate count data for each gene using the alignment results, typically employing featureCounts or similar tools to assign reads to genomic features [41].
- Differential Expression Analysis: Process count data using statistical packages like DESeq2 or limma-voom that model count distributions and test for significant expression changes between conditions [41]. These tools account for biological variability and library size differences.
- Ranking Metric Selection: Extract relevant statistics for ranking genes, most commonly using:
- Log2 fold change (log2FC) values representing magnitude and direction of expression differences
- T-statistics or modified t-statistics that incorporate variance estimates
- P-values or false discovery rates (FDR) representing statistical significance
- Combined metrics such as signed -log10(p-value) * log2FC [40]
The resulting ranked list contains all measured genes ordered by the selected metric, typically with most informative genes at both extremes of the ranking [1].
Protocol 2: Generating Simple Gene Lists from Genomic Variants
Genome and exome sequencing experiments identify genetic variants, including single nucleotide variants (SNVs) and insertions/deletions (indels), producing natural candidates for simple gene lists [1] [39]:
- Variant Calling: Identify genomic variants relative to a reference genome using callers like GATK HaplotypeCaller or Strelka, generating VCF files with variant positions and genotypes [39].
- Variant Annotation and Filtering: Annotate variants with functional predictions using tools like SnpEff or VEP to identify:
- Protein-coding consequences (missense, nonsense, frameshift)
- Non-coding effects (promoter, enhancer, UTR regions)
- Population frequency data from gnomAD or similar databases [39]
- Variant Prioritization: Apply filters to retain likely functional variants:
- Remove common variants (population frequency >0.1%)
- Retain protein-truncating variants (nonsense, frameshift, splice-site)
- Include damaging missense variants (predicted by SIFT, PolyPhen-2)
- Consider non-coding variants in regulatory regions [39]
- Gene-Level Aggregation: Collapse variants to the gene level, generating a final list of genes containing likely functional mutations [39].
This approach was successfully applied in the Pan-Cancer Analysis of Whole Genomes (PCAWG) project, which integrated both coding and non-coding mutations from 2,658 cancers to reveal frequently mutated pathways [39].
Protocol 3: Multi-Omics Data Integration for Enhanced Pathway Discovery
Advanced applications increasingly combine evidence from multiple omics technologies to improve pathway discovery [19] [39] [41]. The ActivePathways method provides a robust framework for such integration:
- Evidence Table Preparation: Create a matrix with genes as rows and different omics datasets as columns, populated with p-values representing statistical significance from each dataset [39].
- Statistical Data Fusion: Apply Brown's method (an extension of Fisher's combined probability test) that combines p-values across datasets while accounting for dependencies between evidence types [39].
- Integrated Gene Prioritization: Rank genes by their combined significance scores, applying lenient thresholds (e.g., unadjusted p < 0.1) to capture sub-significant signals supported by multiple evidence types [39].
- Directional Integration (DPM): For datasets with directional information, use Directional P-value Merging (DPM) to prioritize genes with consistent directional changes across datasets [19]. This method incorporates user-defined constraints (e.g., expecting positive correlation between mRNA and protein expression) to reward consistent genes and penalize inconsistent ones [19].
This integrative approach revealed additional cancer genes and pathways in the PCAWG dataset that were not apparent when analyzing coding or non-coding mutations separately [39].
Figure 2: Multi-omics data integration workflow using statistical fusion methods to combine evidence from diverse molecular profiling technologies.
Table 2: Key Research Reagents and Computational Tools for Preparing Enrichment Analysis Inputs
| Tool/Resource | Function | Application Context |
|---|---|---|
| DESeq2 | Differential expression analysis of RNA-seq count data | Generating ranked lists from transcriptomics |
| STAR | Spliced alignment of RNA-seq reads to reference genome | RNA-seq preprocessing and quantification |
| GATK | Variant discovery and genotyping from sequencing data | Identifying mutated genes for simple lists |
| limma | Differential analysis for microarray and RNA-seq data | Generating ranked lists with modified t-statistics |
| ActivePathways | Multi-omics data integration and pathway analysis | Combining evidence from diverse molecular datasets |
| g:Profiler | Overrepresentation analysis for simple gene lists | Functional interpretation of unordered gene sets |
| GSEA | Gene Set Enrichment Analysis for ranked lists | Pathway analysis of ordered gene lists |
| MSigDB | Collection of annotated gene sets for enrichment testing | Reference pathways and biological signatures |
| EnrichmentMap | Visualization of enriched pathways as networks | Interpreting and communicating results |
Advanced Applications: Directional and Drug Mechanism Enrichment
Directional Constraints in Multi-Omics Integration
The Directional P-value Merging (DPM) method enhances multi-omics integration by incorporating directional biological relationships between datasets [19]. This approach allows researchers to define expected directional associations based on cellular logic or experimental design:
- Central Dogma Constraints: Expect positive correlation between mRNA and protein expression
- Epigenetic Constraints: Expect negative correlation between promoter DNA methylation and gene expression
- Experimental Constraints: Define inverse associations for knockout versus overexpression experiments [19]
DPM implements these constraints through a weighting scheme that rewards genes with consistent directional changes across datasets while penalizing those with conflicting signals [19]. This approach has demonstrated utility in characterizing IDH-mutant gliomas by integrating transcriptomic, proteomic, and DNA methylation datasets, as well as identifying prognostic biomarkers in ovarian cancer with consistent signals at both transcript and protein levels [19].
Drug Mechanism Enrichment Analysis
Drug Mechanism Enrichment Analysis (DMEA) adapts the GSEA algorithm to prioritize therapeutic repurposing candidates by grouping drugs with shared mechanisms of action (MOAs) rather than analyzing individual drugs [8]. This approach:
- Accepts ranked drug lists from various sources: perturbagen signatures, drug sensitivity scores, or molecular classification scores
- Groups drugs by annotated MOAs (e.g., "EGFR inhibitor," "HDAC inhibitor")
- Calculates enrichment scores for each MOA category using a weighted Kolmogorov-Smirnov-like statistic
- Identifies MOAs overrepresented among top candidates, increasing on-target signal while reducing off-target effects [8]
DMEA has successfully identified senescence-inducing and senolytic drug MOAs for primary human mammary epithelial cells, leading to experimental validation of EGFR inhibitors as senolytic agents [8].
Proper preparation of gene lists and ranked lists from omics experiments forms the critical foundation for meaningful pathway enrichment analysis. By selecting appropriate input formats based on experimental data types, implementing robust processing protocols, and leveraging advanced integration methods, researchers can maximize biological insights from complex molecular datasets. The continuous development of multi-omics integration and specialized enrichment methods further enhances our ability to translate molecular measurements into mechanistic understanding of health, disease, and therapeutic interventions.
Pathway enrichment analysis (PEA) is a foundational computational biology method that identifies biological functions overrepresented in a gene group more than expected by chance [12]. This methodology addresses a critical challenge in modern biology: interpreting lists of hundreds or thousands of genes generated by high-throughput genomic experiments like RNA-seq [42]. By measuring the relative abundance of genes pertinent to specific biological pathways using statistical methods, PEA helps researchers translate gene lists into meaningful biological insights [12]. The core principle involves retrieving associated functional pathways from bioinformatics databases and ranking them by relevance, effectively bridging the gap between raw genomic data and biological understanding [12].
Two primary computational approaches dominate the PEA landscape: Overrepresentation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA) [12]. ORA methods identify biological functions overrepresented in a gene set compared to their representation in the genome, typically using a statistical test like Fisher's exact test [12]. In contrast, GSEA approaches detect pathways enriched with genes located at both extreme ends of a ranked gene list, capturing more subtle, coordinated expression changes without requiring arbitrary significance cutoffs [43]. A third category, topology-based PEA (TPEA), incorporates information about interactions between genes and gene products but depends heavily on cell-type-specific gene topologies that remain incomplete [12].
Tool Comparison Table
The following table summarizes the core features, strengths, and limitations of four essential pathway enrichment tools:
| Tool | Primary Analysis Type | Input Requirements | Key Features | Strengths | Limitations |
|---|---|---|---|---|---|
| g:Profiler | ORA, ranked list analysis | Gene list (flat or ranked) | Multi-species support (31 species), ID conversion, ortholog mapping | Integrated toolset, intuitive visualization, handles multiple ID types | Less focused on advanced visualization [44] [45] |
| Enrichr | ORA | Gene list | Extensive library collection (>180,000 gene sets), API access, fuzzy set input | Rapid analysis, comprehensive libraries, crowdsourced signatures | Tabular output lacks network visualization [17] [46] |
| GSEA | GSEA | Ranked gene list with statistics | Permutation-based significance, correlation with expression phenotypes | No arbitrary thresholds, detects subtle coordinated changes | Computationally intensive, long processing times [42] [43] |
| Cytoscape | Network visualization & analysis | Network data + attributes | App ecosystem, complex network visualization, data integration | Powerful visualization, extensible platform | Steep learning curve, requires installation [42] [47] |
| EnrichmentMap: RNASeq (Cytoscape Web) | GSEA + Network visualization | Expression file or RNK file | Web-based, automatic clustering, fast fGSEA implementation | Simplified interface, rapid processing, no installation | Limited to human RNA-seq, fewer advanced features [42] |
Tool-Specific Methodologies and Protocols
g:Profiler Implementation
g:Profiler employs a modified Fisher's exact test to estimate gene abundance in pathways, calculating statistical significance using cumulative hypergeometric P-values [45]. The tool supports three multiple testing correction methods: g:SCS, Bonferroni correction, and Benjamini-Hochberg false discovery rate (FDR) [12]. A distinctive feature is its ability to handle both flat and ranked gene lists, with the latter analyzed through incremental probing of all possible list head sizes to identify functional annotations and statistical cut-points [45].
Experimental Protocol:
- Input Preparation: Compile gene identifiers (HGNC, Ensembl, Entrez, or mixed types) in a simple text file
- Species Selection: Specify from 31 supported species
- Parameter Configuration:
- Select data sources: GO, KEGG, Reactome, TRANSFAC
- Set significance threshold (typically FDR < 0.05)
- Choose output detail level
- Analysis Execution: Submit job via web interface or API
- Result Interpretation:
- Review structured results grouped by domain
- Explore hierarchical relationships in visual graph view
- Download tabular results for further analysis [45]
Enrichr Workflow
Enrichr utilizes a comprehensive Fisher's exact test implementation for enrichment calculations, with recent performance enhancements enabling near-instant results [17] [46]. The platform distinguishes itself through its massive collection of 180,184 annotated gene sets from 102 libraries, including crowd-sourced signatures from GEO data and libraries from NIH Common Fund programs [17].
Experimental Protocol:
- Input Options:
- Standard gene list (copy-paste or upload)
- BED files for genomic regions
- Fuzzy sets for uncertain gene associations
- Library Selection: Choose from categories including Pathways, Ontologies, Cell Types, Diseases, and Drugs
- Analysis Execution: Submit via web interface with automatic background correction
- Result Exploration:
GSEA Methodology
The GSEA algorithm ranks all genes based on their correlation with a phenotype, then calculates an enrichment score representing the degree to which genes in a predefined set are overrepresented at either extreme of the ranked list [43]. Statistical significance is determined through permutation testing, which creates a null distribution by repeatedly scrambling phenotype labels [12]. The method specifically addresses situations where many small, coordinated changes across a pathway collectively produce biological effects without individual genes meeting strict significance thresholds [43].
Experimental Protocol:
- Data Preparation:
- Create ranked gene list using differential expression statistics
- Format according to GSEA specifications (RNK file)
- Gene Set Selection: Choose appropriate collections from MSigDB or custom sets
- Parameter Configuration:
- Set number of permutations (typically 1,000)
- Choose enrichment statistic (weighted or unweighted)
- Define metric for ranking genes
- Analysis Execution: Run through Java application or R package
- Result Interpretation:
- Identify significantly enriched sets (FDR < 25%)
- Examine enrichment plots showing set distribution
- Explore leading edge genes driving enrichment [43]
Cytoscape and EnrichmentMap Implementation
EnrichmentMap: RNASeq implements a streamlined GSEA workflow specifically for human RNA-seq data, utilizing the fGSEA algorithm for faster processing compared to traditional GSEA [42]. The application automatically clusters pathways based on gene overlap similarity and visualizes these clusters using bubble sets, creating interpretable networks where nodes represent pathways and edges connect pathways sharing gene members [42].
Experimental Protocol:
- Input Preparation:
- Upload normalized expression counts (TSV, CSV, Excel) OR
- Provide pre-ranked gene list (RNK file)
- Automated Processing:
- Low-count filtering via edgeR filterByExpr
- TMM normalization for sequencing depth variation
- Differential expression with edgeR
- Pathway enrichment with fGSEA
- Visualization Generation:
- Automatic network layout with up-regulated pathways on one side, down-regulated on the other
- Cluster identification and bubble set visualization
- Result Export:
- Publication-ready network figures
- Shareable web links for collaboration [42]
Workflow Integration Diagram
The following diagram illustrates the relationship between pathway enrichment tools and their position in a typical bioinformatics workflow:
Research Reagent Solutions
The following table details essential computational reagents and resources for pathway enrichment analysis:
| Resource Type | Specific Examples | Function in Analysis | Source/Provider |
|---|---|---|---|
| Gene Set Libraries | Gene Ontology, KEGG, Reactome, WikiPathways | Provide biological context and pathway definitions | Gene Ontology Consortium, KEGG, Reactome, WikiPathways [12] |
| Expression Databases | ARCHS4, GEO, GTEx | Supply reference expression data for comparison | NCBI GEO, GTEx Portal [17] [46] |
| Annotation Databases | Ensembl, MSigDB, Bader Lab Gene Sets | Enable gene identifier mapping and functional annotations | Ensembl, Broad Institute, Bader Lab [42] [43] |
| Statistical Packages | edgeR, fGSEA, clusterProfiler | Perform differential expression and enrichment calculations | Bioconductor, CRAN [42] [43] |
| Visualization Tools | Cytoscape.js, bubble sets | Create interpretable network representations | Cytoscape Consortium [42] |
Analysis Selection Framework
Choosing the appropriate enrichment method depends primarily on your research question and data type. The following diagram illustrates the decision process for selecting the optimal tool:
Pathway enrichment analysis represents a powerful approach for extracting biological meaning from high-throughput genomic data. The four tools discussed—g:Profiler, Enrichr, GSEA, and Cytoscape—each offer unique strengths for different research scenarios. g:Profiler and Enrichr excel at rapid overrepresentation analysis for simple gene lists, while GSEA and its implementation in EnrichmentMap: RNASeq provide more sensitive detection of coordinated expression changes in ranked gene lists. Cytoscape offers unparalleled visualization capabilities for interpreting complex pathway relationships. By understanding the methodological foundations and appropriate applications of each tool, researchers can effectively leverage these resources to translate genomic data into biological insights, ultimately advancing drug development and scientific discovery.
Proximity Extension Assay (PEA) technology represents a revolutionary approach in proteomics, enabling highly sensitive and specific multiplex protein quantification. This technical guide details the standard workflow for transforming raw PEA data into biologically meaningful insights through rigorous preprocessing, statistical analysis, pathway enrichment examination, and advanced visualization. Framed within the broader context of pathway enrichment analysis—a statistical method for identifying biological pathways significantly over-represented in omics data—this workflow provides researchers, scientists, and drug development professionals with a structured framework to elucidate mechanistic insights from protein expression patterns. By integrating robust data processing methodologies with functional interpretation, this pipeline supports biomarker discovery, drug mechanism evaluation, and molecular pathway research.
Pathway enrichment analysis is a foundational bioinformatics method that helps researchers interpret lists of genes or proteins derived from genome-scale (omics) experiments by identifying biological pathways that are statistically over-represented beyond what would occur by chance [1]. This approach addresses a central challenge in modern biology: translating vast molecular datasets into actionable mechanistic understanding of biological processes, disease mechanisms, and therapeutic interventions. In a proteomics context, pathway enrichment analysis reveals how coordinated protein expression changes map onto defined biological processes, providing critical functional context for experimental observations.
Proximity Extension Assay (PEA) technology has emerged as a powerful platform for generating the high-quality proteomic data required for such analyses. PEA is an innovative molecular technique that combines antibody-based immunoassay specificity with DNA amplification sensitivity to detect and quantify proteins [48]. The core principle involves using matched antibody pairs labeled with unique DNA oligonucleotides that bind to the same target protein. When both antibodies bind in close proximity, their DNA tags hybridize and undergo a polymerase-mediated extension reaction, creating a DNA barcode specific to that protein [49]. This barcode is then amplified and quantified using real-time PCR or next-generation sequencing, producing precise protein abundance measurements.
The synergy between PEA technology and pathway enrichment analysis creates a powerful pipeline for proteomic investigation. PEA delivers the high-fidelity, multiplex protein quantification necessary to generate meaningful protein lists for enrichment analysis, while pathway enrichment provides the interpretative framework to extract biological meaning from these lists. This integrated approach is particularly valuable in pharmaceutical development, where understanding a drug's impact on biological pathways is essential for target validation, mechanism of action studies, and biomarker identification.
PEA Technology and Experimental Foundation
Fundamental Principles of PEA
The analytical power of PEA stems from its dual-recognition mechanism and DNA-based signal amplification. The requirement for two independent antibodies to bind the same target molecule simultaneously before signal generation confers exceptional specificity, dramatically reducing non-specific binding and false positives common in traditional immunoassays [50]. This "two-key" system ensures that only correctly bound antibody pairs produce measurable signals, delivering specificity exceeding 99.5% for many protein targets [50].
The signal amplification process provides remarkable sensitivity, enabling detection of low-abundance proteins in minimal sample volumes (as low as 1-3 μL) [49]. By converting protein detection into DNA quantification, PEA leverages the exponential amplification power of PCR, achieving sensitivity down to sub-picogram levels that often exceeds traditional proteomic methods like mass spectrometry for targeted analyses.
Key Research Reagents and Materials
Successful PEA implementation requires carefully designed research reagents and materials. The table below outlines essential components of a standard PEA workflow:
Table 1: Essential Research Reagents for Proximity Extension Assay
| Reagent/Material | Function | Technical Specifications |
|---|---|---|
| Paired Antibody Probes | Dual recognition of target protein epitopes | High-affinity, validated pairs; DNA-oligonucleotide conjugated |
| DNA Polymerase | Extension of hybridized DNA oligonucleotides | High-fidelity, thermal-stable enzyme |
| PCR Master Mix | Amplification of protein-specific DNA barcodes | Optimized for quantitative PCR or NGS library preparation |
| Assay Plates | High-throughput sample processing | 96-well or 384-well format compatible with automation |
| Calibration Standards | Data normalization and quantification | Multipoint dilution series of reference samples |
| Negative Controls | Specificity verification and background assessment | Sample diluent without protein content |
PEA Experimental Workflow
The end-to-end PEA process transforms biological samples into quantitative protein data through a series of meticulously controlled steps. The following workflow diagram illustrates the complete experimental procedure:
PEA Experimental Workflow from Sample to Data
The workflow begins with sample preparation, where minimal volumes (typically 1-3 µL) of biological material are combined with DNA-conjugated antibody pairs in a multiplexed reaction [49]. During the incubation phase, antibodies bind to their specific target proteins, forming immune complexes. When two antibodies bind the same protein molecule, their DNA oligonucleotides are brought into proximity, enabling hybridization. The extension reaction then occurs, where DNA polymerase extends one oligonucleotide using the other as a template, creating a unique DNA barcode quantitatively representing the target protein. These barcodes are amplified and quantified via qPCR or NGS, producing normalized protein expression (NPX) values for downstream analysis [49].
Data Preprocessing and Quality Control
Raw PEA data requires rigorous preprocessing to ensure analytical reliability before biological interpretation. Data preprocessing constitutes approximately 80% of the analytical effort in typical omics workflows, emphasizing its critical importance for generating valid conclusions [51]. For PEA data, this phase encompasses multiple quality assessment and normalization steps to transform raw signal measurements into analytically robust datasets.
Data Quality Assessment and Cleaning
Initial quality evaluation examines both sample-level and protein-level performance metrics. Sample-level quality checks identify outliers potentially resulting from processing errors or biological abnormalities, while protein-level assessments detect analytes with poor detection rates or inconsistent measurements. This quality evaluation incorporates several specific procedures:
- Missing Value Analysis: Identification of proteins with excessive missing measurements across samples. Proteins with >20% missing values typically require removal or imputation depending on analytical context.
- Outlier Detection: Statistical identification of aberrant samples using principal component analysis (PCA) and Mahalanobis distance calculations. Samples exceeding predefined thresholds (e.g., >4 standard deviations from the mean) are flagged for further investigation.
- Signal-to-Noise Evaluation: Assessment of background signal levels through negative controls to determine reliable detection limits.
Missing values in PEA data can result from protein levels below assay detection limits or technical variability. Common handling approaches include removal of proteins with extensive missing data or imputation using methods such as k-nearest neighbors or minimum value replacement, with selection dependent on the presumed missingness mechanism and analysis goals [51] [52].
Normalization and Batch Effect Correction
Normalization addresses technical variability from sample processing, reagent lots, or instrument runs to ensure valid biological comparisons. PEA data typically utilizes internal controls and normalization methods tailored to the platform:
- Internal Standard Normalization: Platform-specific controls spiked into each sample correct for technical variation.
- Inter-Quartile Range Alignment: Scale adjustment based on stable protein expression across samples.
- Batch Effect Correction: Statistical methods like ComBat remove systematic variation between experimental batches while preserving biological signals.
The normalization approach produces Normalized Protein eXpression (NPX) values, a relative quantification unit on a log2 scale where higher values indicate greater protein abundance [49]. NPX values enable direct comparison between samples and analytical runs, forming the foundation for subsequent statistical analyses.
Statistical Analysis for Pathway Enrichment
Differential Expression Analysis
Following quality control and normalization, statistical analysis identifies proteins exhibiting significant abundance changes between experimental conditions. For case-control studies, differential expression analysis typically employs linear models incorporating relevant experimental factors, with empirical Bayes moderation to improve variance estimates for proteins with limited replicates. The analysis generates several key metrics for each protein:
- Fold Change: Magnitude of abundance difference between conditions.
- P-value: Statistical significance of the observed difference.
- False Discovery Rate (FDR): Multiple testing-adjusted probability of false significance.
Results are often visualized through volcano plots displaying fold change against statistical significance, highlighting proteins with both large magnitude and high confidence changes. Differentially expressed proteins meeting significance thresholds (commonly FDR < 0.05 and fold change > 1.5) comprise the candidate list for pathway enrichment analysis.
Pathway Enrichment Analysis Methods
Pathway enrichment analysis evaluates whether differentially expressed proteins collectively associate with specific biological pathways beyond random expectation. Two complementary approaches dominate enrichment analysis methodologies:
Table 2: Pathway Enrichment Analysis Methods
| Method Type | Statistical Approach | Input Data Structure | Key Advantages |
|---|---|---|---|
| Over-Representation Analysis (ORA) | Hypergeometric test/Fisher's exact test | Protein list (significant differentially expressed proteins) | Simple interpretation, straightforward implementation |
| Gene Set Enrichment Analysis (GSEA) | Kolmogorov-Smirnov-like running sum statistic | Ranked protein list (all proteins by significance) | No arbitrary significance thresholds, detects subtle coordinated changes |
Over-representation analysis (ORA) employs hypergeometric testing to evaluate whether proteins annotated to specific pathways occur more frequently in the differentially expressed protein list than expected by chance [1]. While computationally straightforward, ORA requires dichotomizing proteins into significant/non-significant groups, potentially losing information from expression magnitude and statistical confidence.
Gene Set Enrichment Analysis (GSEA) represents a more nuanced approach that considers all measured proteins ranked by their association with the experimental condition [1] [42]. GSEA evaluates whether proteins from predefined pathways cluster at the extremes of this ranked list, identifying pathways with coordinated modest changes that might not reach individual significance thresholds. This method is particularly valuable for detecting subtle but biologically important pathway alterations.
Implementation and Multiple Testing Correction
Practical implementation of pathway enrichment utilizes established bioinformatics tools and databases. Commonly employed resources include:
- g:Profiler: Web-based tool for efficient ORA with comprehensive database integration [1]
- fGSEA: Fast algorithm for GSEA implementation, significantly reducing computation time compared to standard GSEA [42]
- EnrichmentMap: Cytoscape app for visualization and interpretation of enrichment results [42]
Pathway analysis evaluates hundreds to thousands of pathways simultaneously, dramatically increasing false discovery risk. Multiple testing correction methods, particularly false discovery rate (FDR) control, adjust raw p-values to account for these extensive comparisons [1]. Standard practice requires FDR-adjusted p-values (q-values) < 0.05 for declaring significantly enriched pathways, though more stringent thresholds may be appropriate for hypothesis generation versus validation contexts.
Visualization and Interpretation of Results
Enrichment Map Network Visualization
EnrichmentMap creates network-based visualizations that transform tabular enrichment results into interpretable pathway landscapes [1] [42]. In these networks, nodes represent significantly enriched pathways, with size proportional to the number of proteins in the pathway. Edges connect pathways sharing substantial protein overlap (typically Jaccard similarity coefficient > 0.25), visually grouping biologically related processes into functional themes.
The following diagram illustrates the EnrichmentMap visualization architecture:
EnrichmentMap Network Visualization Architecture
Automated clustering algorithms, frequently employing edge-weighted force-directed layout, group highly interconnected pathways into thematic clusters representing broader biological processes [42]. These clusters are often annotated with descriptive labels derived from enriched functional terms within the cluster, facilitating rapid identification of major biological themes affected in the experiment.
Alternative Visualization Strategies
Complementary visualization approaches provide additional perspectives on enrichment results:
- Bubble Plots: Display pathways by statistical significance (y-axis), enrichment strength (x-axis), and protein count (bubble size), creating an intuitive summary visualization.
- Pathway Topology Diagrams: Integrated pathway databases (Reactome, KEGG) enable protein overlay onto established pathway maps, visualizing how altered proteins interact within biological systems.
- Heat Maps with Pathway Annotation: Pair protein expression heatmaps with pathway membership annotations to connect individual protein changes with pathway-level patterns.
Modern implementations like EnrichmentMap: RNASeq (enrichmentmap.org) provide web-based, streamlined workflows that generate publication-quality visualizations with minimal computational expertise required [42]. These tools significantly reduce traditional GSEA processing times from 5-20 minutes to under 1 minute using efficient fGSEA implementation, enhancing analytical accessibility.
Applications in Research and Drug Development
The integrated PEA-pathway enrichment workflow delivers actionable biological insights across multiple research domains, particularly in pharmaceutical development. Key applications include:
- Biomarker Discovery and Validation: Identification of protein signatures and associated pathways that distinguish disease states, predict treatment response, or monitor therapeutic efficacy. A severe brain injury study, for example, measured 1,100 plasma proteins via PEA and identified six novel protein biomarkers linked to inflammatory and neuronal pathways [49].
- Drug Mechanism Elucidation: Comprehensive mapping of compound effects on biological pathways to understand therapeutic mechanisms and off-target effects. Pathway activity scores derived from PEA data have demonstrated >90% concordance with experimentally validated drug mechanisms in patient-derived xenografts and breast cancer cell lines [53].
- Toxicology and Safety Assessment: Evaluation of drug-induced pathway perturbations predictive of adverse effects, supporting early safety assessment in preclinical development.
- Patient Stratification: Identification of pathway-level differences between patient subgroups to inform precision medicine approaches and clinical trial design.
These applications highlight how the PEA-pathway enrichment pipeline bridges analytical measurement and biological interpretation, transforming protein quantification into mechanistic understanding with direct relevance to therapeutic development.
The standardized workflow from PEA data preprocessing through pathway visualization represents a robust framework for extracting biological meaning from complex proteomic datasets. By integrating the analytical sensitivity and specificity of PEA technology with the functional interpretation power of pathway enrichment analysis, researchers can systematically translate protein abundance changes into pathway-level insights. This approach has demonstrated particular utility in pharmaceutical contexts, where understanding drug effects on biological systems is paramount. As proteomic technologies continue evolving toward higher multiplexing capabilities and improved sensitivity, coupled with increasingly sophisticated analytical methods like the gdGSE algorithm that employs discretized expression profiles [53], the integration of experimental measurement and bioinformatic interpretation will remain essential for maximizing the scientific and clinical value of proteomic data.
Pathway enrichment analysis (PEA) is a computational biology method that identifies biological functions overrepresented in a group of genes more than would be expected by chance [12]. This technique has become indispensable for deciphering disease mechanisms and discovering new therapeutic applications for existing drugs [54]. By analyzing gene lists derived from omics experiments, researchers can identify predominant biological pathways driving pathological states and subsequently pinpoint drugs that might reverse these aberrant pathway activities [55]. The method summarizes large gene lists as smaller, more interpretable sets of pathways that provide mechanistic insights into cellular processes disrupted in disease states [1]. For instance, pathway enrichment analysis successfully identified histone and DNA methylation by the polycomb repressive complex as a rational therapeutic target for ependymoma, one of the most prevalent childhood brain cancers [1].
Core Methodologies for Pathway Enrichment Analysis
Statistical Approaches and Their Applications
Pathway enrichment analysis employs several distinct statistical approaches, each with specific strengths for particular research scenarios [26]. The choice of method depends on the type of data available and the specific biological question being addressed.
Table 1: Comparison of Major Pathway Enrichment Analysis Methods
| Method Type | Statistical Basis | Input Data | Key Advantages | Common Tools |
|---|---|---|---|---|
| Over-Representation Analysis (ORA) | Hypergeometric test, Fisher's exact test [12] [40] | Gene list with significance threshold [1] | Simple, intuitive, works with predefined gene lists [12] | g:Profiler [1], Enrichr [12] |
| Gene Set Enrichment Analysis (GSEA) | Permutation-based test [1] | Ranked gene list (no threshold required) [1] | Captures subtle coordinated changes, uses all available data [1] [40] | GSEA [1], fgsea [40] |
| Pathway Topology-Based Methods | Incorporates pathway structure and interactions [26] | Gene list with expression data | More biologically realistic, considers pathway architecture [26] | SPIA [12], PathNet [12] |
Practical Implementation Guide
For Unranked Gene Lists Using g:Profiler
For simple gene lists (e.g., mutated genes or differentially expressed genes with significance thresholds), the g:Profiler web tool provides an accessible option [20]:
- Input Preparation: Compile your gene list in a text file with one gene symbol per line [20]
- Parameter Settings:
- Select appropriate data sources (GO biological processes, Reactome pathways) [20]
- Set functional category size limits (recommended: 5-350 genes) [20]
- Set minimum query/term intersection to 3 [20]
- Enable "Ordered query" if genes are ranked [20]
- Check "No electronic GO annotations" for higher quality annotations [20]
- Execution: Run the analysis and download results in Generic Enrichment Map (GEM) format for visualization [20]
For Ranked Gene Lists Using GSEA
For genome-wide ranked lists (e.g., all genes ranked by fold change), GSEA provides more sensitive detection [20]:
- Input Preparation: Create an RNK file with gene identifiers in the first column and ranking metric (e.g., fold change) in the second [20]
- Gene Set Selection: Choose appropriate gene set database (GMT file) [20]
- Analysis Execution:
- Result Interpretation: Examine enrichment scores, normalized enrichment scores (NES), and false discovery rates (FDR) [1]
Pathway-Centric Drug Repositioning Framework
Integrated Workflow for Drug Repositioning
The following diagram illustrates the multi-stage process of using pathway enrichment analysis for drug repositioning:
Computational Screening and Validation
The pathway-based drug repositioning approach involves identifying drugs that reverse disease-associated pathway signatures [55] [54]:
- Multi-omics Integration: Combine transcriptomic and proteomic data from disease samples to identify consistently dysregulated pathways [55] [19]
- Drug Signature Matching: Screen drug-perturbed gene expression profiles from databases like LINCS and Connectivity Map to identify compounds that reverse disease pathway signatures [55]
- Network Pharmacology Analysis: Construct drug-disease networks and perform additional pathway enrichment on drug targets to elucidate mechanisms of action [55]
- BBB Permeability Screening: Filter candidates based on blood-brain barrier permeability for neurological disorders [55]
- Structural Similarity Analysis: Compare candidate structures with known drugs to identify potentially novel compounds [55]
Table 2: Key Databases for Pathway-Centric Drug Repositioning
| Database | Primary Use | Key Features | Access |
|---|---|---|---|
| LINCS/Connectivity Map | Drug signature matching | Gene expression profiles from drug perturbations [55] | Public web portal |
| DrugBank | Drug target information | Comprehensive drug-target-pathway relationships [54] | Public with registration |
| MSigDB | Pathway database | Curated gene sets including hallmark pathways [1] [40] | Public web portal |
| Reactome | Pathway database | Detailed human pathway information with visualizations [1] | Public web portal |
| PFOCR | Pathway database | Pathway figures extracted from literature with direct citations [4] | Public web portal |
Case Study: Alzheimer's Disease Drug Repositioning
A recent study demonstrated the power of pathway enrichment analysis for Alzheimer's disease drug repositioning through an integrative multi-omics approach [55]:
Experimental Protocol
Data Acquisition and Pre-processing
Transcriptomic and Proteomic Profiling:
- Obtain RNA-seq and proteomics data from post-mortem Alzheimer's disease brain tissues and matched controls
- Perform quality control and normalization using standard pipelines for each data type
- Identify differentially expressed genes and proteins using appropriate statistical tests (e.g., DESeq2 for RNA-seq, linear models for proteomics)
Multi-omics Integration:
- Apply directional integration methods like DPM (Directional P-value Merging) to identify genes with consistent changes across transcriptomic and proteomic datasets [19]
- Define directional constraints based on biological relationships (e.g., positive correlation between mRNA and protein expression)
Pathway Enrichment Analysis
Enrichment Detection:
- Perform over-representation analysis on differentially expressed genes using g:Profiler with Gene Ontology Biological Processes and Reactome pathways [20]
- Conduct GSEA on ranked gene lists using Hallmark gene sets and customized neuronal pathway sets
- Apply multi-omics pathway integration using ActivePathways to identify pathways with complementary evidence across data types [19]
Result Interpretation:
Computational Drug Screening
Signature Reversal Scoring:
- Calculate Reverse Gene Expression Scores (RGES) to quantify the ability of drugs to reverse the Alzheimer's disease signature [55]
- Screen the LINCS database containing drug perturbation profiles using connectivity mapping approaches
Candidate Prioritization:
- Filter candidates based on blood-brain barrier permeability predictions using tools like BBB Predictor
- Perform structural similarity analysis to cluster compounds and identify novel scaffolds
- Review literature and patent databases to exclude previously investigated candidates
Experimental Validation
In Vitro Models:
- Utilize Okadaic acid-induced SH-SY5Y cells as a model of tau hyperphosphorylation and neuronal toxicity
- Employ Lipopolysaccharide-induced BV2 microglial cells as a neuroinflammation model
- Treat models with candidate compounds across a range of concentrations
Outcome Measures:
- Assess cell viability using MTT or Alamar Blue assays in neuronal models
- Measure nitric oxide production as an indicator of neuroinflammatory response in microglial models
- Perform immunocytochemistry for Alzheimer's-relevant markers (e.g., Aβ, p-tau)
Key Findings and Validation
The Alzheimer's drug repositioning study identified TNP-470 and Terreic acid as promising candidates [55]. Network pharmacology analysis revealed that TNP-470 targets were significantly enriched in neuroactive ligand-receptor interaction, TNF signaling, and Alzheimer's disease-related pathways, while Terreic acid targets involved calcium signaling, AD pathway, and cAMP signaling [55]. In vitro validation demonstrated that TNP-470 significantly enhanced viability of OA-induced SH-SY5Y cells at concentrations of 10 μM and 50 μM, and markedly inhibited NO production in LPS-induced BV2 microglial cells [55]. Similarly, Terreic acid promoted survival of OA-treated SH-SY5Y cells and significantly reduced nitric oxide levels [55].
Advanced Applications and Emerging Approaches
Directional Multi-omics Integration
The directional integration of multi-omics datasets represents a significant advancement in pathway analysis [19]. The DPM (Directional P-value Merging) method incorporates user-defined directional constraints to prioritize genes with consistent changes across datasets while penalizing those with inconsistent directions [19]. This approach is particularly valuable for:
- Integrating Epigenetic and Transcriptomic Data: DNA methylation of gene promoters typically correlates negatively with gene expression [19]
- Combining Transcriptomic and Proteomic Data: mRNA and protein expression often show positive correlation based on the central dogma [19]
- Incorporating Clinical Outcomes: Survival information can be directionally integrated with molecular profiles to identify prognostic biomarkers [19]
The following diagram illustrates the directional data fusion process:
Pathway Databases with Enhanced Disease Coverage
Emerging pathway databases are expanding opportunities for disease mechanism discovery and drug repositioning. The Pathway Figure OCR (PFOCR) database deserves special attention as it takes a direct approach to capturing pathway information by extracting published pathway figures from the literature [4]. PFOCR covers 90% of diseases in the Comparative Toxicogenomics Database, significantly outperforming traditional databases like Reactome (17%), WikiPathways (14%), and KEGG (11%) in disease coverage [4]. This extensive coverage makes PFOCR particularly valuable for identifying pathways in rare and understudied diseases.
Table 3: Key Research Reagent Solutions for Pathway-Centric Drug Discovery
| Tool/Category | Specific Solutions | Function in Research | Application Context |
|---|---|---|---|
| Pathway Analysis Tools | g:Profiler [1], GSEA [1], Enrichr [12] | Identify enriched pathways in gene lists | Initial discovery phase for all studies |
| Visualization Platforms | Cytoscape with EnrichmentMap [1] [20], Pathview [40] | Visualize enriched pathways and their relationships | Interpretation and communication of results |
| Drug Signature Databases | LINCS [55], Connectivity Map [55], DSigDB [40] | Connect pathway signatures to drug effects | Drug repositioning studies |
| Multi-omics Integration | ActivePathways with DPM [19], GSVA [40] | Integrate multiple omics datasets with directional constraints | Complex mechanism studies across data types |
| Experimental Validation Systems | SH-SY5Y neuronal model [55], BV2 microglial model [55] | Validate candidate drugs in disease-relevant contexts | Preclinical drug testing for neurological disorders |
Pathway enrichment analysis has evolved from a simple functional interpretation tool to a powerful approach for deciphering disease mechanisms and identifying repositioned therapeutic candidates. By integrating multi-omics data, applying directional analysis methods, and leveraging expansive pathway databases, researchers can systematically connect molecular perturbations to pathological processes and identify drugs that reverse these alterations. The continued development of pathway analysis methodologies and databases promises to further enhance our ability to discover new therapeutic applications for existing drugs across a broad spectrum of human diseases.
Optimizing Your Analysis: Best Practices and Pitfall Avoidance
In the broader context of pathway enrichment analysis, the initial and most critical step is to precisely define your biological question and align it with the appropriate computational method. This decision fundamentally shapes your analytical workflow and the validity of your conclusions. The core methodologies—Over-Representation Analysis (ORA), Functional Class Scoring (FCS), and Pathway Topology (PT)—each have distinct strengths, statistical foundations, and data requirements tailored to different experimental goals [2] [1].
Core Methodologies in Pathway Enrichment Analysis
The table below summarizes the three primary approaches, helping you match your research objective with the correct technique.
| Method | Core Principle | Best For / When to Use | Input Required | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Over-Representation Analysis (ORA) [2] [1] | Statistically tests if genes in a pre-defined list (e.g., DEGs) are overrepresented in a pathway more than expected by chance. | - A pre-selected list of significant genes (e.g., DEGs with p-value & fold-change cutoffs).- Quick, initial functional screening. | A simple list of genes (e.g., differentially expressed genes). | - Simple, intuitive, and fast.- Does not require the entire dataset, just gene identifiers. | - Depends on arbitrary significance thresholds.- Ignores the magnitude and direction of gene expression changes.- Assumes genes are independent. |
| Functional Class Scoring (FCS) [2] [14] | Uses a ranked list of all genes from an experiment to identify pathways enriched at the top or bottom of the list, without relying on hard thresholds. | - Utilizing information from all genes measured in an experiment.- Detecting subtle but coordinated expression changes in pathways. | A ranked list of all genes from an experiment (e.g., ranked by fold-change or statistical significance). | - Does not require arbitrary pre-filtering of genes; more sensitive.- Can identify subtle but coordinated changes. | - Requires the entire gene expression dataset.- More computationally intensive.- Results can be more complex to interpret. |
| Pathway Topology (PT) [2] | Incorporates additional biological information about the pathway structure, such as gene interactions, positions, and roles, into the enrichment model. | - Understanding specific mechanisms and the direction of interactions within a pathway.- When high-quality pathway structure data is available. | Gene list or ranked list, plus pathway topology information. | - More biologically accurate as it uses known pathway structures.- Can provide mechanistic insights. | - Relies on experimental evidence for pathway structures, which is limited for many organisms.- Complex methodology. |
Essential Research Reagents and Tools
Successful pathway analysis relies on a toolkit of curated databases and software. The table below lists key resources for defining gene sets and performing enrichment tests.
| Resource Name | Type | Primary Function / Application |
|---|---|---|
| Enrichr [17] | Software Tool | A web-based platform for rapid ORA, featuring a vast and updated collection of gene set libraries from various domains. |
| GSEA Software & MSigDB [2] [14] | Software Tool & Database | The standard implementation of the FCS method (GSEA) and its accompanying, highly curated collection of gene sets (MSigDB). |
| Gene Ontology (GO) [2] | Database | A foundational resource of standardized terms for biological processes, molecular functions, and cellular components, widely used for ORA. |
| Reactome [2] [1] | Database | A curated, detailed database of human biological pathways, including signaling and metabolism, often used for both ORA and FCS. |
| WikiPathways [56] [1] | Database | A community-driven, open platform for pathway curation, providing a wide array of pathway models. |
| Cytoscape & EnrichmentMap [1] | Software Tool | Used for the visualization and interpretation of enrichment results, helping to identify overarching biological themes from a list of enriched pathways. |
Experimental Protocol: A Step-by-Step Guide
This section outlines the practical workflow for performing pathway enrichment analysis, from data preparation to interpretation.
Stage 1: Define Your Gene List of Interest
The first step is to process your omics data to create a gene list for analysis [1].
- For ORA: Generate a list of differentially expressed genes (DEGs). This typically involves setting statistical thresholds (e.g., adjusted p-value < 0.05 and absolute fold-change > 2) from tools like DESeq2 for RNA-seq data [2] [1].
- For FCS (like GSEA): Create a ranked list of all genes measured in your experiment. Genes are typically ranked by a metric of differential expression, such as the signal-to-noise ratio or the negative log of the p-value multiplied by the sign of the fold-change [1].
- Data Preprocessing: Ensure gene identifiers (e.g., gene symbols) are consistent and updated using resources like the HUGO Gene Nomenclature Committee (HGNC) to avoid conversion errors that can invalidate your analysis [5].
Stage 2: Perform Pathway Enrichment Analysis
- Using ORA with Enrichr:
- Navigate to the Enrichr website [17].
- Paste your list of gene symbols into the input box.
- Select from a wide range of gene set libraries (e.g., GO Biological Process, KEGG, WikiPathways).
- Submit the analysis. Enrichr will return a list of enriched pathways with p-values, adjusted p-values, and combined scores [17].
- Using FCS with GSEA:
- Prepare your expression dataset and phenotype labels file in the required GSEA formats [14].
- Load your data into the GSEA desktop application.
- Select a gene set database from MSigDB (e.g., Hallmark, C2: Curated Gene Sets) [2] [14].
- Run the GSEA algorithm. The output will identify pathways enriched at the top (up-regulated) or bottom (down-regulated) of your ranked list, providing an Enrichment Score (ES), Normalized ES (NES), and False Discovery Rate (FDR) [1].
Stage 3: Visualize and Interpret Results
- Initial Inspection: Review the list of significantly enriched pathways, focusing on their statistical metrics (e.g., FDR-adjusted p-values) [1].
- Reducing Redundancy: Use tools like EnrichmentMap, a Cytoscape app, to create a network of enriched pathways where overlapping genes are represented as connecting edges. This visually clusters related pathways, revealing major biological themes in your data [1].
- Identify Leading-Edge Genes: In GSEA, examine the "leading-edge" subset—the core genes that primarily drive the pathway's enrichment. These genes are prime candidates for further validation and mechanistic studies [1].
Workflow and Logical Relationship Diagram
The following diagram illustrates the critical decision points and paths for selecting the appropriate pathway enrichment analysis method based on your research goal and data.
Pathway enrichment analysis has become a cornerstone of modern genomics and drug discovery, enabling researchers to extract meaningful biological insights from high-throughput omics data. These methods, including Gene Ontology (GO), the Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Set Enrichment Analysis (GSEA), function by statistically evaluating the overrepresentation of predefined gene sets in experimental data [57]. However, a pervasive and often overlooked limitation is that the analytical output is fundamentally constrained by the quality of its input. Effective gene list curation—the process of preparing, validating, and refining gene identifiers—is not a mere preliminary step but a critical determinant of biological validity. This guide provides a comprehensive framework for researchers and drug development professionals to implement robust gene list curation protocols, thereby ensuring the reliability and interpretability of pathway enrichment results within a rigorous scientific context.
Pathway enrichment analysis is a computational biology method that interprets gene expression data by testing for the statistically significant overrepresentation of specific biological pathways or functional categories within a set of genes of interest, typically derived from differential expression analysis [57].
Core Methodologies
The three most widely used enrichment methods are GO, KEGG, and GSEA, each with distinct analytical approaches and outputs [57]:
- GO (Gene Ontology) Enrichment: Classifies genes based on a structured, controlled vocabulary across three independent domains: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC) [57]. It answers the question, "What functions, processes, or locations are statistically overrepresented in my gene list?"
- KEGG (Kyoto Encyclopedia of Genes and Genomes) Enrichment: Maps genes onto specific pathways, such as metabolic or signal transduction pathways, to reveal how genes work together in integrated biological systems [57]. It focuses on systemic, pathway-level insights.
- GSEA (Gene Set Enrichment Analysis): Unlike the hypergeometric test-based methods used by GO and KEGG, GSEA does not require a predefined, significance-based gene cutoff [57]. Instead, it ranks all genes from an experiment based on their expression change and determines whether members of a predefined gene set are randomly distributed throughout the ranked list or concentrated at the top or bottom [14]. This makes it particularly powerful for detecting subtle but coordinated expression changes in situations where individual gene changes are modest.
Table 1: Comparison of Primary Enrichment Analysis Methods
| Feature | GO | KEGG | GSEA |
|---|---|---|---|
| Primary Focus | Functional ontology | Pathway-centric | Coordinated expression shifts |
| Input | A list of DEGs | A list of DEGs | A ranked list of all genes |
| Statistical Method | Hypergeometric test | Hypergeometric/Fisher's test | Kolmogorov-Smirnov-like statistic |
| Key Output | Functional terms | Pathway maps | Enrichment plots & FDR |
| Best For | Detailed functional classification | Understanding pathway interactions | Identifying subtle changes without a clear DEG cutoff |
The Critical Role of Gene List Curation
The principle of "garbage in, garbage out" is acutely relevant to pathway analysis. Even the most sophisticated statistical method cannot produce valid biological conclusions from a poorly curated gene list. Inaccurate curation introduces noise and bias, leading to false positives, missed discoveries, and ultimately, misguided scientific conclusions and costly experimental follow-ups.
Consequences of Inadequate Curation
- False Positives/Negatives: Incorrect gene identifiers can map to wrong pathways, causing irrelevant terms to appear significant (false positives) or obscuring truly enriched pathways (false negatives).
- Loss of Statistical Power: A gene list contaminated with obsolete or unmappable identifiers is effectively smaller, reducing the statistical power of the enrichment test and increasing the risk of Type II errors.
- Irreproducible Results: Poorly documented and inconsistent curation practices are a significant contributor to the reproducibility crisis in genomics.
- Wasted Resources: Basing downstream wet-lab experiments or drug discovery efforts on flawed enrichment results wastes critical time, funding, and resources.
A Protocol for Robust Gene List Curation
The following protocol provides a step-by-step methodology for ensuring input quality prior to enrichment analysis.
Materials and Reagents
Table 2: Essential Research Reagent Solutions for Gene List Curation
| Item Name | Function/Description | Example Tools/Databases |
|---|---|---|
| Gene Annotation Database | Provides current, official gene symbols and functional annotations. Serves as the authoritative reference for identifier mapping. | NCBI Gene, Ensembl |
| ID Mapping Service | A computational tool that systematically converts one type of gene identifier (e.g., microarray probe ID) to another (e.g., official gene symbol). | DAVID, g:Profiler, bioDBnet |
| Functional Database | Provides the gene sets for enrichment testing. The choice dictates the biological insights you can gain. | MSigDB, GO, KEGG |
| Background Gene Set | A custom or default list of all genes that could have been detected in the experiment. Critical for calculating statistical enrichment. | Platform-specific array annotations, all detected genes in RNA-seq |
Step-by-Step Curation Workflow
The following diagram illustrates the logical workflow for a robust gene list curation process, from raw data to a validated list ready for analysis.
Step 1: Identifier Standardization and Gathering
Begin by collecting all gene identifiers from your analysis pipeline. Document the original source (e.g., microarray platform, RNA-seq assembler) and the identifier type (e.g., Ensembl ID, NCBI RefSeq, unofficial symbol). Preserve this original list for audit purposes.
Step 2: Systematic Identifier Mapping
Use a programmatic ID mapping service (e.g., from DAVID or bioDBnet) to convert all identifiers to a stable, universally recognized standard, such as official HGNC gene symbols or Ensembl Gene IDs. Automated tools are superior to manual conversion as they are less error-prone and provide a traceable log.
Step 3: Removal of Invalid and Obsolete Entries
After mapping, a quality control check is essential. Remove any entries that could not be mapped or are flagged as obsolete in the current database. Document the number and type of removed identifiers, as a high failure rate may indicate issues with the original data or outdated platform annotations.
Step 4: Resolution of Many-to-Many Mappings
Some original identifiers may map to multiple official genes (e.g., a single microarray probe targeting homologous genes), or multiple original IDs may map to a single official gene. These cases must be resolved by:
- Deduplication: For enrichment methods requiring a unique gene list (GO, KEGG), collapse multiple identifiers for the same gene into a single entry.
- Informed Selection: For ambiguous probes, consult platform-specific annotation files to determine the primary target or apply a conservative approach by removing the ambiguous entry to prevent false associations.
Step 5: Definition of the Background Set
The background set, or "universe" of genes, is critical for the hypergeometric test used in GO and KEGG analysis [57]. It represents all genes that had a chance of being selected in your experiment.
- Best Practice: Use a custom background set comprising all genes reliably detected and quantified in your experimental setup (e.g., all genes expressed above a threshold in your RNA-seq data).
- Common Pitfall: Using the tool's default background (e.g., all genes in the genome) can lead to severe bias if your experimental technology (e.g., a microarray) only covers a specific subset of the genome.
Step 6: Final Quality Assessment and Logging
Generate a final report for the curation process. This should include:
- Starting number of identifiers.
- Number and type of identifiers successfully mapped.
- Number and list of identifiers removed.
- Final number of unique, curated genes.
- The source and size of the chosen background set. This log ensures full reproducibility and transparency.
Quantitative Impact of Curation: A Scenario-Based Analysis
The impact of curation can be quantified by comparing analysis results from poorly curated and well-curated lists. The following table summarizes potential outcomes across key metrics.
Table 3: Quantitative Impact of Gene List Curation on Analysis Outcomes
| Metric | Poorly Curated List | Well-Curated List | Impact Description |
|---|---|---|---|
| List Size for Analysis | Reduced by 10-30% | Maximized and accurate | Loss of valid genes reduces statistical power. |
| Number of Significant GO Terms/KEGG Pathways | Inflated or deflated; includes false positives. | Biologically relevant and accurate. | Poor curation introduces bias, leading to incorrect conclusions. |
| False Discovery Rate (FDR) | Potentially elevated and unreliable. | More accurately controlled. | Confidence in results is compromised with poor input. |
| Top Enriched Pathways | May include irrelevant or incorrect pathways. | Concordant with experimental design and biology. | Downstream interpretation and hypothesis generation are misdirected. |
| Reproducibility | Low; difficult to replicate with different identifiers. | High; process is documented and repeatable. | Foundation of robust, trustworthy science. |
Case Study: GSEA and the Importance of a Properly Ranked List
While GO and KEGG are sensitive to gene list quality, GSEA has a different vulnerability related to its input. GSEA requires a ranked list of all genes from an experiment, typically by a metric like fold change or signal-to-noise ratio [57]. The quality of this ranking is paramount.
Curation Protocol for GSEA Input
- Ranking Metric Calculation: Calculate the ranking metric (e.g., log2 fold change) for every gene detected in the experiment.
- Gene Identifier Curation: Apply the same rigorous identifier standardization and mapping protocol (as in Section 3.2) to this full list of genes. Remove any gene that cannot be unambiguously mapped.
- Handling Redundancy: If multiple probes/identifiers map to the same official gene symbol, a decision must be made to avoid over-representing that gene in the ranked list. Common strategies include:
- Selecting the identifier with the highest absolute fold change.
- Selecting the identifier with the lowest p-value.
- Taking the average of the ranking metric for all duplicates.
- Final Ranked List Generation: The result is a cleaned, non-redundant list of genes, ranked by the chosen metric, which serves as high-quality input for GSEA.
Visualization of GSEA Input Curation
The diagram below details the specific curation workflow for preparing a gene list for GSEA, highlighting the key step of resolving duplicate mappings before the final ranking.
Pathway enrichment analysis is a powerful lens through which to view complex biological data, but the clarity of that lens depends entirely on the quality of the input. Gene list curation is not a mundane preprocessing task but a foundational scientific practice that directly controls the validity, reproducibility, and biological relevance of research outcomes. By adopting the systematic curation protocols outlined in this guide—including rigorous identifier mapping, background set definition, and process logging—researchers and drug developers can significantly enhance the reliability of their computational findings. In an era of increasingly complex datasets and high-stakes translational research, robust gene list curation is an indispensable component of the rigorous scientific method.
Choosing the Correct Background Set and Accounting for Gene Correlations
Pathway enrichment analysis is a fundamental statistical method for interpreting gene lists generated from genome-scale (omics) experiments, helping researchers identify biological pathways that are enriched beyond what would be expected by chance [1]. However, the validity of its results critically depends on two often-overlookated technical considerations: the appropriate selection of a background gene set and the proper accounting for correlations among genes. The background set defines the universe of possible genes against which statistical enrichment is measured, directly influencing statistical power and specificity [58]. Meanwhile, gene correlations—whether arising from co-regulation, shared biological functions, or chromosomal proximity—can violate the independence assumption underlying many enrichment statistical tests, potentially leading to inflated false discovery rates [53]. This guide provides a technical framework for addressing these challenges, ensuring more biologically meaningful and statistically robust enrichment results for research and drug development applications.
The Critical Importance of Background Set Selection
Conceptual Foundation of Background Sets
In pathway enrichment analysis, the background set represents the reference population of genes from which your gene list of interest is hypothetically drawn. The statistical question being tested is whether genes in your experimental list are over-represented in a particular pathway compared to this background distribution [58]. Using an inappropriate background set introduces substantial bias, potentially leading to both false positives and false negatives.
A commonly used but often incorrect approach is using the entire genome as the background set. This assumes all genes were detectable and equally likely to be selected in the experiment, which is frequently untrue. For example, in RNA-seq experiments, the background should typically comprise only genes expressed above a reliable detection threshold in your experimental system, as non-expressed genes cannot contribute to observed expression differences [1].
Practical Guidelines for Background Set Definition
For RNA-seq and gene expression microarray studies, the background should include genes detected above a minimum expression threshold (e.g., counts per million > 1 in at least a percentage of samples) [1]. This prevents biologically irrelevant enrichment signals from non-expressed genes.
For genomic mutation analyses, the background should be constrained to genes adequately sequenced and covered in the experiment, typically defined by minimum depth-of-coverage and base-quality thresholds [1].
For proteomics and SomaScan data, the background must be limited to proteins actually detectable by the platform used. Specialized resources like SomaModules provide platform-specific background sets tailored to SOMAmer-based proteomic data [59].
For species-specific analyses, background sets should be derived from comprehensive annotations for that particular species. The KEGG database provides organism-specific pathway annotations that serve as appropriate background for many model organisms [58].
Table 1: Background Set Selection Guidelines by Experiment Type
| Experiment Type | Recommended Background | Key Considerations |
|---|---|---|
| RNA-seq | Genes expressed above detection threshold | Avoid non-expressed genes; use CPM/FPKM thresholds |
| Genome Sequencing | Genes with sufficient sequencing coverage | Apply depth/quality filters; consider exome capture efficiency |
| Proteomics (SomaScan) | Platform-detectable proteins | Use specialized resources (e.g., SomaModules) [59] |
| Cross-Species Analysis | Species-specific annotated genome | Use KEGG organism databases or comparative genomics |
Statistical Challenges Posed by Gene Correlations
Genes do not function in isolation but rather in coordinated networks, creating inherent correlations that violate the independence assumption of many statistical tests used in enrichment analysis. These correlations arise from multiple biological and technical sources:
- Co-regulation: Genes in the same pathway are often transcriptionally co-regulated by common transcription factors or signaling pathways [1]
- Chromosomal proximity: Genes located near each other on chromosomes may be co-amplified or co-deleted in copy number variations
- Technical artifacts: Batch effects and platform-specific biases can induce artificial correlations
- Homology: Gene families with sequence similarity may be co-detected due to cross-hybridization or ambiguous mapping
When unaccounted for, these correlations lead to anticonservative p-values in hypergeometric tests and other traditional enrichment methods, as the effective degrees of freedom are overestimated [53]. This problem is particularly pronounced in gene sets with high internal correlation structure.
Methodological Approaches for Addressing Correlations
Several computational strategies have been developed to mitigate the confounding effects of gene correlations:
The gdGSE algorithm employs discretized gene expression profiles rather than continuous values to assess pathway activity, effectively mitigating discrepancies caused by data distributions and correlation structures [53]. This method converts binarized gene expression into a gene set enrichment matrix that demonstrates improved robustness in both bulk and single-cell applications.
Gene Set Enrichment Analysis (GSEA) uses a rank-based permutation approach that preserves gene correlation structure. By permuting sample labels rather than genes, GSEA maintains the inherent correlation structure when generating the null distribution [1].
Sherlock-II, designed for integrating GWAS with eQTL data, addresses correlation through a statistical framework that accounts for linkage disequilibrium (correlation between SNPs) when translating SNP-level associations to gene-level associations [60].
Table 2: Methods Addressing Gene Correlations in Enrichment Analysis
| Method | Statistical Approach | Application Context |
|---|---|---|
| gdGSE [53] | Discrete expression binning | Bulk and single-cell RNA-seq data |
| GSEA [1] | Sample label permutation | Ranked gene lists from omics experiments |
| Sherlock-II [60] | LD-aware integration | GWAS and eQTL integration |
| Hypergeometric Test | Gene permutation (naive) | Basic list enrichment (inflated false positives) |
Integrated Experimental Protocol for Robust Enrichment Analysis
Stage 1: Background Set Definition and Data Preparation
Step 1: Define experiment-appropriate background set
- For transcriptomics: Calculate expression thresholds and filter non-expressed genes
- For genomics: Apply coverage and quality filters to define adequately sequenced genes
- For proteomics: Use platform-specific detectable protein sets (e.g., SomaModules for SomaScan) [59]
- Document inclusion criteria and final background gene count
Step 2: Generate gene list of interest
- For differential expression: Apply appropriate statistical thresholds (FDR < 0.05, fold-change > 2)
- For ranked lists: Calculate ranking metric (e.g., signal-to-noise ratio, t-statistic)
- Ensure all genes in the experimental list are present in the background set
Step 3: Select appropriate pathway database
- Choose biologically relevant database (GO Biological Process, KEGG, Reactome) [1]
- Verify database version and species compatibility
- Download current gene set annotations
Stage 2: Enrichment Analysis with Correlation Awareness
Step 4: Select correlation-appropriate statistical method
- For simple gene lists without strong internal correlations: Hypergeometric test with multiple testing correction
- For ranked gene lists with potential correlations: GSEA with sample permutation [1]
- For single-cell or noisy data: gdGSE with discrete expression binning [53]
- For GWAS integration: Sherlock-II or similar LD-aware methods [60]
Step 5: Execute enrichment analysis
- For hypergeometric test: Use the following parameters:
- N: Total genes in background set
- K: Total genes in pathway within background
- n: Size of experimental gene list
- k: Number of experimental genes in pathway
- Apply multiple testing correction (Benjamini-Hochberg FDR)
- For GSEA: Use 1,000-5,000 permutations for stable p-values
Step 6: Validate results against negative control
- Test enrichment against random gene sets of same size
- Verify known biological expectations are recovered
- Check for platform-specific biases
Diagram 1: Workflow for robust pathway enrichment analysis
Table 3: Key Computational Tools and Databases for Enrichment Analysis
| Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| g:Profiler [1] | Web tool / API | Enrichment analysis with multiple testing correction | Quick analysis of gene lists; multiple database support |
| GSEA [1] | Desktop application | Rank-based enrichment with sample permutation | Pre-ranked gene lists; correlation-aware testing |
| Cytoscape/EnrichmentMap [1] | Visualization platform | Network visualization of enriched pathways | Interpreting multiple related enrichment results |
| MSigDB [1] | Gene set database | Curated collection of pathway gene sets | Background for GSEA; hallmark pathway sets |
| KEGG [58] | Pathway database | Biochemical pathway maps with gene annotations | Species-specific pathway enrichment |
| SomaModules [59] | Specialized resource | SOMAmer-based gene sets for SomaScan data | Proteomics enrichment analysis |
| gdGSE [53] | Algorithm | Discrete expression enrichment method | Single-cell or noisy data analysis |
Advanced Applications and Specialized Considerations
Specialized Data Types and Integration Approaches
Proteomics Data (SomaScan): The SomaModules approach demonstrates how platform-specific background sets significantly improve enrichment detection for SOMAmer-based proteomic data. By creating intracorrelated SOMAmer modules based on 11K SomaScan data, this method generated repositories containing over 40,000 SOMAmer-based gene sets that showed significantly higher enrichment than original gene set counterparts in validation studies [59].
GWAS Integration: The Sherlock-II algorithm provides a framework for addressing correlations in genome-wide association studies by translating SNP-phenotype associations to gene-phenotype associations while accounting for linkage disequilibrium. This method uses a statistical approach that sums log(p-values) of GWAS peaks aligned to eQTL peaks, with background distribution calculated by convolution of log(p-value) distributions across independent LD blocks [60].
Single-Cell RNA-seq: The discrete binning approach of gdGSE is particularly valuable for single-cell data where technical noise and sparse distributions complicate continuous value-based enrichment analysis. This method applies statistical thresholds to binarize gene expression matrices before conversion to gene set enrichment matrices, demonstrating enhanced cell type identification and clustering performance [53].
Diagram 2: Specialized methods for different data types
Choosing the correct background set and properly accounting for gene correlations are not merely statistical technicalities but fundamental requirements for biologically valid pathway enrichment analysis. The appropriate background set ensures that enrichment signals reflect true biological phenomena rather than technical artifacts of the experimental platform, while correlation-aware statistical methods prevent anticonservative results and false discoveries. By implementing the protocols and resources outlined in this guide—selecting experiment-appropriate background sets, applying correlation-robust statistical methods like GSEA or gdGSE, and using specialized approaches for data types like SomaScan or single-cell RNA-seq—researchers can significantly enhance the reliability and interpretability of their pathway enrichment results. These practices form the foundation for generating mechanistically insightful hypotheses that can effectively guide subsequent experimental validation in both basic research and drug development contexts.
Pathway enrichment analysis is a cornerstone of functional genomics, allowing researchers to interpret large-scale 'omics datasets by identifying biological pathways, processes, and functions that are overrepresented in a set of genes of interest [61]. This methodology transforms lists of differentially expressed genes or proteins into biologically meaningful insights about underlying mechanisms [7] [62]. The fundamental challenge in enrichment analysis lies in distinguishing biologically relevant signals from statistical noise, a challenge that is directly addressed through effective filtering strategies for gene set size and coverage.
The statistical power and biological relevance of enrichment results are profoundly influenced by gene set dimensions [63]. Excessively small gene sets may produce unstable results with poor statistical power, while excessively large gene sets often represent broad biological categories that lack specificity and can dominate results due to their size alone [62] [63]. Similarly, the extent of overlap between a gene set and the query gene list (coverage) determines whether findings are robust or potentially spurious. Thus, implementing rigorous filtering strategies for gene set size and coverage is not merely a technical consideration but a fundamental requirement for generating biologically interpretable results from pathway enrichment analysis.
The Impact of Gene Set Size on Statistical Analysis
Statistical Challenges Posed by Gene Set Dimensions
Gene set size directly impacts multiple aspects of enrichment analysis, including statistical significance estimation, multiple testing correction, and biological interpretation. From a statistical perspective, larger gene sets inherently possess greater power to detect modest enrichment signals simply due to their size [63]. This size effect can create a bias where larger pathways consistently appear more significant regardless of their biological relevance to the experimental condition. Conversely, smaller gene sets suffer from reduced statistical power and may fail to reach significance even when biologically relevant, as they require a larger proportion of their genes to be present in the input list to achieve statistical significance [62].
The problem is compounded by the structure of popular biological databases like the Gene Ontology (GO), which contain tens of thousands of terms [63]. Without appropriate filtering, enrichment analyses can generate hundreds or even thousands of statistically significant results, many of which represent overlapping or redundant biological concepts [63]. This multiplicity problem necessitates stringent multiple testing corrections that can obscure truly meaningful findings if not properly managed through strategic filtering.
Empirical Evidence of Size-Related Artifacts
Recent benchmarking studies have quantified the impact of gene set size on enrichment results. The GOAT algorithm development team systematically evaluated how gene set size affects p-value calibration under null conditions, demonstrating that unfiltered analyses can produce biased results across different gene set dimensions [62]. Their work confirmed that proper null hypothesis testing should yield uniform p-values regardless of gene set size, but this ideal is rarely achieved without appropriate filtering or statistical normalization.
Similarly, the developers of ShinyGO explicitly note that "enrichment analysis tends to favor larger pathways" and recommend that users "also consider fold enrichment" rather than relying solely on FDR values when interpreting results [63]. Fold enrichment, which represents the ratio between the observed overlap and the expected overlap by chance, provides an effect size measure that is less dependent on gene set size than raw p-values or FDRs. This combination of statistical significance and effect size measurement represents a crucial filtering strategy in modern enrichment analysis.
Quantitative Guidelines for Gene Set Size Filtering
Established Size Thresholds in Bioinformatics Tools
Various bioinformatics tools and platforms have implemented specific gene set size thresholds based on empirical testing and user experience. The table below summarizes the recommended size filtering parameters from major enrichment analysis tools and resources:
Table 1: Gene set size filtering recommendations across enrichment analysis tools
| Tool/Resource | Minimum Size | Maximum Size | Rationale | Citation |
|---|---|---|---|---|
| ShinyGO | User-defined (default: 10) | User-defined (default: 1000) | Avoid noisy results from small sets; prevent broad categories from dominating | [63] |
| GOAT | 10 | 1000 | Balanced statistical power and specificity | [62] |
| GSEA | 15 | 500 | Standard parameters in default implementation | [64] [61] |
| EnrichmentMap | 3 | 500 | Balance between sensitivity and specificity in network visualization | [42] |
These thresholds represent community standards that have evolved through extensive practical application. The typical minimum size of 10-15 genes ensures sufficient statistical power, while maximum sizes of 500-1000 genes exclude overly broad categories that typically lack biological specificity [62] [63]. For example, extremely large gene sets such as "cellular process" (GO:0009987) or "metabolic process" (GO:0008152) encompass substantial portions of the genome and frequently appear significant even with minimal actual enrichment.
Implementation of Size Filtering in Analysis Workflows
Implementing effective size filtering requires integration at multiple stages of the enrichment analysis workflow. The following diagram illustrates a standardized workflow incorporating size filtering:
Most contemporary tools allow users to customize these parameters based on their specific research context. For hypothesis-generating exploratory analyses, more lenient filters (e.g., 5-2000 genes) may be appropriate to capture novel findings, while confirmatory studies benefit from stricter ranges (e.g., 15-500 genes) to reduce false discoveries [63]. The key consideration is that these parameters should be documented and applied consistently throughout the analysis.
Coverage-Based Filtering Strategies
Defining Coverage Metrics in Enrichment Analysis
While gene set size filtering addresses one dimension of the problem, coverage-based filtering focuses on the relationship between the query gene list and the gene sets being tested. Coverage refers to the extent and quality of overlap between these two sets. The most fundamental coverage metric is the number of overlapping genes (nGenes), which represents the raw count of genes from the input list that are present in a given gene set [63]. However, this absolute count must be interpreted in context, leading to the more informative metric of fold enrichment.
Fold enrichment is calculated as the ratio between the observed proportion of overlapping genes and the expected proportion by chance [63]. This metric is defined as (n/m) / (N/M), where n is the number of overlapping genes, m is the total genes in the query list, N is the size of the gene set, and M is the total number of genes in the background set. Fold enrichment values substantially greater than 1 indicate meaningful overrepresentation, while values close to 1 suggest minimal enrichment regardless of statistical significance.
Implementing Coverage Thresholds in Practice
Establishing appropriate coverage thresholds requires balancing sensitivity and specificity. Most tools implement minimum overlap requirements, typically requiring at least 2-5 genes in the intersection between the input list and gene set [63]. However, relying solely on absolute counts can be misleading, particularly for larger gene sets where small absolute overlaps may represent trivial proportions.
Table 2: Coverage filtering strategies and their applications
| Filtering Approach | Typical Threshold | Use Case | Advantages | Limitations |
|---|---|---|---|---|
| Minimum gene overlap | 2-5 genes | General purpose | Simple to implement and interpret | Favors larger gene sets; insensitive to proportion |
| Fold enrichment | >1.5-2x | Focus on strong effects | Identifies biologically meaningful effects | May miss subtle but important signals |
| Combined filtering | Overlap ≥3 AND fold enrichment ≥2 | High-confidence results | Balanced approach reducing false positives | May exclude true positives with modest effects |
| Leading-edge subset | N/A (GSEA-specific) | Pathway mechanism identification | Identifies core enriched genes within sets | Limited to GSEA methodology [61] |
The leading-edge subset analysis, introduced in the original GSEA methodology, represents a sophisticated coverage-based filtering approach that identifies the core members of high-scoring gene sets that contribute most to the enrichment signal [61]. This method is particularly valuable for distinguishing the central biological mechanisms within broader functional categories.
Integration of Size and Coverage Filtering in Experimental Protocols
Comprehensive Filtering Workflow for Enrichment Analysis
A robust enrichment analysis protocol integrates both size and coverage filtering at appropriate stages. The following experimental protocol outlines a standardized approach applicable to most enrichment analysis tools:
Protocol: Integrated Filtering for Gene Set Enrichment Analysis
Input Preparation: Prepare your ranked gene list or differentially expressed gene set using standard bioinformatics pipelines. For RNA-seq data, this typically involves differential expression analysis using tools like edgeR or DESeq2 [42].
Background Definition: Specify an appropriate background gene set representing all genes considered in the analysis (e.g., all genes detected in the experiment) [63]. This critical step ensures proper normalization of enrichment statistics.
Primary Size Filtering: Apply initial gene set size filters (typically 10-500 genes) to the pathway database before performing enrichment tests [62] [63]. Most tools provide parameters for this during analysis setup.
Enrichment Calculation: Perform the enrichment analysis using your preferred method (overrepresentation analysis, GSEA, etc.). The fGSEA implementation is recommended for preranked gene lists due to its speed and accuracy [42].
Coverage-Based Filtering: Filter results based on both statistical significance (FDR < 0.05) and fold enrichment (>2-fold) [63]. Additionally, require a minimum of 3-5 genes in the overlap between your input list and each significant gene set.
Redundancy Reduction: Apply redundancy filtering to collapse highly overlapping gene sets. The "Remove redundancy" option in ShinyGO eliminates similar pathways that share 95% of their genes and 50% of the words in their names [63].
Visual Validation: Examine the distribution of significant results across different gene set sizes to identify potential size biases. Create visualizations such as enrichment maps to interpret filtered results [42].
The Scientist's Toolkit: Essential Materials and Reagents
Table 3: Research reagent solutions for enrichment analysis
| Tool/Resource | Function | Application Context | Key Features |
|---|---|---|---|
| GSEA Software | Gene set enrichment analysis | Microarray and RNA-seq data interpretation | Computes enrichment scores using rank-based statistics [64] [61] |
| MSigDB Database | Curated gene set collection | Providing biological context for enrichment analysis | Manually curated gene sets with multiple categorization schemes [64] [61] |
| Enrichr Platform | Web-based enrichment analysis | Rapid hypothesis generation from gene lists | Integrates 225+ gene-set libraries with interactive visualization [17] [65] |
| fGSEA R Package | Fast preranked gene set enrichment | Efficient analysis of large datasets | Implements rapid GSEA for preranked gene lists [42] |
| GOAT Algorithm | Efficient gene set enrichment testing | Testing thousands of GO gene sets in seconds | Precomputed null distributions for rapid analysis [62] |
| ShinyGO Web Tool | User-friendly enrichment analysis | Accessibility for wet-lab researchers | Graphical interface with extensive visualization options [63] |
Advanced Considerations and Future Directions
Specialized Filtering Scenarios
Certain research contexts require modified filtering strategies. For novel gene sets with minimal overlap with established pathways, such as those derived from proteomics analyses or studies of under-characterized biological systems, relaxed size and coverage thresholds may be necessary to detect meaningful signals [66]. In such cases, the GeneAgent tool demonstrates how large language models can generate functional descriptions for gene sets with limited database coverage, though this requires careful verification to minimize hallucinations [66].
Single-cell RNA sequencing data presents another specialized scenario where gene set size and coverage considerations differ substantially from bulk analyses. The massive multiple testing burden and sparse expression patterns in single-cell data necessitate modified filtering approaches, with tools like scEnrichr specifically adapted for this context [17] [65].
Emerging Methods and Tool Developments
Recent methodological advances continue to refine how we approach gene set size and coverage in enrichment analysis. The GOAT algorithm introduces a bootstrapping approach to generate competitive null hypotheses that are robust to gene set size variations [62]. Similarly, blitzGSEA provides efficient computation of GSEA through gamma distribution approximation, achieving significant performance improvements while accurately approximating small p-values [65].
The integration of enrichment analysis with knowledge graphs, as implemented in Enrichr-KG, represents another advancement that enables more sophisticated filtering based on network topology rather than simple size thresholds [65]. These systems present enrichment results as subgraphs connecting genes to their enriched terms, allowing researchers to filter based on connectivity patterns and network features in addition to traditional size and coverage metrics.
The future of filtering strategies in enrichment analysis will likely involve increasingly sophisticated integration of multiple filtering dimensions, with machine learning approaches helping to optimize parameter selection based on dataset characteristics [66]. However, the fundamental principles of appropriate size and coverage filtering will remain essential for generating biologically meaningful results from pathway enrichment analysis.
Pathway enrichment analysis is a cornerstone of modern genomic research, allowing scientists to interpret gene lists from high-throughput experiments by identifying biological pathways that are over-represented beyond what would occur by chance [1]. In a typical omics experiment, such as RNA-sequencing or genome-wide association studies, researchers simultaneously test thousands of genes for differential expression or association with traits. This creates a fundamental statistical challenge: when conducting numerous hypothesis tests at a traditional significance threshold (e.g., p < 0.05), the probability of obtaining false positive results increases dramatically [67]. Multiple testing correction methods, particularly those controlling the False Discovery Rate (FDR), have become essential for distinguishing genuine biological signals from statistical noise in pathway enrichment analyses [67] [68]. Without proper correction, researchers risk basing scientific conclusions on false discoveries, potentially leading to futile validation experiments and contaminating the scientific literature with spurious findings [67].
Understanding False Discovery Rate (FDR)
Definition and Statistical Foundation
The False Discovery Rate (FDR) is defined as the expected proportion of false discoveries among all statistically significant findings. Formally, FDR is the expectation of the False Discovery Proportion (FDP), where FDP represents the ratio of false discoveries to total discoveries (with the provision that this ratio is zero when there are no discoveries) [67]. Unlike the Family-Wise Error Rate (FWER), which controls the probability of at least one false discovery, FDR controls the expected proportion of errors among the rejected null hypotheses, making it generally less conservative and more powerful for high-dimensional biological data [67] [68].
The Benjamini-Hochberg (BH) procedure is the most widely used method for FDR control [67]. The BH method operates by:
- Sorting the m p-values from all tests in ascending order: p(1) ≤ p(2) ≤ ... ≤ p_(m)
- Finding the largest k such that p_(k) ≤ (k/m) × q, where q is the desired FDR level (e.g., 0.05 for 5% FDR)
- Rejecting all null hypotheses for j = 1, 2, ..., k
This procedure guarantees that FDR ≤ q when the test statistics are independent or exhibit certain types of positive dependence [67].
Challenges and Counter-Intuitive Behaviors in Omics Data
While FDR methods like BH are popular in omics fields, recent research has revealed counter-intuitive behaviors in datasets with strongly correlated features [67]. In high-dimensional biological data where all null hypotheses are true, the BH procedure still maintains the formal FDR guarantee (resulting in zero findings in >95% of cases). However, in the remaining <5% of cases, the method can report very high numbers of false positives—sometimes as high as 20% of total features in DNA methylation arrays, and up to ~85% in metabolomics data known for high dependency structures [67].
Table 1: Comparison of Multiple Testing Correction Approaches
| Method | Error Rate Controlled | Key Principle | Advantages | Limitations |
|---|---|---|---|---|
| Bonferroni | Family-Wise Error Rate (FWER) | Divides significance threshold (α) by number of tests (m) | Strong control of false positives | Overly conservative; low statistical power |
| Holm (Step-down) | Family-Wise Error Rate (FWER) | Sequentially rejects hypotheses with p-values ≤ α/(m+1-i) | More powerful than Bonferroni while controlling FWER | Still conservative for high-dimensional data |
| Benjamini-Hochberg (BH) | False Discovery Rate (FDR) | Controls expected proportion of false discoveries | More powerful than FWER methods; widely adopted | Can yield high false positives with correlated features [67] |
| q-value | False Discovery Rate (FDR) | Estimates the proportion of false discoveries for each test | Provides FDR estimate for each individual finding | Computational intensity; distributional assumptions |
This phenomenon is particularly pronounced in datasets with a large degree of dependencies between features, such as gene expression data, metabolite data, and epigenome-wide association studies [67]. The variance in the number of rejected features per dataset becomes substantially larger for correlated tests compared to independent data, with BH correction further exaggerating this increase in variance [67].
FDR Control in Pathway Enrichment Analysis
Integration with Pathway Analysis Methods
Pathway enrichment analysis employs three primary methodological approaches, each with distinct implications for FDR control:
3.1.1 Over-Representation Analysis (ORA) ORA statistically evaluates the fraction of genes in a particular pathway found among a set of differentially expressed genes, typically using hypergeometric, Fisher's exact, or binomial tests [2]. These methods determine the probability of observing the overlap between an experimental gene list and a pathway gene set by chance alone. ORA requires an appropriate background gene set for comparison and involves multiple testing across all pathways examined, necessitating FDR correction [2].
3.1.2 Functional Class Scoring (FCS) FCS methods, including Gene Set Enrichment Analysis (GSEA), compute differential expression scores for all measured genes and subsequently compute gene set scores by aggregating the scores of contained genes [1] [2]. GSEA uses a permutational approach to determine significance, which inherently accounts for multiple testing while considering the ranked position of pathway genes across the entire expression profile [2].
3.1.3 Pathway Topology (PT) Methods PT methods incorporate structural information about pathways, including gene product interactions, positions, and roles, which are ignored by ORA and FCS approaches [2]. These methods construct mathematical models that capture entire pathway topology to calculate perturbation factors, combining them into pathway-level statistics with associated p-values [2].
Practical Implementation and Workflow
Table 2: Key Databases for Pathway Enrichment Analysis
| Database | Type | Content Focus | Application in FDR Control |
|---|---|---|---|
| Gene Ontology (GO) | Gene Set | Biological processes, molecular functions, cellular components | Most common resource for ORA; requires FDR correction across thousands of terms [1] [2] |
| Molecular Signatures Database (MSigDB) | Gene Set | Curated gene sets from publications and pathway databases | Used with GSEA; includes Hallmark collection with decreased redundancy [2] |
| Reactome | Pathway | Human biological pathways with detailed molecular interactions | Provides detailed pathway maps; FDR correction needed across pathways [1] [2] |
| KEGG | Pathway | Metabolic and signaling pathways with intuitive diagrams | Licensing restrictions may limit access; FDR essential for pathway analysis [1] |
Figure 1: FDR Control in Pathway Analysis Workflow. This diagram illustrates the integration of FDR correction within the standard pathway analysis pipeline, highlighting its critical position between statistical testing and biological interpretation.
Advanced Considerations and Recent Methodological Developments
Directional Integration in Multi-Omics Data
Recent advances in FDR control address the challenges of multi-omics integration. The Directional P-value Merging (DPM) method incorporates directional constraints when integrating multiple omics datasets, prioritizing genes with consistent directional changes across datasets while penalizing those with inconsistent directions [19]. This approach allows researchers to define expected directional relationships based on biological knowledge (e.g., positive correlation between mRNA and protein expression, negative correlation between DNA methylation and gene expression) [19].
The DPM framework computes a directionally weighted score across k datasets as:
X_DPM = -2(-|Σ(i=1 to j) ln(P_i) × o_i × e_i| + Σ(i=j+1 to k) ln(P_i))
where Pi represents p-values, oi represents observed directional changes, and e_i represents expected directional relationships defined by the user [19]. This method demonstrates enhanced accuracy in identifying consistent pathway regulation while reducing false discoveries arising from discordant multi-omics signals [19].
Novel Algorithms and Approaches
The field continues to evolve with new computational frameworks addressing limitations of conventional FDR methods:
4.2.1 gdGSE Algorithm The gdGSE algorithm employs discretized gene expression profiles rather than continuous values to assess pathway activity, effectively mitigating discrepancies caused by data distributions [53]. This approach demonstrates robust biological insight extraction from diverse datasets, with pathway activity scores showing >90% concordance with experimentally validated drug mechanisms [53].
4.2.2 LD-Aware Multiple Testing in Genetic Studies Quantitative trait locus (QTL) studies face particular challenges due to linkage disequilibrium (LD) between genetic variants. Research has shown that global FDR correction methods like BH are "inappropriate for eQTL studies, as they give inflated (sometimes substantially) FDR that worsens as sample size increases" [67]. This has led to development of LD-aware multiple testing corrections, including efficient permutation testing and hierarchical procedures that incorporate local dependency structures [67].
Experimental Protocols and Best Practices
Protocol for Reliable FDR-Controlled Pathway Analysis
Data Preprocessing and Quality Control
- Perform standard processing specific to omics technology (e.g., normalization for RNA-seq)
- Conduct quality assessment using appropriate tools (FastQC, MultiQC for sequencing data)
- Generate a gene count matrix using tools like featureCounts
Differential Expression Analysis
- Perform statistical testing using established methods (DESeq2 for RNA-seq)
- Apply appropriate variance stabilization methods for high-dimensional data
- Generate raw p-values and effect size estimates (fold changes) for all measured features
Multiple Testing Correction
- Apply Benjamini-Hochberg FDR correction to raw p-values
- Consider dependency structure of data—if high correlations exist, supplement with additional validation approaches
- Use synthetic null data (negative controls) to identify and minimize caveats related to false discoveries [67]
Pathway Enrichment Analysis
- Select appropriate pathway databases based on research question (GO, Reactome, MSigDB)
- Choose enrichment method suited to data structure (ORA for gene lists, GSEA for ranked lists)
- Apply FDR correction at the pathway level to account for testing multiple pathways
Visualization and Interpretation
- Use visualization tools (Cytoscape, EnrichmentMap) to interpret enriched pathways
- Identify main biological themes and their relationships
- Report both statistical significance and effect sizes for biological findings
Research Reagent Solutions
Table 3: Essential Computational Tools for FDR-Controlled Pathway Analysis
| Tool/Resource | Function | Application in FDR Control |
|---|---|---|
| DESeq2 | Differential expression analysis for RNA-seq data | Generates raw p-values for FDR correction [67] [2] |
| g:Profiler | Over-representation analysis for gene lists | Provides FDR-adjusted enrichment p-values [1] [19] |
| GSEA | Gene set enrichment analysis for ranked gene lists | Implements FDR control using permutation testing [1] [2] |
| ActivePathways | Integrative pathway analysis of multi-omics data | Incorporates directional FDR control through DPM method [19] |
| Multiple Testing Correction Tool | Online p-value adjustment | Provides Bonferroni, Holm, Hochberg, and FDR corrections [68] |
Figure 2: FDR Control Mechanism with Caveats. This diagram illustrates the BH FDR control process while highlighting the critical caveat that strongly correlated features can lead to elevated false discoveries despite formal FDR control.
Effective control of the False Discovery Rate is essential for robust pathway enrichment analysis in omics research. While the Benjamini-Hochberg procedure and related FDR methods provide powerful approaches for multiple testing correction, researchers must remain aware of their limitations—particularly in datasets with strongly correlated features where counter-intuitively high numbers of false discoveries can occur [67]. Best practices include using FDR methods in conjunction with suited multiple testing strategies, employing synthetic null data to identify potential caveats, and considering advanced methods like directional integration for multi-omics data [67] [19]. As pathway analysis continues to evolve with novel algorithms and multi-omics integration approaches, appropriate FDR control remains fundamental to deriving biologically meaningful insights from high-dimensional data while minimizing false discoveries.
Validating and Interpreting PEA Results for Robust Biological Insights
Pathway enrichment analysis is a cornerstone of functional genomics, enabling researchers to interpret high-throughput biological data by identifying statistically overrepresented biological processes. For decades, the P-value has served as the primary metric for determining statistical significance, yet its limitations are increasingly apparent within the scientific community. This whitepaper challenges the traditional reliance on binary P-value interpretations and presents a framework incorporating advanced metrics that provide more nuanced, biologically relevant insights. We explore effect sizes, confidence intervals, false discovery rates, and directional analysis methods that collectively offer a more comprehensive approach to significance evaluation. Designed for researchers, scientists, and drug development professionals, this technical guide provides practical methodologies for implementing these advanced metrics, complete with experimental protocols and visualization tools to enhance the rigor and interpretability of enrichment analyses in research settings.
Pathway enrichment analysis is a fundamental technique for interpreting omics datasets (e.g., transcriptomics, proteomics, metabolomics) by identifying biologically meaningful patterns. It examines candidate gene lists from high-throughput experiments to detect statistically enriched biological processes, molecular pathways, or functional categories using established knowledge bases such as Gene Ontology (GO) and Reactome [19]. This approach helps researchers move beyond mere lists of significant genes or proteins to understand systems-level functional implications underlying experimental conditions or disease phenotypes.
Established tools including GSEA (Gene Set Enrichment Analysis), g:Profiler, and Enrichr are widely employed to identify these functional patterns [19]. These methods essentially test whether genes involved in specific biological pathways are overrepresented in a set of differentially expressed genes compared to what would be expected by chance. While traditional enrichment analysis has primarily relied on P-values to determine statistical significance, the field is evolving toward multi-faceted approaches that consider effect magnitude, directionality, and biological context.
The integration of multiple omics datasets presents both opportunities and challenges for enrichment analysis. Combining transcriptomic, proteomic, and epigenomic data can provide complementary biological insights that single-dataset analyses might miss. However, this integration requires sophisticated statistical methods that can handle different data types, experimental biases, and platform-specific technical variations [19]. This whitepaper addresses these challenges by presenting advanced statistical frameworks that move beyond conventional P-value thresholds to deliver more biologically interpretable results.
The Limitations of Statistical Significance
The conventional approach to interpreting research results has been dominated by a binary classification system based primarily on P-values, typically using an arbitrary threshold of P < 0.05 to demarcate "significant" from "non-significant" findings. This practice has been termed the "tyranny of the P-value" and has numerous limitations for scientific interpretation, particularly in enrichment analysis where multiple testing and biological context are crucial considerations [69].
The Misleading Nature of Binary Classification
Treating results as either 'statistically significant' or 'non-significant' fundamentally misrepresents statistical evidence by categorizing a continuous variable. Research has shown that 51% (402/791) of articles from five major journals erroneously interpret statistically non-significant results as indicating "no effect" or "no difference" [69]. Similarly, it is inappropriate to conclude that an association inexorably exists simply because a result was statistically significant. Two studies reporting P-values on opposite sides of the 0.05 threshold are not necessarily in conflict, especially when considering that the point estimates could be identical with differences in statistical power explaining the disparity [69].
The binary significance paradigm has profoundly impacted scientific publishing, contributing to publication bias by deeming studies with non-significant results as unworthy of publication. This selective publication distorts the scientific literature, as the proportion of statistically significant estimates is artificially inflated. Furthermore, a result with high statistical significance (e.g., P < 0.000001) only indicates that the observed finding has a low probability of occurring by chance but reveals nothing about its practical importance or effect size, which may be trivial [70].
Practical Scenarios Where P-Values Fail
In multiple research contexts, reliance solely on P-values leads to misleading conclusions:
- Large sample sizes: In extensive datasets, even minuscule, biologically irrelevant effects can achieve statistical significance. In such cases, statistical significance does not equate to practical importance [70].
- Small effect sizes: A new drug might show a statistically significant improvement in patient outcomes, but if the effect size is minimal, the clinical benefits may not justify costs or potential side effects [70].
- Underpowered studies: Conversely, potentially important findings may be dismissed due to non-significant P-values in studies with limited sample sizes, despite potentially meaningful effect sizes.
The scientific community is increasingly recognizing these limitations, with prominent statisticians and researchers advocating for moving beyond what some have called "the cult of statistical significance" [69]. There is growing consensus that terms such as 'significant', 'statistically significant', 'borderline significant', and their negative expressions should be abandoned in scientific reporting in favor of more nuanced interpretations that consider effect sizes, confidence intervals, and practical implications [69].
Advanced Statistical Frameworks
Effect Sizes and Confidence Intervals
Effect size measures the magnitude of a phenomenon or treatment effect, providing crucial information about practical significance that P-values cannot convey. While P-values indicate whether an effect exists, effect sizes quantify how substantial that effect is. Common effect size measures in enrichment analysis include odds ratios, risk differences, and standardized mean differences.
Confidence intervals (CIs) provide a range of plausible values for an effect size, offering more information than a point estimate alone. A 95% CI indicates that if the same study were repeated multiple times, 95% of the calculated intervals would contain the true population parameter. Wider intervals indicate greater uncertainty, while narrower intervals suggest more precise estimates. The integration of CIs helps researchers assess both statistical significance and practical importance simultaneously [69].
Table 1: Comparison of Statistical Measures Beyond P-values
| Metric | Definition | Interpretation | Advantages |
|---|---|---|---|
| Effect Size | Quantitative measure of the magnitude of a phenomenon | Provides information about the practical importance of results | Not influenced by sample size; allows comparison across studies |
| Confidence Interval | Range of values likely to contain the population parameter | Wider intervals indicate less precision; values outside interval are implausible | Provides information about precision and clinical relevance |
| Minimal Important Difference (MID) | Smallest change in outcome that patients would identify as important | Helps determine clinical relevance of statistical findings | Bridges statistical and clinical significance; patient-centered |
| False Discovery Rate (FDR) | Expected proportion of false positives among significant findings | Controls for multiple testing while maintaining power | Less stringent than family-wise error rate; appropriate for omics data |
Directional Integration in Multi-Omics Analysis
Directional P-value merging (DPM) represents an advanced framework for integrating multi-omics datasets by incorporating both statistical significance and directional changes [19]. This method addresses a critical limitation of conventional approaches that often ignore directional associations between different data types. DPM uses a user-defined constraints vector (CV) to specify expected directional relationships between input datasets, prioritizing genes with consistent directional changes across omics platforms while penalizing those with conflicting signals [19].
The DPM framework calculates a directionally weighted score (X_DPM) across k datasets using the formula:
$${X}{{DPM}}=-2(-{{{{{\rm{|}}}}}}{\Sigma }{i=1}^{j}{\ln}({P}{i}){o}{i}{e}{i}{{{{{\rm{|}}}}}}+{\Sigma }{i=j+1}^{k} {\ln}({P}_{i}))$$
Where Pi represents the P-value from dataset i, oi is the observed directional change, and e_i is the expected direction defined by the constraints vector [19]. This approach allows researchers to test specific biological hypotheses, such as the expected inverse relationship between DNA methylation and gene expression, or the positive correlation between mRNA and protein levels based on the central dogma of molecular biology.
The merged P-value (P'_DPM) is derived from the cumulative χ2 distribution, accounting for gene-to-gene covariation in omics data through the empirical Brown's method for more accurate significance estimation [19]. This directional integration enables more biologically plausible gene prioritization and pathway identification in multi-omics studies.
Methodological Protocols
Implementing Directional P-value Merging
The directional P-value merging workflow consists of four major steps that transform raw omics data into biologically interpretable pathway networks:
Step 1: Data Preparation and Constraints Definition Process upstream omics datasets into a matrix of gene P-values and a corresponding matrix of gene directions (e.g., fold-changes, correlation coefficients, or hazard ratios). Define the constraints vector (CV) based on biological knowledge or experimental design. For example, when integrating DNA methylation and gene expression data, a CV of [-1, +1] would prioritize genes with hypermethylation and downregulation or hypomethylation and upregulation [19].
Step 2: P-value and Direction Merging Apply the DPM algorithm to merge P-values and directions into a single gene list of adjusted P-values. This step prioritizes genes showing significant changes consistent with the predefined directional constraints across multiple omics datasets. The method can incorporate both directional and non-directional datasets, with the latter encoded as zeros in the constraints vector [19].
Step 3: Pathway Enrichment Analysis Analyze the merged gene list for enriched pathways using a ranked hypergeometric algorithm as implemented in the ActivePathways method. This step identifies biological pathways significantly overrepresented in the prioritized gene list and determines which input omics datasets contribute most strongly to each enriched pathway [19].
Step 4: Visualization and Interpretation Visualize resulting pathways as enrichment maps that reveal characteristic functional themes and highlight their directional evidence from omics datasets. These maps facilitate biological interpretation by grouping related pathways and illustrating their statistical support across different data modalities [19].
Determining Minimal Important Differences
Establishing minimal important differences (MIDs) is crucial for contextualizing statistical findings in practical significance. The MID represents the smallest change in a treatment outcome that an individual patient would identify as important and that would indicate a change in patient management [69]. For critical outcomes like mortality, any benefit may be considered important, while for less crucial outcomes, higher thresholds are appropriate.
Protocol for MID determination:
- Identify critical outcomes: Prioritize outcomes based on patient importance and clinical relevance.
- Select determination method: Choose from anchor-based methods (linking change scores to external indicators) or distribution-based methods (using statistical characteristics of the data).
- Establish threshold values: Define absolute and relative effect sizes that constitute meaningful differences.
- Contextualize findings: Compare observed effects against MIDs while considering trade-offs between benefits and harms, costs, and patient values.
The MID threshold should focus on both relative and absolute effects. For example, a 20% relative risk reduction represents dramatically different absolute benefits for patients with 20% versus 1% baseline risk (NNT of 25 versus 500) [69].
Diagram 1: Directional P-value Merging (DPM) workflow for multi-omics data integration.
Essential Research Reagents and Tools
Table 2: Research Reagent Solutions for Advanced Enrichment Analysis
| Tool/Reagent | Function | Application Context |
|---|---|---|
| ActivePathways R Package | Implements DPM for directional multi-omics data fusion | Gene prioritization and pathway analysis across multiple omics datasets [19] |
| Gene Ontology (GO) Database | Provides structured vocabulary of gene functions | Reference knowledge base for pathway enrichment analysis [19] |
| Reactome Pathway Database | Curated database of biological pathways | Pathway annotation for enrichment analysis [19] |
| GSEA Software | Gene Set Enrichment Analysis tool | Identifying enriched gene sets in expression datasets [19] |
| g:Profiler Toolset | Functional enrichment analysis web service | Pathway enrichment analysis with multiple correction methods [19] |
| Enrichr Platform | Integrated enrichment analysis web resource | Gene set enrichment analysis against multiple library databases [19] |
| Empirical Brown's Method | Accounts for gene-gene correlations in P-value merging | Accurate significance estimation in integrated analyses [19] |
Integration of Statistical and Practical Significance
Effectively interpreting enrichment analysis results requires integration of both statistical measures and practical considerations. This integrated approach involves:
Contextualizing Effect Sizes Evaluate the magnitude of enrichment effects against domain-specific knowledge and biologically meaningful thresholds. For example, in drug development, a statistically significant pathway enrichment must be weighed against the anticipated clinical impact and potential side effects. In agricultural biotechnology, a statistically significant effect on crop yield must be assessed against practical farming considerations and economic viability [70].
Considering Trade-offs and Clinical Implications Assess the balance between benefits and potential harms, even for statistically significant findings. An intervention with statistically significant but minimal beneficial effects may not be recommended if associated with serious adverse effects or high costs [69]. The threshold for clinical significance should be more demanding for interventions with greater risks or costs.
Incorporating Certainty of Evidence Utilize structured approaches like GRADE (Grading of Recommendations, Assessment, Development, and Evaluations) to evaluate the certainty of evidence for each outcome, considering study design, risk of bias, consistency, precision, and other factors [69]. This helps contextualize statistically significant findings within the broader evidence landscape.
Diagram 2: Framework for integrating statistical and practical significance in enrichment analysis.
Pathway enrichment analysis is evolving beyond simple P-value-based significance determinations toward more nuanced frameworks that incorporate directionality, effect sizes, and practical relevance. The advanced metrics and methodologies presented in this whitepaper—including directional P-value merging, confidence intervals, minimal important differences, and integrated significance assessment—provide researchers with a more sophisticated toolkit for interpreting enrichment results.
Successful implementation of these approaches requires a cultural shift in scientific practice: abandoning binary thinking, embracing uncertainty through interval estimation, contextualizing findings within domain knowledge, and transparently reporting both precision and practical implications. By adopting these advanced frameworks, researchers can enhance the biological validity and translational potential of their findings, ultimately accelerating scientific discovery and therapeutic development.
As multi-omics technologies continue to advance, the importance of sophisticated analytical frameworks that can integrate diverse data types while respecting biological context will only increase. The methods outlined here represent a significant step toward this future, where statistical rigor and biological relevance converge to drive meaningful scientific insights.
Pathway enrichment analysis is a cornerstone of modern functional genomics, providing a systems-level interpretation of complex omics data. When researchers conduct genome-scale experiments—such as RNA sequencing, proteomics, or genome-wide association studies—they typically generate extensive lists of genes, proteins, or metabolites. Interpreting these lists manually presents a formidable challenge due to the sheer number of molecular entities involved. Pathway analysis addresses this challenge by reducing data complexity through the identification of biologically relevant patterns. Specifically, it tests whether pre-defined sets of genes (pathways) involved in specific biological processes show statistically significant accumulation of experimental signals compared to what would be expected by chance [1].
The fundamental unit in this analysis is the gene set, which represents a collection of genes that work together to carry out a specific biological function, such as a metabolic pathway, signaling cascade, or response to environmental stimulus [1]. These gene sets are obtained from curated databases such as the Molecular Signatures Database (MSigDB), Gene Ontology (GO), KEGG, Reactome, and WikiPathways [1] [71]. The core analytical approach involves testing these gene sets for "enrichment"—statistical over-representation—within the experimental results, thereby translating gene-level statistics into pathway-level insights [1]. This methodology has proven invaluable across diverse applications, from identifying therapeutic targets in cancer research to unraveling the genetic architecture of complex diseases [1] [72].
Core Hypotheses: Competitive vs. Self-Contained
Pathway enrichment methods are fundamentally classified based on the statistical null hypothesis they test, falling into two principal categories: competitive and self-contained tests. This distinction governs both the analytical approach and the interpretation of results [73] [74] [71].
The Competitive Null Hypothesis
Competitive tests, also known as enrichment tests, evaluate whether genes in a pathway of interest are more frequently associated with the experimental phenotype compared to genes not in that pathway [75] [71]. The competitive null hypothesis states that genes in the pathway are at most as often associated with the phenotype as the genes not in the pathway. In essence, competitive approaches test the pathway "against the background" of all other measured genes [74] [71]. Methodologically, these tests treat genes as the sampling units and typically require a comprehensive set of background genes for comparison [73] [71]. Examples of competitive methods include the Hypergeometric test, Fisher's exact test, Gene Set Enrichment Analysis (GSEA), and Correlation Adjusted MEan RAnk (CAMERA) [75] [74] [71].
The Self-Contained Null Hypothesis
Self-contained tests, alternatively called association tests, examine whether the genes in a pathway are jointly associated with the experimental phenotype without reference to other genes [75] [76]. The self-contained null hypothesis states that no genes in the pathway are associated with the phenotype [76] [71]. Unlike competitive tests, self-contained approaches do not require background genes and instead treat biological samples as the sampling units [71]. These methods typically exhibit greater statistical power as they evaluate pathway activity in isolation [75]. Examples of self-contained methods include the multivariate Hotelling T² test, GlobalTest, ROAST (Rotation gene set test), and methods based on minimum spanning trees (MST) [76] [71].
Table 1: Fundamental Differences Between Competitive and Self-Contained Tests
| Feature | Competitive Tests | Self-Contained Tests |
|---|---|---|
| Null Hypothesis | Genes in pathway ≤ associated than genes not in pathway | No genes in pathway are associated |
| Sampling Unit | Genes | Samples/Subjects |
| Background Genes | Required | Not required |
| Statistical Power | Generally lower | Generally higher [75] |
| Interpretation | Relative to other genes | Absolute, for the pathway itself |
| Common Methods | Hypergeometric, GSEA, CAMERA | Hotelling T², ROAST, GlobalTest |
Statistical Foundations and Methodologies
Implementation of Competitive Tests
Competitive tests operate by comparing the statistical evidence for association in pathway genes versus non-pathway genes. The Hypergeometric test and Fisher's exact test are among the simplest competitive approaches, evaluating whether the proportion of significant genes in a pathway exceeds the proportion expected by chance [72] [71]. These methods use a 2×2 contingency table crossing pathway membership with statistical significance, testing the independence between these two classifications [71].
More advanced competitive methods like Gene Set Enrichment Analysis (GSEA) employ a fundamentally different approach. GSEA operates on a ranked list of all genes—typically based on differential expression statistics—and tests whether members of a gene set are non-randomly distributed toward the extremes (top or bottom) of this ranked list [1] [74]. The method calculates an Enrichment Score (ES) representing the maximum deviation from zero of a running sum statistic, which increases when a gene in the set is encountered and decreases otherwise [8]. Statistical significance is assessed through permutation testing, creating a null distribution by repeatedly permuting sample labels or gene set labels [74] [8].
CAMERA (Correlation Adjusted MEan RAnk) represents another competitive approach that incorporates an important adjustment. This method uses a competitive test based on a modified t-test that accounts for the inter-gene correlation, addressing the fact that genes in pathways often exhibit coordinated expression [74] [71].
Implementation of Self-Contained Tests
Self-contained tests evaluate whether all genes in a pathway, considered jointly, show evidence of association with the phenotype. Multivariate tests such as Hotelling's T² represent a direct extension of univariate methods to the multivariate domain, testing the null hypothesis that the mean vectors of gene expression are identical between experimental conditions [76]. These methods explicitly model the covariance structure among genes but require sufficient sample sizes relative to the number of genes tested.
Rotation-based tests like ROAST (Rotation gene set test) employ a different strategy, using rotational permutations of the residual space to assess significance while preserving the correlation structure among genes [71]. This approach remains effective even when the number of samples is smaller than the number of genes in the pathway.
Non-parametric multivariate tests based on Minimum Spanning Trees (MST) offer another self-contained approach. These methods, including multivariate generalizations of the Wald-Wolfowitz and Kolmogorov-Smirnov tests, construct a graph connecting similar samples in the multidimensional gene expression space, then test whether samples from different conditions are well-separated within this graph [76].
Table 2: Representative Methods for Competitive and Self-Contained Testing
| Method | Hypothesis Type | Key Features | Software/Databases |
|---|---|---|---|
| Hypergeometric Test | Competitive | Simple overlap analysis; assumes gene independence | Enrichr [17], g:Profiler [1] |
| GSEA | Competitive | Rank-based; considers entire expression distribution | GSEA, fgsea [71] |
| CAMERA | Competitive | Accounts for inter-gene correlation | limma [74] [71] |
| ROAST | Self-contained | Rotation-based; preserves correlation structure | limma [71] |
| Hotelling T² | Self-contained | Multivariate test of means | Various R packages |
| MST-based tests | Self-contained | Non-parametric; discriminates alternative hypotheses | Custom R code [76] |
Methodological Comparison and Selection Guidelines
Relative Strengths and Limitations
The choice between competitive and self-contained testing frameworks involves important trade-offs with significant implications for interpretation and biological inference.
Competitive tests face a fundamental conceptual criticism: they treat genes as independent sampling units despite the biological reality that genes function within interconnected networks [72]. This approach may also produce misleading results when large proportions of the genome are altered, as the "background" itself becomes significantly changed [71]. Additionally, the hypergeometric test and Fisher's exact test perform poorly in pathway analysis because they assume gene independence and ignore key positional aspects of genes within pathways [72].
Despite these limitations, competitive tests remain widely used, particularly because they can be applied even to studies with limited sample sizes (in extreme cases, even a single sample) [71]. They also answer a question that is often biologically relevant: "Is this pathway more affected than what would be expected by chance?" [75]
Self-contained tests generally demonstrate greater statistical power because they test a less stringent null hypothesis [75]. They also align more naturally with traditional statistical frameworks where samples rather than genes constitute the independent observations [71]. However, these methods typically require multiple samples per condition and may not identify pathways that are genuinely affected but no more so than many other pathways in the system [75].
Empirical Performance Comparisons
Empirical evaluations provide practical insights into method performance. A comprehensive benchmarking study comparing 13 pathway analysis methods across over 1,000 analyses revealed that topology-based methods generally outperform non-topology-based approaches, though no method achieves perfect performance [72]. The Impact Analysis approach, which incorporates pathway topology, demonstrated superior accuracy as measured by Area Under the Curve (AUC) [72].
Another comparative study found that the Adaptive Rank Truncated Product (ARTP) method performed well for both enrichment and association testing, identifying the largest number of enriched pathways across various databases and phenotypes [75]. For self-contained tests, Minimum Spanning Tree (MST)-based non-parametric multivariate tests showed power comparable to conventional approaches while offering enhanced discrimination between different types of alternatives (e.g., mean shifts versus variance changes) [76].
Table 3: Guidelines for Selecting Between Competitive and Self-Contained Approaches
| Consideration | Competitive Tests Recommended | Self-Contained Tests Recommended |
|---|---|---|
| Sample Size | Small sample sizes (even n=1) [71] | Multiple samples per condition [71] |
| Research Question | "Is pathway A more affected than others?" | "Is pathway A affected?" |
| Genomic Context | Focused changes in specific pathways | Widespread changes across genome |
| Statistical Concern | Avoiding absolute claims about pathway activity | Maximizing power to detect any pathway association |
| Implementation | Simple implementation; gene lists sufficient | Requires full expression data |
Advanced Concepts and Emerging Directions
Topology-Aware Methods
Second-generation pathway analysis methods incorporate pathway topology—the structural relationships between genes including their positions, interactions, and regulatory dynamics [73] [72]. These approaches recognize that similarly connected genes often have coordinated functions and that perturbations to centrally positioned "hub" genes may disproportionately impact pathway activity [73].
Topology-based methods consistently demonstrate superior performance compared to non-topology-based approaches according to empirical evaluations [72]. Methods such as Pathway-Express, SPIA (Signaling Pathway Impact Analysis), and NetGSA leverage topological information to improve sensitivity and specificity, particularly for smaller pathway sizes common in metabolomics studies [73]. NetGSA specifically outperforms other methods when analyzing small pathways because it considers both differential expression and changes in interaction strengths between biomolecules [73].
Multi-Omics Integration
The increasing availability of diverse molecular profiling technologies has stimulated development of methods that integrate multiple data types. Directional integration approaches represent a particularly advanced framework for multi-omics pathway analysis [19].
The Directional P-value Merging (DPM) method enables researchers to define expected directional relationships between different omics datasets, then prioritizes genes and pathways showing consistent changes across datasets while penalizing those with inconsistent directionality [19]. For example, researchers can specify that mRNA and protein expression should correlate positively based on the central dogma, while DNA methylation and gene expression should correlate negatively in promoter regions [19]. This approach increases biological plausibility and reduces false positives by testing more specific mechanistic hypotheses.
Specialized Applications
Pathway enrichment methodology continues to evolve to address specialized analytical needs. Drug Mechanism Enrichment Analysis (DMEA) adapts the GSEA framework to group drugs sharing mechanisms of action, facilitating drug repurposing by identifying enriched pharmacological classes in high-throughput screening data [8].
In single-cell RNA sequencing analysis, pathway methods must accommodate unique data characteristics including sparsity and increased noise [71]. Competitive tests like fgsea (fast implementation of GSEA) are commonly applied to differentially expressed genes from cell clusters, while self-contained approaches like vision and AUCell infer pathway activities in individual cells [71].
Experimental Protocols and Practical Implementation
Standard Analytical Workflow
A robust pathway enrichment analysis follows a systematic protocol comprising three major stages [1]:
Gene List Definition: Process omics data to identify genes of interest. For differential expression studies, this involves normalization, statistical testing, and filtering to generate either (a) a simple list of significant genes, or (b) a ranked list based on association statistics [1].
Pathway Enrichment Analysis: Select appropriate competitive or self-contained methods based on experimental design and research questions. Perform statistical testing against pathway databases, applying multiple testing corrections to control false discovery rates [1].
Results Interpretation and Visualization: Interpret significant pathways in biological context, using visualization tools like Cytoscape and EnrichmentMap to identify overarching themes and relationships between enriched pathways [1].
This complete protocol can be performed in approximately 4.5 hours using freely available software such as g:Profiler, GSEA, Cytoscape, and EnrichmentMap [1].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Resources for Pathway Enrichment Analysis
| Resource | Type | Function | Access |
|---|---|---|---|
| g:Profiler | Web tool / API | Competitive enrichment analysis for gene lists | https://biit.cs.ut.ee/gprofiler/ |
| Enrichr | Web tool / API | Competitive analysis with extensive library support | https://maayanlab.cloud/Enrichr/ |
| GSEA/fgsea | Software package | Competitive rank-based enrichment analysis | https://www.gsea-msigdb.org/ |
| limma (ROAST, CAMERA) | R package | Self-contained and competitive tests with correlation adjustment | Bioconductor |
| ActivePathways | R package | Integrative analysis including directional multi-omics | CRAN |
| MSigDB | Database | Curated collection of gene sets for enrichment testing | https://www.gsea-msigdb.org/ |
| Reactome | Database | Manually curated pathway knowledgebase | https://reactome.org/ |
| Cytoscape/EnrichmentMap | Visualization | Network-based visualization of enrichment results | https://cytoscape.org/ |
Visualizing Analytical Concepts and Workflows
Diagram 1: Pathway enrichment analysis workflow comparing competitive and self-contained approaches
Diagram 2: Fundamental differences in null hypotheses between competitive and self-contained tests
The distinction between competitive and self-contained null hypotheses represents a fundamental conceptual division in pathway enrichment methodology, with significant implications for study design, analytical approach, and biological interpretation. Competitive tests ask whether a pathway is more affected than the genomic background, while self-contained tests ask whether a pathway is affected at all. This methodological dichotomy extends throughout the analytical workflow, from experimental design through to biological interpretation.
The evolving landscape of pathway analysis continues to incorporate more sophisticated approaches including topological information, directional multi-omics integration, and specialized applications in drug discovery and single-cell biology. As these methods advance, they offer increasingly powerful frameworks for translating high-dimensional molecular measurements into biologically meaningful insights. Researchers should select methods based on their specific experimental context, biological questions, and data characteristics, while remaining mindful of the underlying statistical assumptions and limitations of each approach.
Pathway Enrichment Analysis (PEA) is a computational biology method that identifies biological functions overrepresented in a group of genes more than would be expected by chance [12]. As a critical component of omics research, PEA helps researchers move beyond mere lists of significant genes to understand systems-level biological phenomena. However, the output of PEA typically generates extensive lists of enriched pathways that can be challenging to interpret without appropriate visualization techniques. The sheer volume of results, coupled with inherent redundancy and relationships between pathways, creates a significant interpretation bottleneck [77]. Enrichment maps and pathway networks address this challenge by providing powerful visual frameworks that transform tabular data into biological insights, enabling researchers to identify broader biological themes and patterns that might otherwise remain obscured in extensive statistical outputs [20].
Foundations of Pathway Enrichment Analysis
Algorithmic Approaches to Enrichment Analysis
Understanding the fundamental algorithms behind pathway enrichment is crucial for proper visualization and interpretation. Three primary classes of enrichment algorithms exist, each with distinct characteristics and visualization needs [77]:
- Singular Enrichment Analysis (SEA): This traditional approach iteratively tests annotation terms one at a time against a list of significant genes. While simple and useful, SEA often produces hundreds of results with significant redundancy due to hierarchical relationships between terms [77].
- Gene Set Enrichment Analysis (GSEA): GSEA considers all genes in an experiment (not just those deemed significant) by analyzing their ranked distribution. It determines if genes sharing a particular annotation are randomly distributed throughout the ranked list or clustered at the extremes, indicating association with phenotypic classes [12] [77].
- Modular Enrichment Analysis (MEA): This advanced approach considers relationships between different annotation terms during enrichment analysis, reducing redundancy and preventing dilution of important biological concepts [77].
From Analysis Results to Biological Insight
The transition from statistical results to biological insight represents the central challenge that visualization addresses. A typical PEA output identifies numerous significantly enriched pathways, but understanding how these pathways interact and collectively contribute to the biological phenomenon under investigation requires synthesis across multiple related terms [20]. Enrichment maps facilitate this synthesis by creating network-based visualizations where connections represent biological relationships, allowing researchers to quickly identify functional modules and overarching themes in their data [20].
Enrichment Maps: Principles and Implementation
Conceptual Framework of Enrichment Maps
Enrichment maps provide a network visualization of PEA results where nodes represent enriched terms or pathways, and edges connect related terms based on genetic similarity [20]. This approach transforms long, redundant lists of enriched pathways into structured networks that reveal functional modules and biological themes. The fundamental principle involves representing similarity between enriched terms through spatial proximity and visual connections, enabling researchers to quickly identify major functional categories in their data without navigating extensive tabular output [20].
Table: Key Components of an Enrichment Map
| Component | Description | Visual Representation |
|---|---|---|
| Nodes | Individual enriched pathways, terms, or gene sets | Size typically indicates number of genes in the pathway |
| Edges | Connections between related pathways | Thickness indicates degree of gene overlap between pathways |
| Clusters | Groups of highly interconnected nodes | Spatial proximity and often color-coding |
| Layout | Spatial arrangement of nodes and edges | Force-directed algorithms for clear visualization |
Construction Workflow for Enrichment Maps
The process of creating enrichment maps follows a systematic workflow that integrates analysis tools with visualization platforms. The following diagram illustrates the key steps in this process:
The enrichment map workflow begins with preparing two essential inputs: a gene list of interest and a pathway database in GMT format [20]. The GMT file is a tab-separated text file where each line represents a pathway containing a pathway ID, descriptive name, and associated genes [20]. For the analysis step, researchers must select the appropriate enrichment method based on their data type: g:Profiler for thresholded gene lists or GSEA for complete ranked gene lists [20]. These tools generate statistical results that are subsequently imported into Cytoscape with the EnrichmentMap app to create the network visualization [20]. The final interpretation stage involves identifying functional modules and biological themes within the visualized network.
Practical Implementation Guide
g:Profiler Analysis for Thresholded Gene Lists
For flat, unranked gene lists, g:Profiler provides an accessible web-based tool [20]. The analysis requires specific parameterization to generate optimal results for enrichment maps:
- Input Preparation: Paste the gene list into the Query field and select the "Ordered query" option when working with partially ranked lists [20].
- Annotation Selection: Initially include only Biological Processes (BP) from Gene Ontology and molecular pathways from Reactome to reduce redundancy [20].
- Pathway Filtering: Set the size of functional categories to between 5 and 350 genes to exclude overly broad or narrowly specific pathways [20].
- Intersection Threshold: Require at least 3 genes in the query/term intersection to ensure statistical reliability [20].
- Output Format: Select "Generic Enrichment Map (TAB)" format to generate files compatible with Cytoscape visualization [20].
GSEA Analysis for Ranked Gene Lists
For complete ranked gene lists (such as all genes from an expression experiment), the GSEA desktop application provides appropriate analysis [20]:
- Input Format: Prepare an RNK file containing gene identifiers in the first column and ranking metric (e.g., fold change) in the second column [20].
- Method Selection: Use the "Run GSEAPreranked" tool with the loaded RNK and GMT files [20].
- Parameter Configuration: Employ default parameters initially, including 1000 permutations for significance testing [20].
- Result Export: The analysis generates enrichment scores and significance values for each pathway in the database [20].
Advanced Network Visualization with Cytoscape
Cytoscape Setup and Workflow
Cytoscape serves as the primary platform for creating and analyzing enrichment maps, requiring specific configuration for optimal results [20]:
- Software Installation: Install Java Runtime Environment, Cytoscape (version 3.6.0 or higher), and the necessary apps including EnrichmentMap (version 3.1+), clusterMaker2, WordCloud, and AutoAnnotate [20].
- Data Import: Load enrichment results from either g:Profiler or GSEA analysis directly into Cytoscape [20].
- Network Generation: Use the EnrichmentMap app to automatically create networks from the enrichment results, with nodes representing pathways and edges indicating gene overlaps [20].
- Cluster Identification: Apply clustering algorithms (such as those in clusterMaker2) to identify functional modules within the enrichment map [20].
- Annotation: Use AutoAnnotate to label clusters based on common functional themes, facilitating biological interpretation [20].
Visual Optimization and Interpretation
Effective enrichment maps require careful visual optimization to maximize interpretability. The following aspects should be considered:
- Node Attributes: Size nodes according to the number of genes in each pathway to emphasize larger functional units [20].
- Color Coding: Apply consistent color schemes to represent statistical significance (e.g., FDR values) or functional categories [20].
- Edge Weighting: Set edge thickness proportional to the degree of gene overlap between connected pathways [20].
- Layout Selection: Use force-directed layouts that naturally group highly interconnected nodes, revealing functional modules [20].
Pathway Networks: Beyond Enrichment Maps
From Functional Enrichment to Biological Pathways
While enrichment maps visualize relationships between enriched terms, pathway networks represent the actual biological interactions between molecular components within and between pathways. These networks illustrate how genes and proteins interact in coordinated ways to accomplish biological functions [12]. Different pathway databases represent these interactions with varying conventions: for example, KEGG signaling pathways use nodes to represent genes or gene products with edges defining activation or inhibition signals, while metabolic pathways typically represent biochemical compounds as nodes and reactions as edges [12].
Constructing Pathway Networks
Building meaningful pathway networks requires careful consideration of biological context and data representation:
Pathway network construction begins with integrating significantly enriched pathways from PEA with molecular interaction data, including protein-protein interactions, signaling relationships, and metabolic conversions [12]. Experimental data such as gene expression changes or mutational status are then overlaid onto this framework [12]. Topological analysis identifies key nodes and interaction points between pathways, revealing potential crosstalk mechanisms [12]. The resulting visualization provides mechanistic insight into how multiple pathways coordinately drive the biological phenotype under investigation.
Applications in Drug Discovery and Development
Drug Mechanism Enrichment Analysis (DMEA)
The enrichment principle extends beyond genes to drug repurposing through Drug Mechanism Enrichment Analysis (DMEA), which adapts GSEA to identify enriched drug mechanisms of action (MOAs) in rank-ordered drug lists [8]. This approach groups drugs with shared MOAs to improve prioritization of drug repurposing candidates, increasing on-target signal and reducing off-target effects compared to individual drug analysis [8]. DMEA follows the same statistical framework as GSEA but applies it to sets of drugs rather than genes, identifying MOAs overrepresented at either end of a ranked drug list [8].
Table: Comparison of Enrichment-Based Drug Discovery Approaches
| Method | Input Data | Statistical Approach | Key Output | Limitations |
|---|---|---|---|---|
| DMEA | Rank-ordered drug list with MOA annotations | GSEA algorithm adapted for drugs | Enriched MOAs with NES and FDR | Requires predefined MOA annotations |
| CMap L1000 Query | Gene expression signatures | Pattern matching to reference database | Similar drug perturbations | Limited to CMap database |
| DrugEnrichr | Unranked drug list | Fisher's exact test | Enriched drug terms | Limited statistical rigor |
| DSEA | Unranked drug list | Enrichment analysis | Associated gene sets | Queries gene sets, not MOAs |
Case Study: Identifying Senolytic Drug MOAs
A practical application of this approach successfully identified potential senescence-inducing and senolytic drug mechanisms for primary human mammary epithelial cells [8]. Researchers applied DMEA to rank-ordered drug lists based on molecular classification scores, which identified EGFR inhibitors as significantly enriched for senolytic activity [8]. Subsequent experimental validation confirmed the senolytic effects of EGFR inhibitors, demonstrating how enrichment-based approaches can prioritize candidates for further investigation [8].
The Scientist's Toolkit: Essential Research Reagents
Table: Key Research Reagents for Enrichment Analysis and Visualization
| Reagent/Resource | Type | Function | Example Sources |
|---|---|---|---|
| Pathway Databases | Knowledgebase | Provide curated gene-pathway associations for enrichment testing | KEGG, Reactome, WikiPathways, Gene Ontology [12] |
| GMT Files | Data Format | Standardized file format containing pathway-gene associations | Baderlab, MSigDB, custom-generated files [20] |
| Enrichment Analysis Tools | Software | Perform statistical enrichment analysis | g:Profiler, GSEA, Enrichr [12] [20] |
| Network Visualization Platform | Software | Create and analyze enrichment maps and pathway networks | Cytoscape with EnrichmentMap app [20] |
| Gene Expression Data | Experimental Data | Input for generating gene lists for enrichment analysis | RNA-seq, microarray datasets [78] |
| Drug MOA Annotations | Knowledgebase | Provide drug-mechanism relationships for DMEA | PRISM, DrugBank, custom annotations [8] |
Best Practices and Technical Considerations
Method Selection Guidelines
Choosing the appropriate enrichment method and visualization approach depends primarily on input data characteristics [20]:
- For thresholded gene lists (e.g., significant differentially expressed genes): Use g:Profiler or similar ORA tools followed by enrichment map visualization [20].
- For complete ranked gene lists (e.g., all genes from an expression experiment): Use GSEA with preranked analysis to preserve expression magnitude information [20].
- For drug repurposing applications: Use DMEA with rank-ordered drug lists annotated with mechanism of action information [8].
Quality Control and Validation
Robust enrichment analysis and visualization require careful attention to quality metrics:
- Input Gene List Quality: Ensure proper identifier mapping and consider background population appropriate for the experimental context [12] [77].
- Pathway Database Selection: Use databases relevant to the biological context and organism under study [12].
- Multiple Testing Correction: Always apply appropriate multiple testing correction (e.g., Benjamini-Hochberg FDR) to account for false discoveries [12] [79].
- Statistical Power Considerations: Filter pathways by size (typically 5-350 genes) to ensure meaningful interpretation [20].
- Experimental Validation: Where possible, confirm key findings through orthogonal experimental approaches [8].
The field of pathway enrichment visualization continues to evolve with several promising directions. Integration of multi-omics data into unified network representations will provide more comprehensive biological insights [78]. Temporal enrichment analysis approaches can capture dynamic pathway alterations across experimental time courses [77]. Machine learning methods are being incorporated to improve cluster identification and automated annotation of enrichment maps [20]. Additionally, interactive web-based visualization platforms are making enrichment analysis more accessible to researchers without bioinformatics expertise [8].
Visualization through enrichment maps and pathway networks represents an essential component of modern pathway enrichment analysis, transforming statistical outputs into biological understanding. By implementing the principles and methods outlined in this guide, researchers can effectively interpret complex enrichment results, identify overarching biological themes, and generate testable hypotheses for further investigation. These approaches have proven particularly valuable in drug discovery applications, where they help prioritize candidate therapeutics for repurposing by identifying enriched mechanisms of action across multiple drugs [8]. As enrichment methodology continues to advance, visualization techniques will remain critical for extracting meaningful biological insights from increasingly complex genomic datasets.
This technical guide elucidates the process of validating text-mined genetic targets for drug discovery through pathway enrichment analysis. We present a structured framework that integrates literature-derived gene sets with functional genomics and experimental validation, using a published study on Connective Tissue Disease-Associated Pulmonary Arterial Hypertension (CTD-PAH) as a primary case study. This in-depth analysis demonstrates how pathway enrichment techniques transform unstructured literature data into testable therapeutic hypotheses, providing researchers and drug development professionals with validated methodologies for systematic drug repurposing and novel target identification.
Pathway enrichment analysis represents a cornerstone of modern bioinformatics, providing systematic methods to interpret high-dimensional biological data within the context of existing molecular knowledge. For drug discovery, these techniques bridge the gap between genomic findings and therapeutic applications by identifying biologically coherent patterns that are statistically unlikely to occur by chance alone [78] [2].
The fundamental premise involves testing whether genes associated with a particular condition or drug response disproportionately map to specific biological pathways, molecular functions, or cellular components [2]. When applied to text-mined gene sets, pathway enrichment provides mechanistic plausibility to computational predictions, prioritizing targets with established biological context for experimental validation. This approach has evolved from simple over-representation analysis to sophisticated multi-omics integration methods that capture complex biological relationships [39].
Methodological Framework: From Text to Therapeutic Hypotheses
The validation pipeline from text-mined genes to novel drug discoveries follows a sequential workflow with distinct analytical phases, each requiring specific tools and statistical approaches.
Literature Mining and Gene Set Acquisition
The initial phase involves extracting disease-gene associations from biomedical literature using automated text mining tools. In the CTD-PAH case study, researchers utilized the pubmed2ensembl database, an extension of the BioMart system containing over 2,000,000 PubMed articles and approximately 150,000 Ensembl genes [80] [81]. Two separate queries were performed: one for "pulmonary arterial hypertension" (PAH) returning 797 genes, and another for "connective tissue diseases" (CTD) returning 441 genes. The intersection of these gene sets identified 179 overlapping genes implicated in both conditions, establishing the candidate gene list for subsequent analysis [80].
Functional Enrichment Analysis
The 179 overlapping genes underwent comprehensive functional annotation using DAVID, with statistical significance threshold set at p < 0.05 [80] [81]. This analysis identified significantly enriched Gene Ontology terms and Kyoto Encyclopedia of Genes and Genomes pathways, providing biological context for the gene set.
Table 1: Significant Functional Enrichments in CTD-PAH Gene Set
| Analysis Type | Category | Significantly Enriched Terms | Statistical Threshold |
|---|---|---|---|
| Gene Ontology | Biological Process | Regulation of response to organic substance, cell proliferation, positive regulation of response to stimulus | p < 0.05 |
| Gene Ontology | Cellular Component | Extracellular region, extracellular region part, extracellular space | p < 0.05 |
| Gene Ontology | Molecular Function | Receptor binding, identical protein binding, enzyme binding | p < 0.05 |
| KEGG Pathways | Signaling Pathways | Cancer pathways, cytokine-cytokine receptor interaction, PI3K-Akt signaling pathway | p < 0.05 |
Protein-Protein Interaction Network Analysis
To identify functionally coherent gene modules within the candidate set, the 179 genes were uploaded to STRING (version 11.0) with a high-confidence interaction threshold (minimum score > 0.9) [80] [81]. This produced a protein-protein interaction network comprising 149 nodes and 1,205 edges. Subsequent analysis using the Molecular Complex Detection app in Cytoscape identified two significant gene modules:
- Module 1: 25 nodes with 180 edges
- Module 2: 20 nodes with 104 edges [80]
Module 2 was selected for further drug-gene interaction analysis based on its cohesive network properties.
Drug-Gene Interaction Mapping
The 20 genes in Module 2 were analyzed using the Drug Gene Interaction Database to identify existing drugs targeting these genes [80] [81]. To ensure high-confidence predictions, stringent filtering criteria were applied (Query Score ≥5 and Interaction Score ≥1), yielding 13 candidate drugs targeting six key genes.
Table 2: Validated Drug Candidates for CTD-PAH Identified Through Text Mining
| Target Gene | Number of Drugs | Drug Examples | Interaction Types | FDA Approval Status |
|---|---|---|---|---|
| IL6 | 1 | Siltuximab | Antagonist, antibody | Approved for other indications |
| IL1B | 2 | Canakinumab, Rilonacept | Inhibitor | Approved for other indications |
| MMP9 | 1 | Marimastat | Inhibitor | Approved for other indications |
| VEGFA | 3 | Bevacizumab, Aflibercept, Sunitinib | Antibody, inhibitor | Approved for other indications |
| TGFB1 | 1 | Metelimumab | Antibody | Approved for other indications |
| EGFR | 5 | Gefitinib, Erlotinib, Cetuximab | Inhibitor, antibody | Approved for other indications |
Experimental Design and Validation Protocols
Rigorous validation is essential to establish translational potential for computationally predicted drug-disease relationships.
In Vitro Validation of Predicted Compounds
For experimentally testing predicted compounds, the following protocol provides a standardized approach:
Cell-Based Proliferation Assay Protocol
- Cell Culture: Maintain relevant cell lines (e.g., primary human cells or established cell models) under standard conditions appropriate for the cell type.
- Compound Treatment: Prepare serial dilutions of candidate compounds in appropriate vehicle controls. Include both positive and negative controls.
- Dose-Response Analysis: Treat cells with compounds across a concentration range (typically 0.1-100 μM) for 24-72 hours.
- Viability Assessment: Measure cell proliferation using standardized assays (e.g., MTT, XTT, or ATP-based assays).
- Data Analysis: Calculate IC50 values using non-linear regression analysis. Perform statistical testing with appropriate multiple comparison corrections [82].
Orthogonal Validation Methods
- Literature Validation: Search for independent evidence supporting predicted relationships in subsequent publications not included in the original analysis [82].
- Clinical Data Mining: Interrogate electronic health records or pharmacovigilance databases for evidence of efficacy in real-world patient populations.
- Mechanistic Studies: Employ targeted knockdown of identified genes to confirm pathway necessity using siRNA or CRISPR-based approaches.
Advanced Pathway Enrichment Methodologies
Beyond conventional over-representation analysis, several advanced methods enhance discovery potential for drug target identification.
Multi-Omics Integration with ActivePathways
ActivePathways employs data fusion techniques to integrate significance values from multiple omics datasets [39]. The method follows a three-step process:
- Statistical Data Fusion: Combine p-values from different omics datasets using Brown's extension of Fisher's combined probability test, which accounts for dependencies between datasets.
- Pathway Enrichment Analysis: Perform ranked hypergeometric testing on the integrated gene list against pathway databases.
- Evidence Analysis: Determine contributing evidence from individual datasets for each significantly enriched pathway.
In pan-cancer analysis, ActivePathways identified pathways supported by both coding and non-coding mutations that were undetectable when analyzing either dataset separately [39].
Drug Mechanism Enrichment Analysis (DMEA)
DMEA adapts Gene Set Enrichment Analysis to evaluate whether drugs sharing mechanism of action are enriched in rank-ordered drug lists [8]. The method:
- Calculates enrichment scores using a weighted Kolmogorov-Smirnov-like statistic
- Determines significance via empirical permutation testing
- Computes normalized enrichment scores and false discovery rates DMEA improves prioritization of drug repurposing candidates by aggregating signal across multiple drugs with shared mechanisms [8].
Visualization of Analytical Workflows
The following diagram illustrates the complete text mining to validation pipeline:
Text Mining to Drug Discovery Workflow
Successful implementation requires specific computational tools, databases, and experimental reagents.
Table 3: Essential Resources for Text Mining and Pathway Analysis
| Resource Category | Specific Tools/Databases | Primary Function | Application Context |
|---|---|---|---|
| Literature Mining | Pubmed2Ensembl, CoPub | Extract gene-disease associations from literature | Initial gene set discovery |
| Functional Enrichment | DAVID, g:Profiler, Enrichr | GO and pathway enrichment analysis | Biological interpretation of gene sets |
| Pathway Databases | KEGG, Reactome, WikiPathways | Curated pathway knowledge bases | Reference for enrichment analysis |
| Protein Interactions | STRING, BioGRID, NDEx | Protein-protein interaction networks | Identify functional modules |
| Drug-Gene Interactions | DGIdb, DrugBank, PharmGKB | Map genes to targeting drugs | Therapeutic hypothesis generation |
| Visualization | Cytoscape, Enrichment Map | Network visualization and analysis | Interpret complex relationships |
| Experimental Validation | Cell lines, compounds, assay kits | In vitro confirmation of predictions | Biological validation of predictions |
Discussion and Future Perspectives
The integrated approach of text mining and pathway enrichment analysis represents a powerful paradigm for accelerating drug discovery. The CTD-PAH case study demonstrates how systematically extracted literature knowledge can yield mechanistically grounded therapeutic hypotheses with reduced development timelines compared to traditional approaches [80] [81].
Future methodological developments will likely focus on enhanced multi-omics integration, incorporation of artificial intelligence for relationship extraction, and dynamic pathway analysis that considers temporal and spatial cellular contexts [78] [39]. As these methods mature, their integration with electronic health records and real-world evidence will further strengthen the translational potential of computationally predicted drug-disease relationships.
For researchers implementing these approaches, rigorous validation remains paramount. Computational predictions must be viewed as hypothesis-generating rather than conclusive evidence, with biological validation serving as an essential component of the discovery pipeline. When properly implemented, this framework provides a systematic methodology for uncovering novel therapeutic applications from existing knowledge, potentially yielding new treatment options for diseases with unmet medical needs.
Pathway Enrichment Analysis (PEA) is a cornerstone bioinformatics method for interpreting gene lists generated from genome-scale (omics) experiments. It helps researchers move from a simple list of genes to a mechanistic understanding of underlying biology by identifying biological pathways that are statistically over-represented more than would be expected by chance alone [1]. This process is fundamental for discovering functional insights in diverse areas, from disease mechanism investigation to drug repositioning strategies [1] [83].
The core principle of PEA involves statistically testing all pathways in a given database for enrichment in an experimentally-derived gene list. This relies on the availability of curated pathway databases and robust statistical methods to distinguish true biological signal from random chance, often corrected for multiple hypothesis testing [1]. Effective PEA has led to significant biomedical advances, such as identifying histone and DNA methylation as a therapeutic target in childhood brain cancer and clarifying gene-deletion pathways in autism [1].
Key PEA Methodologies and Tools
PEA tools employ different statistical approaches tailored to the nature of input data—either a simple gene list or a ranked list. Benchmarking requires understanding these core methodologies.
Core Statistical Approaches
- Over-representation Analysis (ORA): Used with simple gene lists (e.g., mutated genes, interacting proteins). Tools like g:Profiler apply hypergeometric tests or Fisher's exact test to calculate the probability of observing the overlap between an input gene list and a pathway gene set by chance [1]. The standard formula calculates a P-value representing this probability, followed by multiple testing correction [1].
- Gene Set Enrichment Analysis (GSEA): Designed for ranked gene lists (e.g., by differential expression). GSEA uses an enrichment score (ES) computed by walking down the ranked list, increasing a running sum when a gene is in the pathway and decreasing it otherwise. The ES reflects whether pathway members are randomly distributed or clustered at the top/bottom of the list. A leading-edge subset of genes often accounts for the enrichment signal [1].
Quantitative Benchmarking of PEA Tools
The table below summarizes primary PEA tools, their methodologies, and key characteristics for benchmarking.
Table 1: Core PEA Tools and Methodologies for Benchmarking
| Tool Name | Core Methodology | Input Data Type | Key Statistical Metric | Primary Application Context |
|---|---|---|---|---|
| g:Profiler [1] | Over-representation Analysis (ORA) | Gene List | P-value (hypergeometric/Fisher's exact test) | General-purpose functional enrichment |
| GSEA [1] | Gene Set Enrichment Analysis | Ranked Gene List | Enrichment Score (ES), Normalized ES (NES) | Discovering subtle, coordinated expression changes |
| ClusterProfile [83] | ORA & Functional Profiling | Gene List / Ranked List | P-value | Functional profiling of biological themes |
| gdGSE [53] | Discretized Expression Analysis | Gene Expression Matrix | Gene Set Enrichment Score | Robust pathway activity from bulk/single-cell data |
| EnrichmentMap [1] | Visualization & Integration | Enrichment Results | N/A (Visualization) | Interpreting and clustering multiple enriched pathways |
Emerging Metrics and Algorithms
Newer algorithms and metrics are being developed to address limitations of traditional P-value approaches.
- Inverse Pathway Frequency (IPF) Metrics: IPF addresses that traditional P-values assume equal probability for each gene, which is often inaccurate. Housekeeping genes appear in many pathways, while specific genes are functionally unique. IPF assigns higher weight to genes appearing in fewer pathways, increasing their contribution to enrichment scores for more specific biological processes [83].
- gdGSE Algorithm: This framework uses discretized gene expression profiles instead of continuous values, mitigating discrepancies from data distributions. It binarizes gene expression matrices before conversion to enrichment matrices, showing >90% concordance with experimentally validated drug mechanisms [53].
Experimental Design for PEA Benchmarking
A robust benchmarking experiment requires standardized data, a clear workflow, and defined evaluation criteria to ensure fair, interpretable tool comparisons.
Experimental Workflow for Benchmarking
The following diagram visualizes the standard workflow for designing and executing a PEA tool benchmarking study.
Benchmarking Input Data Preparation
Standardized input data is critical. A typical approach uses a published dataset with known biological outcomes.
- Sample Dataset: A common paradigm uses a drug-centric resource. For example, 31,118 PubMed abstracts for the drug rapamycin can be processed to extract gene sets [83].
- Gene List Generation: Apply multiple text-mining methods to the same corpus to generate different gene sets for evaluation [83]:
- Co-occurrence (ABSTRACT/SENTENCE): Extract genes co-mentioned with the drug in abstracts or sentences.
- Syntactic (DEPENDENCY): Use parsers to identify gene subjects/objects in a sentence dependency tree.
- Semantic (TEES): Apply trained event extraction systems to find genes with specific interactions.
- Ground Truth Establishment: Use curated pathway databases like KEGG to define known drug-related pathways as a "gold standard" for benchmarking [83].
Key Performance Metrics for Evaluation
Benchmarking assesses both statistical performance and practical utility.
- Statistical Sensitivity and Specificity: Evaluate the ability to correctly identify known true pathways (sensitivity) while minimizing false positives (specificity) using the established ground truth [83].
- Novelty and Robustness: Assess the ability to replicate known biological discoveries (e.g., rapamycin's efficacy in breast cancer) from historical data, demonstrating real-world predictive power [83].
- Concordance with Experimental Data: For methods like gdGSE, a high concordance (>90%) with patient-derived xenografts or cell line validation is a strong indicator of accuracy [53].
- Computational Efficiency: Measure runtime and resource requirements, especially for large datasets or single-cell analyses.
Successful PEA implementation relies on specific computational tools, databases, and reagents.
Table 2: Essential Research Reagents and Resources for PEA
| Category | Resource Name | Primary Function in PEA | Key Features / Application Notes |
|---|---|---|---|
| Pathway Databases | Gene Ontology (GO) [1] | Provides standardized terms and gene annotations for biological processes, molecular functions, and cellular components. | Hierarchically organized; biological process annotations are most commonly used. |
| Molecular Signatures Database (MSigDB) [1] | A comprehensive collection of gene sets from various sources, including curated pathways and expression signatures. | Includes 'hallmark' gene sets, a relatively non-redundant collection. | |
| KEGG [1] [83] | A database of pathway maps for molecular interactions and reaction networks. | Known for intuitive pathway diagrams; useful for metabolic pathways. | |
| Reactome [1] | An open-access, manually curated database of human pathways and reactions. | Most actively updated general-purpose human pathway database. | |
| Software & Platforms | Cytoscape [1] | An open-source platform for visualizing complex networks and integrating with enrichment data. | Essential for creating visualizations of enriched pathways and their interactions. |
| EnrichmentMap [1] | A Cytoscape app that visually clusters and interprets enrichment results. | Helps identify main biological themes from a long list of enriched pathways. | |
| R/Bioconductor (ClusterProfile) [83] | A programming environment and package for ORA and functional profiling. | Offers high flexibility for custom analysis and integration into computational pipelines. | |
| Experimental Reagents | Patient-Derived Xenografts (PDXs) & Cell Lines [53] | Biologically relevant models for experimentally validating pathway activity predictions. | High concordance (>90%) with computational predictions indicates strong algorithm performance. |
Benchmarking studies reveal that no single PEA tool is universally superior. The choice depends on the data type (list vs. ranked), biological question, and need for novel discovery versus robust confirmation. Traditional ORA methods like g:Profiler are straightforward for predefined gene lists, while GSEA is powerful for detecting subtle effects across entire expression datasets [1]. Emerging methods like gdGSE and metrics like IPF show promise in increasing robustness and biological specificity by addressing statistical limitations of earlier approaches [83] [53].
Future development will likely focus on better integration of heterogeneous data types, improved statistical models that account for gene-specific properties and pathway structures, and enhanced visualization techniques for clearer interpretation. As the field progresses, rigorous and standardized benchmarking will remain essential for guiding researchers toward the most effective analytical strategies for their specific research contexts in drug development and basic biology.
Conclusion
Pathway Enrichment Analysis has evolved into an indispensable bioinformatics technique that transforms complex gene lists into actionable biological insights. By understanding its foundational principles, correctly applying methodological approaches like ORA and GSEA, adhering to troubleshooting best practices, and rigorously validating results, researchers can reliably uncover the mechanistic underpinnings of disease and treatment. The future of PEA is integration—spanning single-cell multi-omics data, incorporating more sophisticated network biology, and powering AI-driven drug discovery. As pathway databases and algorithms continue to advance, PEA will remain a cornerstone for translating genomic-scale data into meaningful clinical and therapeutic breakthroughs.
References
- 1. Pathway enrichment analysis and visualization of omics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6607905/]
- 2. Gene ontology and pathway analysis - Bioinformatics [https://bioinformatics.ccr.cancer.gov/docs/bioinformatics-for-beginners-2025/Module3_Pathway_Analysis/Lesson15/]
- 3. Pathway Analysis vs Gene Set Analysis [https://advaitabio.com/science/pathway-analysis-vs-gene-set-analysis/]
- 4. Using published pathway figures in enrichment analysis and ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09816-1]
- 5. A web-based tool that integrates data visualization and ... [https://www.nature.com/articles/s41598-023-34163-2]
- 6. Pathway Enrichment Analysis through Network UTilization [https://pmc.ncbi.nlm.nih.gov/articles/PMC12311273/]
- 7. QIAGEN Ingenuity Pathway Analysis (IPA) [https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/]
- 8. Drug mechanism enrichment analysis improves prioritization ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05343-8]
- 9. Pathway analysis for genome-wide genetic variation data [https://www.sciencedirect.com/science/article/abs/pii/S1673852721000412]
- 10. How to use Bonferroni correction for multiple hypothesis ... [https://www.statsig.com/perspectives/bonferroni-correction-multiple-testing]
- 11. Bonferroni Correction | Amplitude Experiment [https://amplitude.com/docs/feature-experiment/advanced-techniques/bonferroni-correction]
- 12. Nine quick tips for pathway enrichment analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9371296/]
- 13. Pathways and gene sets: What is functional enrichment ... [https://bioinformatics.ccr.cancer.gov/btep/pathways-and-gene-sets-what-is-functional-enrichment-analysis/]
- 14. GSEA [https://www.gsea-msigdb.org/]
- 15. Gene set enrichment analysis [https://en.wikipedia.org/wiki/Gene_set_enrichment_analysis]
- 16. Pathway Enrichment Analysis - TRANSIT's documentation! [https://transit.readthedocs.io/en/v3.3.12/method_pathway_enrichment.html]
- 17. Enrichr - Ma'ayan Lab – Computational Systems Biology [https://maayanlab.cloud/enrichr/]
- 18. Pathway Enrichment Analysis | Reactome [https://www.ebi.ac.uk/training/online/courses/reactome-quick-tour/pathway-enrichment-analysis/]
- 19. Directional integration and pathway enrichment analysis ... [https://www.nature.com/articles/s41467-024-49986-4]
- 20. Pathway enrichment analysis and visualization of omics ... [https://cytoscape.org/cytoscape-tutorials/protocols/enrichmentmap-pipeline/]
- 21. KEGG Mapper Color [https://www.genome.jp/kegg/mapper/color.html]
- 22. Analysis Tools [https://reactome.org/userguide/analysis]
- 23. Reactome Pathway Database: Home [https://reactome.org/]
- 24. Help with Investigating Gene Sets [https://www.gsea-msigdb.org/gsea/msigdb/help_annotations.jsp]
- 25. Help:Guidelines [https://classic.wikipathways.org/index.php/Help:Guidelines]
- 26. Interpreting omics data with pathway enrichment analysis [https://www.sciencedirect.com/science/article/abs/pii/S0168952523000185]
- 27. Hyperpathway: Visualizing Organization of Pathway ... [https://www.preprints.org/manuscript/202508.2227/v1]
- 28. Longitudinal Omics Data Analysis: A Review on Models, ... [https://arxiv.org/html/2506.11161v1]
- 29. Integrating multi-omics and machine learning for disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204941/]
- 30. View topic - 2025-Gupta et al-preprint [https://cytoforum.stanford.edu/viewtopic.php?f=10&t=6465&sid=569281472c2ea69dec31c295d3c40931]
- 31. Integrating Machine Learning and GGE Biplot for ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1647903/full]
- 32. Pathway centric analysis for single-cell RNA-seq and ... [https://www.nature.com/articles/s41467-023-44206-x]
- 33. A critical comparison of topology-based pathway analysis methods - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5784953/]
- 34. SEMgsa: topology-based pathway enrichment analysis with ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04884-8]
- 35. GSEA vs ORA: Two key pathway analysis approach... [https://pluto.bio/resources/Learning%20Series/gsea-vs-ora-two-key-pathway-analysis-approaches-for-next-gen-sequencing-data]
- 36. NeXus: An Automated Platform for Network Pharmacology ... [https://www.mdpi.com/1422-0067/26/22/11147]
- 37. Prediction of response to anti-cancer drugs becomes ... [https://www.nature.com/articles/s41598-019-39019-2]
- 38. Gene Set Enrichment Analysis of Interaction Networks ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7943873/]
- 39. Integrative pathway enrichment analysis of multivariate ... [https://www.nature.com/articles/s41467-019-13983-9]
- 40. GSEA Analysis Tutorial: Gene Set Collections and Best ... [https://bigomics.ch/blog/a-short-guide-to-gene-set-and-pathway-enrichment-analysis/]
- 41. MOMIC: A Multi-Omics Pipeline for Data Analysis, ... [https://www.mdpi.com/2076-3417/12/8/3987]
- 42. Gene-set enrichment analysis and visualization on the web ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12373637/]
- 43. Gene set enrichment analysis - RNA-seq - GitHub Pages [https://alexslemonade.github.io/refinebio-examples/03-rnaseq/pathway-analysis_rnaseq_02_gsea.html]
- 44. g:Profiler – a web server for functional enrichment analysis ... [https://biit.cs.ut.ee/gprofiler/page/docs]
- 45. g:Profiler—a web-based toolset for functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1933153/]
- 46. Enrichr: a comprehensive gene set enrichment analysis web ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4987924/]
- 47. Cytoscape: An Open Source Platform for Complex Network ... [https://cytoscape.org/]
- 48. Proximity Extension Assay (PEA) [https://www.mtoz-biolabs.com/proximity-extension-assay-pea.html]
- 49. Proximity Extension Assay (PEA) – A Guide to Protein ... [https://olinkpanel.creative-proteomics.com/knowledge/pea-technology.html]
- 50. What is PEA? [https://olink.com/technology/what-is-pea]
- 51. Data Preprocessing in Machine Learning: Steps & Best ... [https://lakefs.io/blog/data-preprocessing-in-machine-learning/]
- 52. Data preprocessing: a comprehensive step-by-step guide [https://www.future-processing.com/blog/data-preprocessing-a-comprehensive-step-by-step-guide/]
- 53. gdGSE: An algorithm to evaluate pathway enrichment by ... [https://www.sciencedirect.com/science/article/pii/S200103702500159X]
- 54. Pathway Analysis for Drug Repositioning Based on Public ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3956470/]
- 55. Integrative multi-omics approach for drug repositioning in ... [https://www.sciencedirect.com/science/article/pii/S2950523225000363]
- 56. Pathway Analysis: What It Is and How to Conduct It [https://olvtools.com/en/rnaseq/help/pathway-enrichment-analysis]
- 57. GO vs KEGG vs GSEA: How to Choose the Right ... [https://www.metwarebio.com/go-vs-kegg-vs-gsea-enrichment-analysis/]
- 58. Fundamentals and Visualization of Enrichment Analysis in ... [https://www.protocols.io/view/enrichment-analysis-fundamentals-and-visualization-c5rxy57n.html]
- 59. A Pathway Enrichment Approach Tailored to SomaScan Data [https://pubmed.ncbi.nlm.nih.gov/40789203/]
- 60. Identifying the genetic basis and molecular mechanisms ... [https://elifesciences.org/reviewed-preprints/107957]
- 61. Gene set enrichment analysis: A knowledge-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1239896/]
- 62. GOAT: efficient and robust identification of gene set ... [https://www.nature.com/articles/s42003-024-06454-5]
- 63. ShinyGO 0.85 [https://bioinformatics.sdstate.edu/go/]
- 64. Gene Set Enrichment Analysis (GSEA) User Guide [https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideTEXT.htm]
- 65. Enrichment Analysis Tools | Ma'ayan Laboratory ... [https://labs.icahn.mssm.edu/maayanlab/resources/enrichment/]
- 66. GeneAgent: self-verification language agent for gene-set ... [https://www.nature.com/articles/s41592-025-02748-6]
- 67. Beware of counter-intuitive levels of false discoveries in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12359981/]
- 68. Multiple Testing Correction: Home [https://multipletesting.com/]
- 69. There is life beyond the statistical significance - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8052638/]
- 70. BEYOND THE P-VALUE - Statistical Significance vs ... [https://vsni.co.uk/beyond-the-p-value-statistical-significance-vs-practical-significance/]
- 71. 18. Gene set enrichment and pathway analysis [https://www.sc-best-practices.org/conditions/gsea_pathway.html]
- 72. Pathway Analysis Methods Review [https://advaitabio.com/science/best-pathway-analysis/]
- 73. A comparative study of topology-based pathway enrichment ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3146-1]
- 74. Gene set analysis methods: a systematic comparison [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-018-0166-8]
- 75. Comparison of methods for competitive tests of pathway ... [https://pubmed.ncbi.nlm.nih.gov/22859961/]
- 76. Gene set analysis for self-contained tests: complex null and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3509490/]
- 77. An introduction to effective use of enrichment analysis software [https://humgenomics.biomedcentral.com/articles/10.1186/1479-7364-4-3-202]
- 78. Drug target inference through pathway analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3672337/]
- 79. Pathway Enrichment Analysis - TRANSIT's documentation! [https://transit.readthedocs.io/en/v3.3.11/method_pathway_enrichment.html]
- 80. Text Mining-Based Drug Discovery for Connective Tissue ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2022.743210/full]
- 81. Text Mining-Based Drug Discovery for Connective Tissue ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8971927/]
- 82. Literature Mining for the Discovery of Hidden Connections ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000943]
- 83. A Novel Metric to Quantify the Effect of Pathway Enrichment ... [https://medinform.jmir.org/2021/6/e28247/]