RNA-Seq Validation with qPCR: A Strategic Guide for Biomedical Researchers
This article provides a comprehensive framework for researchers and drug development professionals on the critical role of qPCR in validating RNA-Seq data.
RNA-Seq Validation with qPCR: A Strategic Guide for Biomedical Researchers
Abstract
This article provides a comprehensive framework for researchers and drug development professionals on the critical role of qPCR in validating RNA-Seq data. It explores the foundational reasons for validation, from confirming subtle gene expression changes in heterogeneous diseases like osteoarthritis to meeting publication requirements. The content delves into methodological best practices, including robust experimental design and the use of novel bioinformatics tools for reference gene selection. It further addresses common troubleshooting scenarios and optimization strategies, supported by recent large-scale benchmarking studies. Finally, the article offers a balanced perspective on when validation is essential versus when it may be redundant, empowering scientists to make informed decisions that enhance the reliability and clinical translatability of their transcriptomic findings.
The Unavoidable Why: Core Reasons for qPCR Validation in the RNA-Seq Era
RNA sequencing (RNA-Seq) has become a cornerstone of modern transcriptomics, offering an unbiased, genome-wide view of RNA expression. However, the sophisticated nature of this technology means its results are not infallible; they are contingent upon a complex chain of technical steps, each a potential point of failure. This guide examines the critical technical limitations that can compromise RNA-Seq data reliability and underscores the necessity of orthogonal validation, particularly with qPCR, to ensure robust and reproducible findings, especially in critical fields like drug discovery and clinical diagnostics.
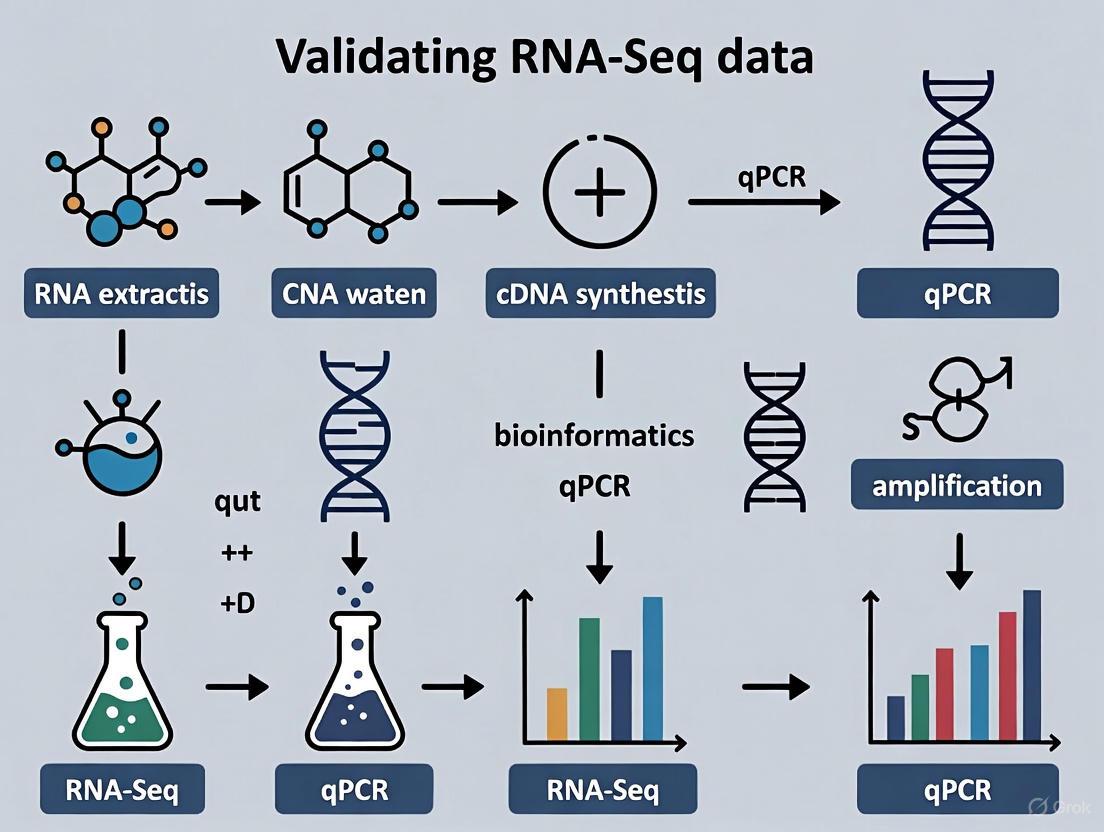
The RNA-Seq Workflow and Its Inherent Vulnerabilities
The process of transforming biological RNA into interpretable sequencing data is a multi-stage pipeline where technical artifacts can be introduced at every step. Understanding these vulnerabilities is the first step toward mitigating their impact. The following diagram outlines a typical bulk RNA-Seq workflow and highlights key points where reliability can falter.
Critical Technical Limitations and Failure Points
RNA-Seq data can be skewed by numerous factors, from the initial sample quality to the final computational decisions. This section details the primary sources of technical bias and error.
Sample Quality and RNA Integrity
RNA quality is the foundational element of a successful sequencing study, and its degradation is a problem that cannot be rectified in downstream analysis [1].
- RNA Integrity Number (RIN): A commonly used metric for RNA quality. While a RIN greater than 7 is generally recommended for high-quality sequencing, this can be challenging to achieve with certain sample types like blood [1]. Degraded RNA, with a low RIN, leads to 3' bias in transcript coverage and poor detection of longer transcripts.
- Impact on Poly-A Selection: Standard mRNA sequencing workflows that use oligo-dT beads to capture polyadenylated RNA are particularly unsuitable for degraded samples, as they rely on an intact poly-A tail [1]. For such samples, methods utilizing random priming and ribosomal RNA (rRNA) depletion are preferred.
- Contamination: The accuracy of RNA quantification and sequencing can be severely affected by contaminants. The 260/280 and 260/230 ratios should be assessed during extraction to ensure minimal protein or DNA contamination [1].
Library Preparation Biases
The process of converting RNA into a sequenceable library is a major source of technical variability.
- Stranded vs. Unstranded Libraries: A key early decision is whether to use a stranded protocol. Stranded libraries preserve the information about which DNA strand was transcribed, which is critical for identifying overlapping genes on opposite strands and for accurate annotation of long non-coding RNAs [1]. While unstranded protocols are simpler and cheaper, they discard this valuable information.
- rRNA Depletion and Off-Target Effects: Ribosomal RNA can constitute up to 80% of cellular RNA. If sequenced, it consumes most of the sequencing reads, dramatically increasing the cost to obtain meaningful data on messenger and non-coding RNAs [1]. Depletion techniques (e.g., using RNAseH) are used to remove rRNA. However, this step is not without its own issues; it can be highly variable between labs and may have off-target effects, inadvertently depleting some genes of interest while enriching others [1].
- Input Material and PCR Amplification: Low input RNA can lead to over-amplification during the library construction PCR. This results in a high duplication rate, where many sequencing reads are PCR copies of the same original fragment, reducing the complexity of the library and potentially skewing quantitative measurements [2].
Normalization and Analytical Challenges
After sequencing, the raw data must be processed and normalized, a step fraught with statistical pitfalls that can lead to incorrect biological conclusions.
- The Normalization Imperative: Raw read counts from RNA-Seq are not directly comparable between samples because the total number of sequenced reads (sequencing depth) varies. A gene in a sample with more total reads will naturally have a higher count, even if its true expression level is identical [3]. Normalization is the mathematical process of correcting for this and other biases.
- Common Normalization Methods: Different methods correct for different types of biases. Simple methods like Counts per Million (CPM) only correct for sequencing depth, while RPKM/FPKM and Transcripts per Million (TPM) correct for both sequencing depth and gene length, making expression levels comparable between different genes within a sample [3]. For differential expression analysis, more advanced methods like the median-of-ratios (DESeq2) and TMM (edgeR) are designed to be robust against the presence of a few highly expressed genes that can distort the count distribution [3].
- Inadequate Experimental Design: The reliability of differential expression analysis is strongly dependent on thoughtful experimental design. A lack of sufficient biological replicates is a common weakness. While three replicates per condition is often considered a minimum, this may be insufficient if biological variability is high [3]. With only two replicates, the ability to estimate variability and control false discovery rates is greatly reduced, and a single replicate does not allow for any robust statistical inference [3].
Table 1: Common Normalization Methods and Their Properties
| Method | Sequencing Depth Correction | Gene Length Correction | Library Composition Correction | Suitable for DE Analysis? | Notes |
|---|---|---|---|---|---|
| CPM | Yes | No | No | No | Simple scaling; heavily affected by highly expressed genes [3]. |
| RPKM/FPKM | Yes | Yes | No | No | Allows within-sample gene comparison; not ideal for cross-sample comparison [3]. |
| TPM | Yes | Yes | Partial | No | An improvement over RPKM/FPKM; better for cross-sample comparisons [3]. |
| median-of-ratios (DESeq2) | Yes | No | Yes | Yes | Robust against composition bias; used for differential expression [3]. |
| TMM (edgeR) | Yes | No | Yes | Yes | Robust against composition bias; used for differential expression [3]. |
Limitations in Transcriptome Characterization
Despite its power, standard short-read RNA-Seq has inherent limitations in resolving complex aspects of transcriptome biology.
- Incomplete Transcriptome Annotation: Probe-based methods (like microarrays) and short-read sequencing can only detect RNAs that have been previously annotated and for which specific probes exist. Novel RNAs, such as those appearing in intergenic and intronic regions, may be sequenced but remain unannotated [1].
- The Isoform Resolution Problem: A significant limitation of short-read RNA-Seq is its difficulty in accurately resolving full-length splice isoforms. While it can detect alternative splicing events, short reads (typically 50-300 bp) must be computationally assembled to reconstruct the complete transcript, which is error-prone for long or complex isoforms [4]. This is a key area where long-read RNA sequencing (e.g., from PacBio or Oxford Nanopore) is transformative, as it enables the end-to-end sequencing of full-length transcripts, providing unambiguous isoform information [4].
The Indispensable Role of qPCR Validation
Given the multitude of technical vulnerabilities in the RNA-Seq pipeline, validation of key results is not merely a best practice but a fundamental requirement for rigorous science.
qPCR serves as a robust orthogonal method to confirm RNA-Seq findings. Its strengths lie in its high sensitivity, specificity, and dynamic range. Unlike RNA-Seq, which provides a relative snapshot of the entire transcriptome, qPCR can be optimized for absolute quantification of a smaller set of critical targets with high precision. Validating a subset of differentially expressed genes identified by RNA-Seq using qPCR boosts confidence in the overall dataset and helps filter out false positives arising from the technical issues described above [5].
A Protocol for Orthogonal Validation
A structured approach to validation ensures that results are comparable and meaningful.
- Gene Selection: Select a panel of target genes (e.g., 5-10) from the RNA-Seq results, including both significantly up-regulated and down-regulated genes.
- Reference Gene Validation: The accuracy of qPCR relies on stable reference genes for normalization. Do not assume traditional "housekeeping" genes (e.g., ACTB, GAPDH) are stable under your specific experimental conditions. Their expression can vary, leading to inaccurate results [5]. Stability should be determined empirically using algorithms like geNorm or NormFinder. RNA-Seq data itself can be mined to identify new, more stable candidate reference genes for a given study context [5].
- cDNA Synthesis: Use the same RNA samples that were submitted for RNA-Seq (or aliquots from the same extraction) to reverse-transcribe RNA into cDNA. This controls for variability originating from the biological source itself.
- qPCR Execution: Perform qPCR in technical replicates for each biological sample. The use of probe-based chemistry (e.g., TaqMan) can offer greater specificity than intercalating dyes.
- Data Analysis and Correlation: Calculate relative expression changes (e.g., using the 2^(-ΔΔCt) method) normalized to the validated reference genes. A strong positive correlation between the fold-change values obtained by RNA-Seq and qPCR confirms the technical reliability of the primary sequencing data.
Essential Research Reagent Solutions
The following table lists key reagents and materials used in RNA-Seq and validation workflows, along with their critical functions.
Table 2: Key Research Reagents for RNA-Seq and Validation
| Reagent / Material | Function | Technical Considerations |
|---|---|---|
| RNA Stabilization Reagents | Preserve RNA integrity immediately upon sample collection (e.g., PAXgene for blood) [1]. | Essential for preserving high-quality RNA, especially from sensitive tissues; prevents degradation-driven bias. |
| rRNA Depletion Kits | Remove abundant ribosomal RNA to enrich for coding and non-coding RNAs of interest [1]. | Choice between probe-based (magnetic beads) and RNase H-based methods involves trade-offs in enrichment efficiency and reproducibility [1]. |
| Stranded Library Prep Kits | Create libraries that retain strand-of-origin information for transcripts [1]. | Preferred for most applications, especially when studying anti-sense transcription or complex genomes. |
| Spike-in Control RNAs | Exogenous RNA added to samples in known quantities [6]. | Used to monitor technical performance, assess dynamic range, sensitivity, and normalize for sample-specific biases. |
| qPCR Assays | Target-specific primers and probes for validating gene expression [5]. | Design should be optimized for efficiency and specificity. Probe-based assays are generally more specific. |
| Validated Reference Genes | Genes with stable expression used for qPCR normalization [5]. | Must be empirically validated for each experimental condition; failure to do so is a major source of error. |
RNA-Seq is a powerful but imperfect tool. Its reliability can falter due to factors ranging from degraded starting material and biased library preparation to improper statistical normalization and inadequate experimental design. In a research landscape increasingly driven by genomic data, particularly in drug discovery and clinical applications, these technical limitations carry significant consequences. Therefore, orthogonal validation of RNA-Seq results, primarily through qPCR, is a non-negotiable step in the scientific process. It transforms a potentially noisy high-throughput dataset into a verified, trustworthy foundation for biological discovery and translational application.
The Imperative for Orthogonal Confirmation in Peer-Reviewed Publication
Next-generation RNA sequencing (RNA-Seq) has unequivocally established itself as the gold standard for whole-transcriptome gene expression analysis in research and clinical applications. Its unparalleled capacity for novel transcript discovery, detection of splice variants, and broad dynamic range has positioned it as a superior alternative to microarray technology [7] [8]. However, this technological supremacy raises a critical methodological question: in an era of sophisticated sequencing platforms, does orthogonal confirmation—particularly through quantitative real-time PCR (qPCR)—remain an essential requirement for peer-reviewed publication? The scientific community exhibits divided opinions on this issue; some researchers consider validation an indispensable step for verifying key findings, while others view it as an unnecessary relic from the microarray era [9]. This guide examines the technical and methodological evidence supporting the continued necessity of orthogonal confirmation, providing researchers with a structured framework for determining when validation is imperative and how to execute it with scientific rigor.
The Concordance Debate: Quantitative Evidence from Platform Comparisons
Establishing the Baseline Correlation Between Platforms
Multiple independent studies have systematically evaluated the correlation between RNA-Seq and qPCR expression measurements, revealing generally high but imperfect concordance. A comprehensive benchmark analysis utilizing whole-transcriptome RT-qPCR data for over 18,000 protein-coding genes demonstrated high expression correlation across five common RNA-Seq workflows, with Pearson correlation coefficients (R²) ranging from 0.798 to 0.845 [7]. Fold-change correlations between RNA-Seq and qPCR were even stronger, with R² values between 0.927 and 0.934, indicating excellent agreement when comparing expression differences between sample conditions [7].
However, these encouraging overall correlations mask critical discrepancies in specific gene subsets. The same study revealed that 15-19% of genes showed non-concordant differential expression results between RNA-Seq and qPCR [7]. While most discrepancies occurred with low fold changes (<2), approximately 1.8% of genes exhibited severe non-concordance, with these problematic genes typically being shorter, having fewer exons, and showing lower expression levels [7].
Comparative Analysis of RNA-Seq and qPCR Performance
Table 1: Concordance Analysis Between RNA-Seq and qPCR Technologies
| Performance Metric | Concordance Level | Problematic Gene Characteristics | Recommended Action |
|---|---|---|---|
| Overall Expression Correlation | R² = 0.80-0.85 [7] | Lower expressed genes | Consider platform-specific bias |
| Fold-Change Correlation | R² = 0.93-0.93 [7] | Genes with FC < 2 | Interpret small fold-changes cautiously |
| Differential Expression Concordance | 81-85% of genes [7] | Shorter genes with fewer exons | Prioritize validation for key short genes |
| Severe Non-Concordance | ~1.8% of genes [7] | Low expression + short length | Essential validation for story-critical genes |
Recent technological comparisons extend beyond qPCR. When evaluating RNA-Seq against established NanoString technology in Ebola-infected non-human primates, researchers observed strong correlation with Spearman coefficients ranging from 0.78 to 0.88 across most samples [10]. This demonstrates that discordance issues are not unique to qPCR but represent broader challenges in transcriptomic measurement consistency.
When Is Orthogonal Confirmation Non-Negotiable?
High-Risk Scenarios Mandating Validation
The collective evidence supports a nuanced, risk-based approach to validation rather than a universal mandate. The following scenarios represent circumstances where orthogonal confirmation becomes essential:
Low-Expression Genes with Critical Findings: When a study's central conclusion depends on differential expression patterns in low-abundance transcripts, qPCR validation is strongly recommended. The benchmark study by Everaert et al. revealed that approximately 93% of non-concordant genes between RNA-Seq and qPCR exhibited fold changes lower than 2, with the most severely discordant genes typically expressed at low levels [9] [7].
Minimal Expression Differences with Biological Significance: Genes displaying small but biologically crucial fold changes (typically <1.5) represent high-risk candidates for misinterpretation without orthogonal confirmation [9].
Foundation of Entire Narratives on Few Genes: When a research story depends entirely on expression patterns of a limited number of genes—particularly if they exhibit the problematic characteristics outlined in Table 1—validation becomes indispensable [9].
Extension to Additional Samples/Conditions: qPCR provides an efficient method to verify RNA-Seq-identified expression patterns across expanded sample sets, additional time points, or related experimental conditions not included in the original sequencing [9].
Validation Exceptions: When RNA-Seq Stands Alone
In contrast, orthogonal confirmation may be unnecessary under these conditions:
State-of-the-Art Experimental and Computational Workflows: When RNA-Seq experiments employ rigorous methodologies, adequate biological replication, and validated analysis pipelines, the resulting data is generally reliable without confirmation [9].
Genome-Wide Discovery Studies: Research focusing on overall transcriptomic patterns rather than individual genes may not require validation, particularly when findings are supported by strong statistical evidence across gene sets [9].
High-Expression Genes with Large Fold Changes: Genes with robust expression levels and substantial differential expression (typically >4-fold) demonstrate high inter-platform concordance and may not necessitate confirmation [7].
Methodological Framework for Technically Sound Validation
Reference Gene Selection: Moving Beyond Traditional Housekeeping Genes
The critical foundation of reliable qPCR validation rests on appropriate reference gene selection. Traditional housekeeping genes (e.g., GAPDH, ACTB) often demonstrate unacceptable expression variability across biological conditions, potentially introducing systematic errors [11] [12]. A superior approach leverages RNA-Seq data itself to identify optimally stable reference genes.
The Gene Selector for Validation (GSV) software implements a rigorous filtering algorithm to identify optimal reference genes based on five criteria applied to Transcripts Per Million (TPM) values from RNA-Seq data [11]:
- Expression >0 TPM across all samples
- Standard variation of log₂(TPM) <1
- No exceptional expression (>2× average of log₂ expression)
- Average log₂ expression >5
- Coefficient of variation <0.2
This methodology successfully identified STAU1 as the most stable reference gene for endometrial decidualization studies, outperforming conventionally used references like β-actin [5]. Similarly, in canine gastrointestinal tissue research, ribosomal protein genes RPS5 and RPL8 demonstrated superior stability compared to traditional references [12].
Table 2: Strategic Selection of Reference Genes for qPCR Normalization
| Selection Method | Advantages | Limitations | Implementation Example |
|---|---|---|---|
| RNA-Seq Based Selection (GSV) | Data-driven, condition-specific | Requires computational processing | Identified STAU1 for decidualization studies [5] |
| Traditional Housekeeping | Familiar, established | Often unstable across conditions | GAPDH, ACTB frequently variable [11] |
| Global Mean Normalization | No single gene bias | Requires large gene sets (>55 genes) | Optimal for profiling 81 genes in canine tissue [12] |
| Ribosomal Proteins | Often highly stable | Potential co-regulation | RPS5, RPL8 best in canine GI study [12] |
Technical Execution of qPCR Validation
The following protocol outlines a rigorous approach for validating RNA-Seq results via qPCR:
Sample Preparation:
- Use identical RNA samples for both RNA-Seq and qPCR validation to eliminate preparation variability [13].
- For formalin-fixed paraffin-embedded (FFPE) samples, use specialized extraction kits (e.g., AllPrep DNA/RNA FFPE Kit) with integrated DNase digestion [14].
- Rigorously assess RNA quality using metrics such as RNA Integrity Number (RIN) prior to analysis [14].
cDNA Synthesis and qPCR Setup:
- Reverse transcribe 0.5μg total RNA using oligo(dT) primers and Supreme Script II reverse transcriptase in 10μL reactions: 42°C for 60 minutes, 70°C for 15 minutes [13].
- Dilute cDNA to 25μL and store at -20°C [13].
- Perform qPCR reactions in 20μL volumes containing: 10μL 2× SYBR Green PreMix, 0.6μL each forward/reverse primer (10μM), 8.7μL RNase-free water, and 0.7μL cDNA template [13].
- Implement three technical replicates for each biological sample to assess technical variability [13].
Primer Design and Validation:
- Design primers with melting temperatures of 57-63°C (optimized to 60°C) and amplicon sizes of 90-180bp [13].
- Validate primer specificity through melt curve analysis, accepting only primers producing single peaks [13].
- Calculate PCR efficiency via standard curve using serial cDNA dilutions [13].
Data Analysis:
- Apply the 2^(-ΔΔCt) method for relative quantification using stable reference genes identified through RNA-Seq analysis [13] [11].
- For large gene sets (>55 genes), consider global mean normalization as a superior alternative to reference genes [12].
Integrated Workflow for Validation Decision-Making
The diagram below illustrates a systematic approach to determining when orthogonal confirmation is necessary:
Essential Research Reagent Solutions
Table 3: Critical Reagents for RNA-Seq Validation Studies
| Reagent/Category | Specific Examples | Function in Workflow | Technical Considerations |
|---|---|---|---|
| RNA Extraction Kits | AllPrep DNA/RNA (Qiagen), EZ1 Advanced XL | Nucleic acid isolation with DNA contamination control | Assess DNA contamination via RSeQC percentage of sense strand reads [14] |
| Library Prep Kits | TruSeq Stranded mRNA (Illumina), SureSelect XTHS2 (Agilent) | RNA-Seq library construction | Quality control via TapeStation, Qubit, LightCycler [14] |
| qPCR Master Mixes | Talent qPCR Premix (SYBR Green) | Amplification detection with SYBR Green chemistry | Verify PCR efficiency (80-110%) [13] [12] |
| Reverse Transcriptase | Superscript II (Thermo Fisher) | cDNA synthesis from RNA templates | Use oligo(dT) priming for mRNA [13] |
| NMD Inhibitors | Cycloheximide (CHX) | Block nonsense-mediated decay for truncating variants | Confirm efficacy via SRSF2 NMD-sensitive transcript [15] |
| Reference Gene Software | GSV, NormFinder, GeNorm | Identify stable reference genes from RNA-Seq data | Apply multiple algorithms for consensus [11] [12] |
Orthogonal confirmation of RNA-Seq findings represents a fundamental principle of rigorous scientific methodology rather than a redundant technical exercise. The evidence clearly demonstrates that while RNA-Seq technologies have achieved remarkable sophistication, strategic validation remains essential for specific high-risk scenarios—particularly when research narratives hinge on few genes, low-expression transcripts, or minimal fold changes with biological significance. By implementing the structured framework, methodological protocols, and analytical tools outlined in this guide, researchers can significantly enhance the reliability, reproducibility, and credibility of their transcriptomic findings. In an era of increasing scrutiny regarding scientific reproducibility, targeted orthogonal confirmation stands as a hallmark of rigorous, publication-ready research.
Bolstering Findings from Small-Scale or Low-Replicate RNA-Seq Studies
RNA sequencing (RNA-seq) has become the cornerstone technology for transcriptome-wide gene expression profiling. However, studies conducted with a small number of biological replicates or on a limited scale present unique challenges for reliable data interpretation. Such studies are often constrained by sample availability, technical resources, or cost, leading to potential issues with statistical power and reproducibility. Within the broader thesis on why validate RNA-Seq with qPCR research, this guide addresses the critical methodologies for bolstering confidence in findings from such constrained experimental designs. The fundamental rationale for validation stems from the distinct technical biases and limitations inherent in both RNA-seq and qPCR methodologies. While RNA-seq provides an unbiased, genome-wide snapshot of transcription, its accuracy can be compromised by factors like alignment errors, sequencing depth, and normalization methods, particularly when biological replication is low. qPCR validation serves as an independent verification using a different technical principle, thereby strengthening the biological conclusions drawn from the initial RNA-seq discovery phase.
Why Validation is Crucial for Underpowered Studies
The Perils of Low Replication
RNA-seq experiments with a small number of biological replicates suffer from reduced statistical power, making it difficult to distinguish true biological variation from technical noise. One study demonstrated that when replication is low, the false-negativity rates of some differential expression analysis methods, such as DESeq2 and the Two-stage Poisson Model (TSPM), can be exceptionally high [16]. This means truly differentially expressed genes (DEGs) are often missed. Conversely, other tools like Cuffdiff2 showed a high false-positivity rate, leading to erroneous identification of DEGs [16]. Validation with qPCR on independent biological samples is the preferred method to confirm true-positive DEGs between biological conditions, as it moves beyond in silico analyses or technical replication using the same RNA samples [16].
The Limits of Sample Pooling
In an effort to reduce costs, some researchers pool biological replicate RNA samples before sequencing. However, experimental evidence has shown that this strategy can introduce a "pooling bias" and often results in a low positive predictive value for the DEGs identified [16]. While pooling may retain biological averaging, it eliminates the ability to estimate biological variance from the sequencing data itself. Compared to sequencing individual biological replicates, analyses of RNA-pools showed weak agreement, undermining their ability to reliably predict true-positive DEGs [16]. Therefore, validation becomes paramount when pooling is used as a cost-saving measure in a study.
Designing a Robust Validation Experiment
When is qPCR Validation Appropriate?
qPCR validation is particularly critical in two key scenarios common to small-scale RNA-seq studies. First, it is essential when a second method is necessary to confirm an observation for which there may be skepticism, such as during the peer-review process for publication. Second, it is highly appropriate when the RNA-seq data is based on a small number of biological replicates where proper statistical tests cannot be robustly applied [17]. In this "cost-savings" mindset, using qPCR to focus on a few interesting targets across more samples is an excellent method for validating the RNA-seq results and building out the study.
The Gold Standard: Independent Biological Replication
The most powerful validation design involves performing qPCR on a new set of RNA samples derived from independent biological replicates, not the same samples used for the RNA-seq [18] [17]. Performing qPCR on the same RNA samples only validates the technology, confirming that two different techniques yield the same result from the same source material. In contrast, performing qPCR on a new set of samples validates not only the technology but also the underlying biological response, providing significantly more confidence in the findings [17].
Table 1: Key Considerations for qPCR Validation of RNA-seq Results
| Consideration | Suboptimal Approach | Recommended Approach |
|---|---|---|
| Sample Selection | Using the same RNA samples for both RNA-seq and qPCR. | Using independent biological replicate samples for qPCR validation [18]. |
| Reference Genes | Selecting traditional "housekeeping" genes (e.g., Actin, GAPDH) based on convention. | Systematically identifying stable, highly-expressed reference genes from the RNA-seq data itself [11]. |
| Candidate Gene Choice | Validating only the most significantly differentially expressed genes. | Including a random selection of DEGs to avoid cherry-picking and assess the false discovery rate [16]. |
Experimental Protocols and Methodologies
Selecting Optimal Reference Genes for qPCR
A critical, often neglected step in qPCR validation is the selection of appropriate reference genes (also known as endogenous controls). Traditionally, housekeeping genes (e.g., actin and GAPDH) and ribosomal proteins have been used based on their presumed stable expression. However, recent work shows these genes can be modulated depending on the biological condition, leading to misinterpretation of results if they are unstable [11]. The development of software like "Gene Selector for Validation" (GSV) allows researchers to systematically identify the most stable and highly expressed genes directly from their RNA-seq dataset to serve as optimal reference genes [11]. The GSV algorithm uses TPM (Transcripts Per Million) values from the RNA-seq data and applies a series of filters to identify genes that are consistently expressed across all samples with low variation, while also filtering out stable genes with low expression that might fall below the detection limit of qPCR [11].
Workflow for End-to-End Validation
The following diagram illustrates a robust workflow for validating a small-scale RNA-seq study, from initial sequencing to final confirmation, incorporating best practices for qPCR validation.
Prioritizing Candidate Genes for Validation
With a typically limited budget for qPCR assays, prioritizing which genes to validate is essential. A novel pipeline has been developed that uses evolutionary conservation and preferential expression of genes across brain tissues to prioritize candidate genes, increasing the translational utility of RNA-seq in model organisms [19]. Furthermore, when selecting variable genes for validation, tools like GSV can filter for genes that are within the detection limit of RT-qPCR and show a considerable difference between samples, ensuring that the chosen candidates are suitable for downstream experimental confirmation [11].
Table 2: Comparison of Common Differential Gene Expression (DEG) Analysis Methods for Low-Replicate Studies
| Method | Reported Performance in Low-Replicate Scenarios | Sensitivity | Specificity | Key Consideration |
|---|---|---|---|---|
| edgeR | High sensitivity and specificity; overall agreement with qPCR was good with a false positivity rate of ~9% [16]. | 76.67% | ~91% | Considered a robust choice for studies with limited replicates [16]. |
| Cuffdiff2 | High false-positivity rate; contributed 87% of false positive DEGs in one validation study [16]. | 51.67% | N/A | Use with caution; high risk of identifying false DEGs [16]. |
| DESeq2 | High specificity but very low sensitivity; identified only a single DEG in one 8-replicate study [16]. | 1.67% | 100% | High false-negativity rate; may miss many true DEGs [16]. |
| TSPM | High false-negativity rate; performance is highly dependent on the number of replicates [16]. | ~5% | ~91% | Not recommended for studies with very low replication [16]. |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for RNA-seq Validation
| Item | Function / Application | Key Considerations |
|---|---|---|
| Total RNA Isolation Kit | Extraction of high-quality RNA from biological samples. | Ensure high RNA Integrity Number (RIN >7.0) [20]. Use kits that effectively remove genomic DNA. |
| mRNA Enrichment Kit | Selection of polyadenylated mRNA for RNA-seq library prep. | Poly(A) selection is common but can introduce 3' bias. rRNA depletion provides broader transcriptome coverage. |
| Stranded cDNA Library Prep Kit | Construction of sequencing-ready libraries from RNA. | Stranded protocols preserve information on the originating strand of the transcript. |
| qPCR Master Mix | Amplification and fluorescence-based quantification of cDNA. | Use kits with high efficiency and a wide dynamic range. SYBR Green or probe-based chemistries are standard. |
| Molecular Grade Water | A nuclease-free solvent for preparing RNA and PCR reagents. | Essential for preventing RNase-mediated degradation and ensuring reaction specificity. |
| Validated Primers or Probes | Sequence-specific amplification of target and reference genes. | Design for high amplification efficiency (~90-110%). Test for specificity (e.g., single peak in melt curve). |
Findings from small-scale or low-replicate RNA-seq studies can be significantly bolstered through a rigorous and well-designed qPCR validation strategy. This involves moving beyond the same samples used for sequencing to test independent biological replicates, systematically selecting stable reference genes from the transcriptomic data, and being aware of the performance characteristics of different DEG analysis tools. By integrating these practices, researchers can enhance the reliability, credibility, and translational potential of their research, transforming a preliminary transcriptomic finding into a robust biological conclusion.
Enhancing Confidence for Clinical Application and Drug Development
RNA sequencing (RNA-seq) has become a foundational tool in biomedical research for genome-wide expression profiling. However, its transition from a research tool to a method informing clinical decisions and drug development pipelines demands rigorous validation to ensure results are reliable, reproducible, and actionable. Orthogonal validation, particularly using reverse transcription quantitative PCR (RT-qPCR), provides this critical confidence. While RNA-seq is robust, studies reveal that a small but significant fraction of results can be non-concordant with RT-qPCR findings, especially for lowly expressed genes or those with small fold-changes [9]. This technical guide outlines the necessity, frameworks, and methodologies for validating RNA-seq data, providing researchers and drug development professionals with a roadmap to enhance the credibility of their transcriptomic findings for preclinical and clinical applications.
Why Validate RNA-Seq? Evidence from the Field
The assumption that RNA-seq is inherently reliable requires careful examination, as the consequences of inaccurate data are magnified in clinical and drug development contexts. A comprehensive benchmark study analyzing over 18,000 human genes found that depending on the bioinformatics pipeline, 15–20% of genes were "non-concordant" between RNA-seq and RT-qPCR results [9]. Although the vast majority of these non-concordant cases involved genes with low expression or small fold-changes (<2), approximately 1.8% of genes showed severe discrepancies. This evidence underscores that RNA-seq, while powerful, is not infallible.
Validation becomes paramount in specific scenarios:
- For Key Findings: When a biological conclusion or clinical hypothesis rests on the differential expression of a handful of genes.
- For Low-Abundance Transcripts: When targeting genes with low expression levels, where technical noise is more pronounced.
- For Small Fold-Changes: When biological effects are subtle but purported to be significant.
- For Bridging Studies: When RNA-seq findings from discovery cohorts need to be confirmed in larger validation cohorts using a more accessible and cost-effective method [9].
The transition of RNA-seq into the clinical diagnostic arena further highlights its validated utility. For instance, in oncology, combining RNA-seq with whole exome sequencing (WES) in a cohort of 2,230 tumor samples improved the detection of clinically actionable gene fusions and recovered variants missed by DNA-only testing [14]. In rare Mendelian disorders, clinical RNA-seq tests have been developed that can provide a functional basis for reclassifying variants of uncertain significance, thereby increasing diagnostic yields [21] [22]. These advanced clinical applications were contingent upon extensive analytical and clinical validation, establishing a precedent for any serious translational research endeavor.
Clinical Validation Frameworks for RNA-Seq
Implementing RNA-seq in a regulated environment requires a structured validation framework that moves beyond simple correlation studies. The following table summarizes key performance metrics and benchmarks from established clinical RNA-seq studies:
Table 1: Analytical Performance Benchmarks from Clinical RNA-Seq Validations
| Validation Component | Sample Type(s) | Key Metrics and Benchmarks | Reference |
|---|---|---|---|
| Comprehensive Diagnostic Test | Fibroblasts, Blood (130 samples) | Established gene-/junction-specific reference ranges from control data; tested on 40 positive controls with known diagnostic findings. | [22] |
| Integrated Tumor Portrait | Fresh Frozen and FFPE Tumors (2230 samples) | Analytical validation using reference samples with 3042 SNVs and 47,466 CNVs; orthogonal confirmation in patient samples. | [14] |
| Minimally Invasive Rare Disease | Peripheral Blood Mononuclear Cells (PBMCs) | Expression of ~80% of intellectual disability/epilepsy panel genes; ability to detect splicing defects and NMD. | [21] |
These studies demonstrate that a robust clinical validation strategy typically involves multiple steps:
- Analytical Validation: Using reference materials and cell lines to determine the accuracy, precision, and sensitivity of the assay in detecting expression outliers and splicing defects [14] [22].
- Orthogonal Confirmation: Using an independent method (like RT-qPCR) on a subset of patient samples to verify key findings [14].
- Clinical Utility Assessment: Applying the assay to large, real-world patient cohorts to demonstrate its ability to uncover biologically and clinically relevant alterations that would have been missed otherwise [14] [21].
A critical challenge in diagnostic RNA-seq is tissue-specific gene expression. For example, one study found that even in commonly used clinically accessible tissues like blood and fibroblasts, over 37% and 48% of coding genes, respectively, can have low expression (TPM < 1), potentially limiting their assessability [22]. This underscores the need for validation studies to be performed in the specific tissue relevant to the disease or drug target.
A Technical Protocol for RT-qPCR Validation
RT-qPCR remains the gold standard for gene expression validation due to its high sensitivity, specificity, reproducibility, and wide adoption in clinical settings [23] [9]. The following workflow outlines the key steps for a robust validation experiment.
Selection of Reference and Target Genes
The selection of appropriate genes is the most critical step for a successful validation.
- Reference Gene Selection: Traditional housekeeping genes (e.g.,
GAPDH,ACTB) are often unstable across different biological conditions. Software tools like Gene Selector for Validation (GSV) can systematically identify the most stable and highly expressed reference genes directly from the RNA-seq dataset itself [11]. Ideal reference genes should have low variability (standard deviation of log2(TPM) < 1), high expression (average log2(TPM) > 5), and a low coefficient of variation (< 0.2) across all samples in the study [11]. - Target Gene Selection: For validating differentially expressed genes, select candidates that represent a range of expression levels and fold-changes. The GSV tool can also identify highly variable genes suitable for validation [11]. In a colorectal cancer study, the genes
HPGD,PACS1, andTDP2were selected from RNA-seq data and successfully validated using Taqman qPCR as prognostic biomarkers in patient plasma [23].
Detailed Experimental Workflow
- RNA Extraction and QC: Use dedicated kits for your sample type (e.g., miRNeasy Serum/Plasma Kit for cell-free RNA [23], RNeasy kits for tissues/cells [24] [22]). Assess RNA quantity and quality using a fluorometer (e.g., Qubit) and an instrument like the TapeStation to ensure RNA Integrity Number (RIN) ≥ 7 [24] [22].
- Reverse Transcription: Synthesize complementary DNA (cDNA) from 500 ng of total RNA using a high-quality kit like the SuperScript VILO cDNA Synthesis Kit, which includes primers for random hexamers and oligo(dT) to ensure comprehensive coverage [25].
- qPCR Reaction:
- Chemistry: Use either SYBR Green or TaqMan chemistry. SYBR Green is more cost-effective but requires careful optimization and validation of primer specificity. TaqMan probes offer greater specificity and are preferred in clinical settings [23].
- Protocol: Perform reactions in a 20-μL volume using a master mix like PowerUp SYBR Green or TaqMan Fast Advanced Master Mix. Run samples in technical replicates on a real-time PCR instrument (e.g., QuantStudio 3) [25].
- Primers/Probes: Use commercially available TaqMan Gene Expression Assays or carefully designed, validated primers [23] [25].
Data Analysis and Interpretation
The standard method for analysis is the comparative Ct (ΔΔCt) method [23] [25]:
- Calculate ΔCt = Ct(target gene) - Ct(reference gene)
- Calculate ΔΔCt = ΔCt(test sample) - ΔCt(control sample)
- The fold change is expressed as 2^(-ΔΔCt)
Finally, use statistical tests (e.g., one-sample t-tests on log2 fold-change values against a test value of zero) to determine if the observed expression changes are significant [23]. A successful validation is demonstrated by a strong correlation between the fold-changes observed in RNA-seq and those confirmed by RT-qPCR.
The Scientist's Toolkit: Essential Reagents and Kits
Table 2: Key Research Reagent Solutions for RNA-seq Validation
| Item | Function | Example Products & Kits |
|---|---|---|
| RNA Extraction Kits | Isolate high-quality, intact total RNA from diverse sample types. | RNeasy Mini/Fibrous Tissue Kits (Qiagen) [24] [22], miRNeasy Serum/Plasma Kit (Qiagen) [23], AllPrep DNA/RNA FFPE Kit (Qiagen) [14] |
| Reverse Transcription Kits | Synthesize stable cDNA from RNA templates for downstream qPCR. | SuperScript VILO cDNA Synthesis Kit (Thermo Fisher) [25], PrimeScript RT Master Mix (Takara) [23] |
| qPCR Master Mixes | Provide optimized buffers, enzymes, and dyes for efficient and specific amplification. | PowerUp SYBR Green Master Mix (Thermo Fisher) [25], TaqMan Fast Advanced Master Mix (Thermo Fisher) [23] |
| Gene Expression Assays | Ensure specific detection and quantification of target transcripts. | TaqMan Gene Expression Assays (Applied Biosystems) [23], designed primer pairs for SYBR Green |
| Nucleic Acid QC Instruments | Accurately assess RNA concentration, purity, and integrity. | Qubit Fluorometer (Thermo Fisher) [14] [22], TapeStation System (Agilent) [14] [24], Fragment Analyzer (Agilent) [24] |
In the high-stakes fields of clinical application and drug development, assuming the absolute accuracy of a single omics technology is a significant risk. A robust framework that integrates RNA-seq discovery with RT-qPCR confirmation creates a foundation of verifiable data upon which sound biological conclusions, diagnostic tests, and therapeutic decisions can be built. By adhering to structured validation protocols, leveraging appropriate bioinformatic tools for gene selection, and utilizing trusted reagent solutions, researchers can enhance confidence in their data, ultimately accelerating the translation of genomic discoveries into tangible clinical benefits.
From Data to Validation: A Methodological Roadmap for Reliable qPCR
The emergence of RNA sequencing (RNA-seq) has revolutionized transcriptomics, providing an unprecedented platform for genome-wide expression profiling without the probe-specific biases that historically limited microarray technologies [26] [9]. However, this powerful technique introduces new analytical challenges, particularly regarding the validation of findings through orthogonal methods like quantitative real-time PCR (qPCR). While some researchers argue that RNA-seq's probe-independent nature eliminates the need for validation, evidence indicates that significant technical variability can occur throughout the extended RNA-seq workflow, from sample preparation through data analysis [26] [9]. This variability necessitates a rigorous approach to confirming results, especially when studies rely on the differential expression of a limited number of genes or when findings have substantial clinical or therapeutic implications.
Within this validation framework, the selection of appropriate reference genes (also termed housekeeping genes) for qPCR normalization emerges as a critical pre-analytical step that fundamentally determines the reliability and interpretability of validation results. Reference genes serve as internal controls to correct for technical variations in RNA integrity, cDNA synthesis efficiency, and enzymatic amplification [27] [28]. The fundamental assumption is that these genes maintain constant expression across all experimental conditions and tissue types. However, numerous studies have conclusively demonstrated that no single reference gene displays universal stability [29] [28]. The expression of commonly used housekeeping genes, such as β-actin (ACTB) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), can vary significantly across different tissues, developmental stages, and experimental conditions [30] [28]. Consequently, the improper selection of reference genes represents a pervasive source of inaccuracy that can compromise the validation of RNA-seq data, potentially leading to false conclusions and irreproducible findings.
This technical guide provides a comprehensive framework for the identification and validation of stable reference genes derived directly from RNA-seq data, ensuring the reliability of downstream qPCR validation experiments. By establishing rigorous pre-validation protocols, researchers can enhance the credibility of their transcriptomic studies and strengthen the biological conclusions drawn from integrated genomic analyses.
Computational Identification of Candidate Reference Genes from RNA-Seq Data
The initial phase of selecting stable reference genes begins with a systematic computational analysis of RNA-seq data. This process leverages the comprehensiveness of transcriptomic datasets to identify genes with inherently stable expression patterns across the specific experimental conditions under investigation.
Data Preprocessing and Quality Control
Before evaluating gene expression stability, raw RNA-seq data must undergo stringent quality control and processing. The standard workflow includes adapter trimming, quality filtering, and alignment of reads to a reference genome using tools such as STAR aligner [22] [14]. Following alignment, gene-level quantification is performed using tools like HTSeq or RNA-SeQC to generate raw count data or normalized expression values such as Transcripts Per Million (TPM) [20] [22]. These steps are crucial for ensuring that subsequent stability analyses are based on accurate and reliable expression measurements. Researchers should also assess RNA integrity numbers (RIN), sequence coverage depth, and alignment rates to confirm data quality before proceeding to stability analysis [22].
Selection Criteria for Candidate Genes
When identifying potential reference genes from RNA-seq data, several key characteristics should be considered:
Moderate Expression Levels: Candidates should exhibit neither extremely high nor extremely low expression, as both extremes can introduce normalization artifacts. Genes with average TPM values between 100 and 1000 often represent suitable candidates [27].
Low Inter-Sample Variation: Look for genes with consistently stable expression across all samples in the dataset, as measured by low coefficient of variation (CV) in TPM or count values.
Established Housekeeping Genes: Include traditionally used reference genes (e.g., ACTB, GAPDH, ribosomal proteins) for comparative analysis, while recognizing they may not be optimal in all contexts [30] [29].
Biological Function: Prefer genes involved in core cellular processes such as cytoskeletal maintenance, basic metabolism, or protein synthesis, as these are more likely to maintain stable expression [29].
Table 1: Example Candidate Reference Genes Identified from RNA-Seq Studies Across Species
| Organism | Stable Genes Identified | Unstable Genes | Citation |
|---|---|---|---|
| Sweet Potato | IbACT, IbARF, IbCYC | IbGAP, IbRPL, IbCOX | [27] |
| Honeybee | arf1, rpL32 | α-tubulin, GAPDH, β-actin | [30] |
| Guava | PgTUB1, PgEF1a, PgEF2 | PgRBP47 | [29] |
| Human PBMCs | RPL13A, S18, SDHA | IPO8, PPIA | [31] |
| Small Ruminants | B2M, PPIB, BACH1, ACTB | RPS15, RPLP0, TBP | [28] |
Statistical Analysis for Stability Ranking
After identifying an initial set of candidate genes, researchers should employ dedicated algorithms to quantitatively assess and rank their expression stability. The following statistical tools are widely used in combination for this purpose:
GeNorm: This algorithm calculates a gene expression stability measure (M) for each candidate gene based on the average pairwise variation between all genes in the analysis. Genes with lower M values demonstrate higher stability. GeNorm also determines the optimal number of reference genes required for accurate normalization [27] [29].
NormFinder: This method employs a model-based approach to evaluate expression stability while considering both intra-group and inter-group variations, making it particularly valuable for studies involving multiple sample groups or treatments [27] [28].
BestKeeper: This algorithm utilizes pairwise correlation analysis to assess the stability of candidate genes based on the geometric mean of their Cq values, providing a complementary perspective to variance-based methods [27] [31].
ΔCt Method: This comparative approach evaluates expression stability by calculating the pairwise variability between different candidate genes, with lower variability indicating higher stability [31] [30].
RefFinder: This comprehensive tool integrates results from all the aforementioned algorithms (GeNorm, NormFinder, BestKeeper, and ΔCt method) to generate a overall stability ranking, providing a robust consensus for candidate gene selection [27] [30].
The following diagram illustrates the complete computational workflow for identifying candidate reference genes from raw RNA-seq data:
Experimental Validation of Selected Reference Genes
Following the computational identification of candidate reference genes, laboratory-based validation is essential to confirm their stability under specific experimental conditions. This multi-stage process transitions from in silico predictions to empirical verification.
Primer Design and Validation
The initial wet-lab phase requires careful primer design and validation for each candidate reference gene:
Design Specifications: Primers should amplify 80-200 bp products spanning exon-exon junctions where possible to minimize genomic DNA amplification. The amplicon should have a Tm of approximately 60°C with minimal primer-dimer formation or secondary structure [29].
Validation Protocol: Each primer pair requires validation through a standard curve analysis using serial dilutions of cDNA. Key parameters include:
- Amplification Efficiency: Ideally between 90-110% [29]
- Correlation Coefficient (R²): >0.980 indicating linearity
- Specificity: Confirmed by melt curve analysis with a single peak
Documentation: Comprehensive records of primer sequences, amplification conditions, and validation parameters should be maintained in accordance with MIQE guidelines [9].
qPCR Experimental Design and Execution
The validation experiment must be carefully designed to accurately assess reference gene stability:
Sample Selection: Include representative samples spanning all experimental conditions, tissues, and time points relevant to the planned studies. Biological replicates are essential – typically at least three independent replicates per condition [26].
qPCR Protocol: Perform qPCR reactions using consistent thermal cycling conditions across all candidate genes. Include appropriate controls (no-template controls, reverse transcription controls) to identify potential contamination or amplification artifacts.
Data Collection: Record quantification cycle (Cq) values using consistent threshold settings across all plates. Manual inspection of amplification curves is recommended to identify any irregularities that might affect Cq accuracy [27] [29].
Table 2: Essential Research Reagents for Reference Gene Validation
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| RNA Isolation Kits | RNeasy Mini Kit (Qiagen), AllPrep DNA/RNA Kit (Qiagen), PicoPure RNA Isolation Kit (Thermo Fisher) | High-quality RNA extraction from various sample types including cells, tissues, and FFPE samples [20] [22] [14] |
| Reverse Transcription Kits | NEBNext Poly(A) mRNA Magnetic Isolation Module, High-Capacity cDNA Reverse Transcription Kit | cDNA synthesis from RNA templates with high efficiency and reproducibility [20] [22] |
| qPCR Master Mixes | SYBR Green Master Mix, TaqMan Gene Expression Master Mix | Fluorescence-based detection of amplified DNA during qPCR cycles [31] [28] |
| Library Prep Kits | TruSeq Stranded mRNA Kit (Illumina), NEBNext Ultra DNA Library Prep Kit | Preparation of sequencing libraries for RNA-seq analysis [20] [14] |
Stability Confirmation and Final Selection
The final validation stage involves analyzing the qPCR data to confirm the stability of candidate reference genes:
Re-analysis with Validation Algorithms: Process the experimentally derived Cq values using the same stability algorithms employed for the RNA-seq data (GeNorm, NormFinder, BestKeeper, RefFinder) [27] [31]. This generates an empirical stability ranking based on actual qPCR data.
Concordance Assessment: Compare the computationally predicted stability rankings from RNA-seq data with the experimentally derived rankings from qPCR. High concordance between these datasets validates the computational approach and confirms the suitability of selected reference genes.
Validation with Target Genes: As a functional test, use the top-ranked reference genes to normalize the expression of target genes with known expression patterns. Successful reproduction of expected expression patterns confirms the utility of the selected reference genes [30] [28].
The following workflow diagram outlines the complete experimental validation process:
Implementation Guidelines for Reliable Gene Expression Studies
The successful identification and validation of stable reference genes culminates in their practical implementation for normalizing qPCR data in target gene expression studies. This section outlines evidence-based recommendations for optimal utilization of reference genes across diverse research contexts.
Determining the Optimal Number of Reference Genes
A critical consideration in reference gene implementation is determining how many are necessary for reliable normalization. The geNorm algorithm provides a systematic approach to this question by calculating the pairwise variation (Vn/Vn+1) between sequential normalization factors [27] [29]. A commonly applied threshold is V < 0.15, indicating that the inclusion of an additional reference gene does not significantly improve normalization accuracy. Most studies find that 2-3 validated reference genes are sufficient for robust normalization across diverse experimental conditions [27] [28]. Using a single reference gene is generally discouraged unless its stability has been extensively documented in the specific experimental system under investigation.
Context-Dependent Selection and Application
Reference gene stability is inherently context-dependent, necessitating careful consideration of experimental variables:
Tissue-Specific Considerations: Genes stable in one tissue type may be unsuitable for others. For example, in sweet potato, IbACT demonstrated high stability across multiple tissues, while IbCOX showed significant variability [27]. Similarly, different gene combinations were optimal for antennae, hypopharyngeal glands, and brains in honeybee studies [30].
Experimental Conditions: Environmental factors, treatments, and developmental stages profoundly influence gene stability. In hypoxic conditions, RPL13A, S18, and SDHA emerged as stable reference genes for PBMCs, while IPO8 and PPIA performed poorly [31]. Physiological adaptations in small ruminants reared at high-altitudes necessitated distinct reference gene panels (B2M, PPIB, BACH1, ACTB) compared to traditional options [28].
Species-Specific Factors: Cross-species application of reference genes requires validation. While some genes (e.g., elongation factors, ribosomal proteins) frequently demonstrate stability across taxa, empirical confirmation is essential [30] [29] [28].
Integration with RNA-Seq Validation Frameworks
The selection of stable reference genes represents a foundational element in comprehensive RNA-seq validation protocols. When determining whether qPCR validation is necessary for RNA-seq findings, researchers should consider these evidence-based guidelines:
Validation Recommended: When studies rely on a limited number of key genes for biological conclusions; when RNA-seq identifies subtle expression changes (less than 2-fold); when investigating low-abundance transcripts; or when extending findings to additional sample types not included in the original RNA-seq experiment [26] [9].
Validation Optional: When RNA-seq data are derived from multiple biological replicates (minimum of three) showing strong concordance; when studying highly abundant transcripts with large expression differences; or when conducting purely exploratory analyses without immediate functional implications [26] [9].
Recent comprehensive analyses indicate that approximately 1.8% of genes show severe non-concordance between RNA-seq and qPCR results, with these typically being lower expressed, shorter transcripts [9]. This underscores the particular importance of validation for studies focusing on such problematic genes.
The systematic approach to selecting and validating stable reference genes outlined in this technical guide provides a critical foundation for robust gene expression studies. By leveraging RNA-seq data as a starting point for identifying candidate genes, followed by rigorous experimental validation using multiple algorithmic approaches, researchers can significantly enhance the reliability of qPCR-based confirmation of transcriptomic findings. This pre-validation paradigm represents a essential component of methodologically sound molecular research, ensuring that biological conclusions rest upon technically solid analytical frameworks. As transcriptomic technologies continue to evolve and find new applications in both basic research and clinical diagnostics, the principles of rigorous reference gene selection will remain fundamental to generating reproducible, scientifically valid gene expression data.
The reliability of any RNA sequencing (RNA-Seq) study, and by extension the justification for its validation via quantitative PCR (qPCR), rests upon a foundation of rigorous experimental design. A poorly designed RNA-Seq experiment can yield misleading results, rendering subsequent qPCR validation inefficient or scientifically questionable. This guide details the core principles of experimental design power—specifically focusing on biological replication, controls, and sample splitting—to ensure that RNA-Seq data is robust, reproducible, and worthy of downstream validation. The relationship between RNA-Seq and qPCR is not merely sequential but deeply interconnected; a well-powered RNA-Seq experiment provides the credible differential expression targets that make qPCR validation a meaningful confirmatory step [32]. Challenges such as technical biases in RNA-seq [32] and the inherent complexity of transcriptome-wide data [3] make a strategic design not just beneficial, but essential for generating actionable biological insights, particularly in critical fields like drug discovery [6].
The Cornerstone of Power: Biological Replication
Why Biological Replicates Are Non-Negotiable
In the context of RNA-Seq, a "biological replicate" is defined as an RNA sample collected from an independently processed biological unit within a treatment group. For example, cells from different animals, separately passaged cell cultures, or distinct human donors all constitute biological replicates [6] [33]. Their primary purpose is to capture the natural biological variability that exists within the population being studied, allowing researchers to distinguish consistent treatment effects from random individual variation [6] [33].
The power of a statistical test is its probability of correctly detecting a true effect, such as a genuinely differentially expressed gene. Underpowered experiments, often due to insufficient replication, are a primary cause of false negatives and irreproducible results [34]. Biological replication is the single most critical factor for improving statistical power in RNA-Seq experiments [35]. Simulations and empirical studies have consistently shown that allocating resources to increase the number of biological replicates provides a greater boost to power than increasing sequencing depth beyond a reasonable level [35]. One study found that sequencing depth could be reduced to as low as 15% in some scenarios without a substantial negative impact on false positive or true positive rates, provided sufficient biological replication was maintained [35].
Determining the Number of Biological Replicates
The choice of the number of biological replicates is a balance between statistical ideals and practical constraints. While two replicates per condition is the absolute minimum for any statistical comparison, it provides very low power and poor estimation of variability [3]. As shown in the table below, a minimum of three biological replicates is often considered a baseline, but larger numbers are strongly recommended for reliable results.
Table 1: Guidelines for Biological Replication in RNA-Seq Experiments
| Scenario | Recommended Minimum Replicates | Rationale and Considerations |
|---|---|---|
| General Standard/Pilot Studies | 3-5 per condition [3] [6] | Provides a baseline for estimating variability and enables rudimentary statistical testing. |
| Experiments with High Biological Variability | 6-12 per condition [6] | Necessary for complex tissues, human patient samples, or heterogeneous cell populations to achieve sufficient power. |
| Experiments with Low Variability | 4-8 per condition [6] | Inbred animal models, cell lines, or clonal populations may require fewer replicates, but more is always beneficial. |
| For Robust Detection of Small Effect Sizes | 10+ per condition [33] | Detecting subtle expression changes requires greater power, which is directly achieved by increasing replicates. |
Control Strategies and Sample Splitting
Designing Effective Experimental Controls
Controls are the benchmark against which experimental effects are measured. A carefully considered control strategy is vital for attributing observed changes in gene expression to the experimental intervention rather than confounding factors.
- Treatment vs. Control Groups: The most fundamental design compares a treated group to an untreated control. "No treatment" controls should be handled identically to the treated samples, while "mock" controls (e.g., adding a solvent like DMSO) account for the vehicle's effects [6].
- Spike-In Controls: Synthetic RNA molecules (e.g., SIRVs) added in known quantities to each sample before library preparation are invaluable. They serve as an internal standard to monitor technical performance across the entire workflow, allowing for assessment of sensitivity, dynamic range, and quantification accuracy [6]. This is particularly crucial for large-scale studies where batch effects are a concern.
- Pilot Studies: A small-scale pilot experiment is highly recommended before committing to a full-scale study. It provides critical preliminary data on biological variability, which is essential for performing a formal sample size calculation. It also allows for validation of wet-lab and data analysis workflows [6] [33].
Sample Splitting, Randomization, and Batch Effects
How samples are assigned to processing groups and sequenced is as important as the samples themselves. Failure to properly split and randomize samples can introduce "batch effects"—systematic technical variations that are confounded with biological groups and can utterly invalidate results.
- Randomization: The assignment of samples to treatment groups, as well as the order of all downstream processes (library preparation, sequencing lane assignment), must be randomized [33]. For example, all control samples should not be processed on one day and all treated samples on another. Bench scientists must randomize the order of sample processing in the lab to avoid confounding effects with time or location (e.g., well position in a multi-well plate) [33].
- Blocking and Batch Correction: When full randomization is impossible (e.g., due to large sample numbers), a blocked design should be used. This involves distributing samples from all experimental groups evenly across processing batches (e.g., different sequencing lanes or days). This design allows for statistical methods to later "correct" for the technical batch effect during data analysis [6].
- Sample Splitting Diagram: The following workflow visualizes the key steps for properly splitting and processing samples to minimize bias.
Table 2: Key Research Reagent Solutions for RNA-Seq Experimental Design
| Tool / Reagent | Primary Function | Application in Experimental Design |
|---|---|---|
| RNA Extraction Kit (e.g., RNeasy, AllPrep) [32] [22] | Isolation of high-quality RNA from cells or tissues. | The choice of kit depends on sample type (e.g., FFPE, blood, cells) and whether concurrent DNA extraction is needed. Consistent use is critical. |
| Spike-In RNA Controls (e.g., SIRVs, ERCC) [6] | Exogenous RNA transcripts added to each sample. | Provides an internal standard for normalizing technical variation and assessing assay performance across batches and runs. |
| Stranded mRNA/Total RNA Library Prep Kit [14] [22] | Converts RNA into a sequencing-ready library. | Selection depends on RNA integrity (e.g., FFPE vs. fresh frozen), need for ribosomal RNA depletion, and the RNA species of interest (e.g., mRNA vs. non-coding). |
| Quality Control Instruments (Qubit, TapeStation, Bioanalyzer) [14] [22] | Quantifies and assesses the integrity of nucleic acids. | Essential quality gates before proceeding to costly library preparation; ensures input material is of sufficient quality and quantity. |
| Statistical Power Analysis Software (e.g., Scotty, pwr) [3] [35] | Calculates necessary sample size prior to the experiment. | Uses pilot data or estimates of effect size and variability to determine the number of biological replicates needed to avoid underpowered studies. |
Connecting Experimental Design to qPCR Validation
The ultimate test of RNA-Seq data quality is often its concordance with an orthogonal, sensitive method like qPCR. The correlation between RNA-Seq and qPCR expression estimates is not always perfect, with studies reporting moderate correlations (e.g., Spearman's rho between 0.2 and 0.53 for HLA genes) [32]. This highlights that technical differences between the platforms can influence results.
A powerful and well-controlled RNA-Seq design directly addresses these challenges and strengthens the validation phase in several ways:
- Identifying True Positives: By adequately powering the study with biological replicates, the list of differentially expressed genes sent for qPCR validation is enriched with true biological effects, rather than technical artifacts or false discoveries.
- Providing a Biological Context: The replication and blocking design that captures biological variability ensures that the expression changes measured by qPCR are representative of the population, not just idiosyncratic to a single sample.
- Informing qPCR Design: The RNA extraction methods, sample types, and biological conditions optimized for the RNA-Seq study can be directly mirrored in the qPCR validation, ensuring a fair and meaningful comparison between the two platforms.
In conclusion, a rigorous focus on biological replication, strategic controls, and unbiased sample splitting is not merely a preliminary step but the very foundation upon which credible RNA-Seq results are built. This robust foundation is what makes the subsequent investment in qPCR validation a scientifically justified and valuable endeavor, ultimately leading to more reliable and translatable biological conclusions.
In the modern genomics landscape, RNA sequencing (RNA-Seq) has become the cornerstone technology for comprehensive gene expression profiling. However, the journey from raw biological sample to robust, interpretable data begins long before sequencing commences. The initial wet lab phase—encompassing RNA extraction, handling, and quality control (QC)—is a critical determinant of success for all downstream applications, from discovery-phase RNA-Seq to targeted validation using quantitative PCR (qPCR). This foundational stage establishes the integrity of the transcriptional snapshot, ensuring that the resulting data accurately reflects the biological state under investigation.
The imperative for rigorous QC is further amplified when research aims to bridge high-throughput discovery with focused validation. Within the context of a broader thesis on validating RNA-Seq with qPCR, the reliability of the initial RNA sample is the common thread that unites these techniques. High-quality RNA extracted with precision provides a solid substrate not only for a successful RNA-Seq library but also for the subsequent qPCR assays that will confirm key findings. This guide provides an in-depth technical overview of the core principles and practices for navigating the wet lab workflow from RNA extraction to quality assessment, providing researchers with the knowledge to generate data that is both technically sound and biologically meaningful.
RNA Quality Metrics: RIN and DV200
The assessment of RNA integrity is a non-negotiable first step in any transcriptomic study. Two primary metrics, the RNA Integrity Number (RIN) and the DV200 value, are routinely used to quantify RNA quality, each with distinct strengths and optimal applications.
The RNA Integrity Number (RIN) is an algorithm-assigned score ranging from 1 (completely degraded) to 10 (perfectly intact). It is generated by an Agilent Bioanalyzer system and evaluates the entire electrophoretic trace of an RNA sample, including the presence and ratios of ribosomal RNA peaks. Traditionally, a RIN value greater than 7.0 is considered suitable for standard RNA-Seq workflows [36].
The DV200 metric represents the percentage of RNA fragments that are longer than 200 nucleotides. This metric has gained prominence, particularly for partially degraded samples, such as those derived from formalin-fixed paraffin-embedded (FFPE) tissues or post-mortem sources, because it focuses on the size distribution of fragments that are actually usable in library construction [36] [37]. Recent research highlights DV200 as a more accurate predictor of successful RNA-seq outcomes in degraded or post-mortem samples compared to RIN [36].
The table below summarizes the typical quality thresholds for different downstream applications:
Table 1: RNA Quality Thresholds for Downstream Applications
| Application | Recommended RNA Input | Recommended Quality Metric | Minimum Threshold | Ideal Range |
|---|---|---|---|---|
| Stranded mRNA Seq | ≥800 ng total [38] | RIN [38] | RIN > 5.5 [38] | RIN > 7.0 [38] |
| Total RNA Seq | ≥500 ng total [38] | RIN [38] | RIN > 3.5 [38] | Not Specified |
| Transcriptome Capture(e.g., for FFPE/low-quality RNA) | ≥1 µg total [38] | DV200 [38] | DV200 > 30% [38] | Higher DV200 values correlate with greater sequencing output [36] |
A comparative study on post-mortem human liver tissue demonstrated that samples with a mean DV200 of 63.81% and a mean RIN of 7.14—harvested within 10 hours post-mortem—were consistently suitable for next-generation RNA sequencing [36]. Furthermore, the study found a significant positive correlation between higher DV200 values (70-80%) and the total number of bases sequenced, highlighting its utility as a predictive metric for sequencing efficiency [36].
RNA Extraction and QC: Detailed Experimental Protocols
RNA Extraction Methodologies
A robust RNA extraction protocol is fundamental. The methodology must be tailored to the sample type (e.g., fresh tissue, blood, FFPE). Below is a generalized protocol, with notes on adaptations.
Protocol: Guanidinium-Thiocyanate Phenol-Chloroform Extraction (e.g., TRIzol)
This method is effective for a wide variety of sample types, including cells and tissues, due to its ability to rapidly inactivate RNases.
- Homogenization: Homogenize 50-100 mg of tissue or cell pellet in 1 mL of TRIzol reagent. Use a mechanical homogenizer for tough tissues. For liquid samples like plasma, a dedicated kit like the miRNeasy Serum/Plasma Kit is more appropriate [23].
- Phase Separation: Incubate the homogenate for 5 minutes at room temperature to dissociate nucleoprotein complexes. Add 0.2 mL of chloroform per 1 mL of TRIzol used. Cap the tube securely, shake vigorously for 15 seconds, and incubate at room temperature for 2-3 minutes.
- Centrifugation: Centrifuge the mixture at 12,000 × g for 15 minutes at 4°C. The solution will separate into three phases: a red organic phase (phenol-chloroform), an interphase (DNA), and a colorless upper aqueous phase (RNA).
- RNA Precipitation: Carefully transfer the aqueous phase to a new tube without disturbing the interphase. Precipitate the RNA by mixing with 0.5 mL of isopropyl alcohol per 1 mL of TRIzol used. Incubate at room temperature for 10 minutes.
- RNA Pellet: Centrifuge at 12,000 × g for 10 minutes at 4°C. The RNA will form a gel-like pellet on the side and bottom of the tube.
- Wash: Carefully remove the supernatant. Wash the RNA pellet with 1 mL of 75% ethanol (in RNase-free water) per 1 mL of TRIzol used. Vortex briefly and centrifuge at 7,500 × g for 5 minutes at 4°C.
- Redissolution: Air-dry the pellet briefly for 5-10 minutes (do not let it dry completely, as this reduces solubility). Dissolve the RNA in 20-50 µL of RNase-free water.
Quality Control Assessment Workflow:
The following diagram illustrates the logical workflow for assessing RNA quality post-extraction, leading to the decision on its suitability for downstream applications.
Determining DV200 Values
The DV200 value is calculated using automated electrophoresis systems from Agilent Technologies. The general procedure is as follows [37]:
Protocol: DV200 Determination on Agilent Systems
- System Setup: Use either the 2100 Bioanalyzer system (with 2100 Expert software), TapeStation systems (with TapeStation Analysis software), or the Fragment Analyzer systems (with ProSize data analysis software).
- Load and Run Sample: Follow the manufacturer's instructions for the specific RNA assay (e.g., RNA Nano, RNA Pico) to load the RNA sample and run the analysis.
- Apply DV200 Calculation:
- For Bioanalyzer: Import the appropriate DV200 assay file (.xsy) into the software to apply the calculation to your data file. The DV200 value will be displayed in the results [37].
- For TapeStation: In the "Regions" settings, define a new region with a lower limit of 200 nucleotides and an upper limit (e.g., 10,000 nt). Name the region "DV200". The value is provided as a percentage of the total signal in this region [37].
- For Fragment Analyzer: In the ProSize software, use the "Edit Configurations" menu to perform a "Smear Analysis" between 200 nt and the upper limit. The DV200 value will be displayed in the "% total" column [37].
The Research Reagent Toolkit
Successful execution of RNA workflows relies on a suite of essential reagents and kits. The following table details key solutions and their specific functions in the process.
Table 2: Essential Research Reagents for RNA Workflows
| Reagent / Kit Name | Function / Application | Specific Example or Note |
|---|---|---|
| TRIzol LS Reagent | RNA isolation from liquid samples like plasma or serum; maintains RNA integrity during storage [23]. | Used for stabilizing plasma samples before cfRNA extraction in biomarker studies [23]. |
| miRNeasy Serum/Plasma Kit | Spin-column based purification of cell-free RNA (cfRNA) and microRNA from plasma/serum [23]. | Critical for isolating cfRNA for circulating biomarker research, as used in colorectal cancer studies [23]. |
| Agilent RNA Assays | Quality control using the Bioanalyzer, TapeStation, or Fragment Analyzer to generate RIN and DV200 metrics [37]. | The DV200 metric is particularly crucial for assessing FFPE-derived or partially degraded RNA [37]. |
| PrimeScript RT Master Mix | Reverse transcription for cDNA synthesis from purified RNA templates [23]. | Used in validation workflows to prepare samples for qPCR analysis [23]. |
| TaqMan Gene Expression Assays | Target-specific qPCR probes and primers for precise gene expression quantification [23]. | Enables validation of RNA-Seq results for specific genes of interest (e.g., HPGD, PACS1) [23]. |
| TaqMan Fast Advanced Master Mix | qPCR reaction mix optimized for fast, sensitive, and reliable detection of target genes [23]. | Used with TaqMan assays for high-quality qPCR data generation in validation studies [23]. |
Connecting RNA QC to RNA-Seq and qPCR Validation
The quality of the starting RNA material has a direct and profound impact on the reliability of both RNA-Seq and qPCR results. High-quality RNA ensures that the transcriptomic profile generated by RNA-Seq is an accurate representation of the biological sample. When RNA integrity is compromised, biases can be introduced; for example, degradation can lead to under-representation of the 5' ends of transcripts during library preparation, skewing expression estimates [36].
This established link is the very reason why qPCR validation remains a critical step in many research pipelines. While RNA-Seq is powerful for hypothesis generation, qPCR serves as an orthogonal method to confirm key findings with high sensitivity and specificity. The relationship between these techniques, underpinned by initial RNA quality, is summarized in the following workflow:
qPCR validation is particularly appropriate in two key scenarios: first, when a second, orthogonal method is required to confirm an observation for publication, and second, when the initial RNA-Seq data is based on a small number of biological replicates, making statistical conclusions less robust [17]. Using qPCR on a new set of samples with proper biological replication not only validates the technology but also confirms the underlying biological response [17]. A 2025 study on colorectal cancer biomarkers successfully exemplified this pipeline: they identified candidate cfRNAs via RNA-Seq and then validated their prognostic significance using Taqman qPCR in a larger, independent patient cohort [23].
Navigating the wet lab from RNA extraction to quality control is a disciplined process that forms the bedrock of any credible transcriptomic study. A thorough understanding and meticulous application of quality metrics like RIN and DV200 enable researchers to make informed decisions about sample suitability, directly influencing the success of downstream RNA-Seq and the reliability of subsequent qPCR validation. By adhering to robust protocols and utilizing the appropriate reagent toolkit, scientists can ensure that their data, from high-throughput sequencing to targeted quantification, is built upon a foundation of technical rigor, thereby maximizing the biological insights and impact of their research.
The validation of RNA-Sequencing (RNA-seq) findings through reverse transcription quantitative PCR (RT-qPCR) remains a critical step in ensuring the reliability of gene expression studies. A major bottleneck in this process is the robust selection of reference genes, which are traditionally chosen based on their presumed stable expression, often leading to misinterpretation of results. This whitepaper details how the bioinformatics software Gene Selector for Validation (GSV) automates and optimizes the selection of both reference and variable candidate genes from transcriptome data. We provide a comprehensive technical guide on GSV's methodology, present experimental protocols for its validation, and frame its utility within the broader context of RNA-seq verification, offering drug development professionals and researchers a standardized, time-efficient pipeline to enhance the accuracy of their gene expression analyses.
RNA Sequencing (RNA-seq) has become the method of choice for transcriptome-wide gene expression profiling, generating vast and complex datasets. While the technology is powerful, the question of whether its results require independent verification is a persistent consideration in the research community. Orthogonal validation, particularly using RT-qPCR, is often employed to confirm key findings, as it provides high sensitivity, specificity, and reproducibility [9]. However, the reliability of RT-qPCR is entirely contingent upon the use of properly validated reference genes—genes with stable, high expression across the biological conditions under study [39] [11].
The conventional practice of selecting reference genes based solely on their function as housekeeping genes (e.g., ACTB, GAPDH) is fraught with risk. Numerous studies have demonstrated that the expression of these traditional genes can be significantly modulated under different experimental or pathological conditions [11]. When an unstable reference gene is used for normalization, it introduces errors in the quantification of target genes, potentially leading to erroneous biological interpretations [39] [9]. The Gene Selector for Validation (GSV) tool was developed to address this fundamental challenge. By leveraging pre-existing RNA-seq data, GSV provides a data-driven, automated approach to identify the most stable reference genes and the most promising variable genes for validation, thereby strengthening the entire gene expression analysis pipeline [39] [11] [40].
GSV is a software tool developed in Python that transforms a quantitative transcriptome (in the form of Transcripts Per Million, or TPM, values) into curated lists of optimal candidate genes for RT-qPCR validation [11] [41].
Core Algorithm and Workflow
The algorithm of GSV follows a filtering-based methodology that operates on log2-transformed TPM values. Its workflow bifurcates to select for two distinct types of candidate genes: reference candidates (highly expressed, stable genes) and validation candidates (highly expressed, variable genes) [11]. The logical flow of the algorithm is illustrated below.
Input and Output Specifications
GSV is designed for practicality, accepting multiple common file formats.
- Input: The software requires a table of TPM values where rows represent genes and columns represent different RNA-seq libraries or samples. It accepts single files in
.xlsx,.xls,.csv, or.txtformats. It can also process multiple output files (.sf) directly from the Salmon quantification software, automatically handling technical replicates if they are appropriately named [41]. - Output: GSV generates two primary outputs: 1) a ranked list of reference candidate genes that exhibit high stability and expression, and 2) a list of validation candidate genes that show high expression and significant variability across conditions. These results can be saved in
.xlsx,.xls, or.txtformat for further analysis [41].
Filtering Criteria and Mathematical Foundation
The power of GSV lies in its sequential application of stringent filters. The table below details the mathematical criteria used for selecting reference genes, which ensure the identification of genes that are both stable and highly expressed enough to be reliably detected by RT-qPCR.
Table 1: GSV Filtering Criteria for Reference Gene Selection [11]
| Filter Step | Criterion | Mathematical Formula | Biological & Technical Rationale |
|---|---|---|---|
| 1. Presence | Expression > 0 | TPM_i > 0 for all libraries (i) |
Ensures the gene is expressed in all analyzed conditions. |
| 2. Variability | Low Variation | σ(log2(TPM_i)) < 1 |
Selects genes with low standard deviation in log2 expression across samples. |
| 3. Outlier | No Exceptional Expression | |log2(TPM_i) - mean(log2(TPM))| < 2 |
Removes genes with extreme expression in any single library. |
| 4. Expression | High Expression Level | mean(log2(TPM)) > 5 |
Guarantees the gene is expressed at a level easily detectable by RT-qPCR. |
| 5. Consistency | Low Coefficient of Variation | σ(log2(TPM_i)) / mean(log2(TPM)) < 0.2 |
A relative measure of stability, further refining the candidate list. |
For variable gene selection, the process is more streamlined, focusing on high expression (mean(log2(TPM)) > 5) and high variability (σ(log2(TPM_i)) > 1) to find genes suitable for testing differential expression [11].
Experimental Protocol: From RNA-seq to qPCR Validation
The following section provides a detailed, step-by-step protocol for using GSV to select and validate candidate genes, using the published Aedes aegypti case study as a guide [39] [11].
Phase 1: RNA-seq Data Preprocessing and GSV Analysis
- Transcriptome Quantification: Process raw RNA-seq reads (from any major sequencing platform [42]) through a standard bioinformatics pipeline. Align reads to the reference genome and perform transcript-level quantification using tools like Salmon or kallisto to generate a count matrix. Normalize raw counts to Transcripts Per Million (TPM).
- Data Formatting: Format the TPM matrix into a table where rows are genes, columns are biological samples (averaging technical replicates if necessary), and cells contain TPM values. Save this table in a
.csvor.xlsxformat. - GSV Execution:
- Download the GSV executable for Windows 10 from the GitHub repository [41].
- Launch
GeneSelectorforValidation.exeand load the formatted TPM table. - Configure the input settings (e.g., column name for gene identifiers).
- Run the analysis using the recommended default filter values for optimal results.
- Save the two output lists: stable reference candidates and variable validation candidates.
Phase 2: Wet-Lab Validation via RT-qPCR
- Candidate Gene Selection: From the GSV output, select the top 3-5 ranked reference genes and a set of 5-10 variable genes of biological interest for experimental validation.
- RNA Extraction and cDNA Synthesis: Using the same biological samples from the RNA-seq experiment, extract total RNA, ensuring high purity (A260/A280 ~1.8-2.0). Treat with DNase I to remove genomic DNA contamination. Synthesize cDNA using a reverse transcriptase kit with oligo(dT) and/or random hexamer primers.
- qPCR Assay Design and Optimization: Design and validate primer pairs for each candidate gene with an efficiency between 90% and 110%. Perform qPCR reactions in triplicate on a real-time PCR instrument.
- Stability Analysis: Input the resulting quantification cycle (Cq) values for the reference candidates into stability analysis software such as geNorm, NormFinder, or BestKeeper. These programs will calculate a stability measure (M) and rank the genes from most to least stable [11].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents and Tools for RNA-seq Validation via GSV and RT-qPCR
| Item | Function / Description | Example Kits / Software |
|---|---|---|
| RNA Extraction Kit | Isolates high-quality, intact total RNA from biological samples. | Qiagen RNeasy Kit, TRIzol Reagent |
| RNA-seq Library Prep Kit | Prepares sequencing libraries from RNA; choice depends on input amount and platform. | NEBNext Ultra II Directional RNA Library Prep [42] |
| Sequencing Platform | Generates raw RNA-seq reads. | Illumina NovaSeq Series, Element Biosciences AVITI, Singular Genomics G4 [42] |
| Quantification Software | Generates TPM values from raw sequencing reads. | Salmon, kallisto |
| Gene Selection Software | Analyzes TPM data to select optimal reference and validation genes. | Gene Selector for Validation (GSV) [41] |
| Reverse Transcriptase Kit | Synthesizes cDNA from RNA template for qPCR. | High-Capacity cDNA Reverse Transcription Kit |
| qPCR Master Mix | Contains enzymes, dNTPs, and buffer optimized for quantitative PCR. | SYBR Green or TaqMan Master Mix |
| Stability Analysis Software | Analyzes Cq values to confirm the stability of reference genes. | geNorm, NormFinder, BestKeeper [11] |
Case Study and Performance Benchmarking
Application inAedes aegyptiResearch
In a real-world application, GSV was used to analyze an Aedes aegypti transcriptome. The software successfully identified eIF1A and eIF3j as the top-ranked stable reference genes. Subsequent RT-qPCR analysis confirmed that these GSV-selected genes were more stable than traditionally used reference genes like ribosomal proteins (e.g., RpL32, RpS17) [39] [11]. This finding highlights a critical point: genes presumed to be stable can be suboptimal in specific experimental contexts, and their use can compromise data interpretation. GSV's data-driven approach mitigates this risk.
Comparative Analysis with Other Tools
GSV was benchmarked against other software using synthetic datasets. Its key advantage lies in its integrated filtering for both stability and expression level. Unlike other tools such as NormFinder or geNorm, which are designed to analyze Cq values from RT-qPCR and can only handle a limited number of genes, GSV is built specifically for RNA-seq data and can process entire transcriptomes [11]. Crucially, GSV's requirement for a high mean log2(TPM) (Filter 4) automatically filters out stable but lowly expressed genes that would be poor candidates for RT-qPCR due to assay detection limits. This results in a more reliable and practical list of reference genes.
Table 3: Comparison of GSV with Other Gene Selection and Analysis Tools
| Feature | GSV | geNorm / NormFinder | OLIVER |
|---|---|---|---|
| Primary Data Input | RNA-seq (TPM values) | RT-qPCR (Cq values) | Microarray or RT-qPCR (Cq values) |
| Scope of Analysis | Whole transcriptome | Limited, user-predefined set of genes | Larger than geNorm, but not whole transcriptome |
| Key Differentiator | Integrated filtering for expression level, preventing selection of stable, low-expression genes. | Analyzes stability only, regardless of initial expression level in the transcriptome. | Command-line based, less user-friendly interface. |
| Output | Ranked lists of reference AND variable genes. | Stability ranking of input reference genes. | Ranking of candidate genes from input data. |
Integration into a Broader Thesis on RNA-seq Validation
The implementation of GSV directly addresses a core question in the thesis of RNA-seq validation: When and how is validation necessary? While RNA-seq is a robust technology, a 2021 study noted that approximately 1.8% of genes, typically those with low expression and small transcript size, can show severe "non-concordance" between RNA-seq and RT-qPCR results [9]. This justifies targeted validation, especially when a study's conclusions hinge on the expression patterns of a few key genes.
GSV enhances the validation workflow by making it systematic and cost-effective. It moves validation away from a perfunctory, often poorly executed step, to a rigorous, data-driven process. By ensuring the selection of optimal reference genes, GSV directly improves the accuracy and reliability of the RT-qPCR data used for validation. Furthermore, by creating a shortlist of variable genes, it focuses costly and time-consuming wet-lab experiments on the most promising candidates, maximizing research efficiency [39] [40]. For drug development professionals, this translates into increased confidence in gene expression biomarkers and therapeutic targets identified via RNA-seq.
The bioinformatics software GSV represents a significant advancement in the pipeline for gene expression analysis. It automates a critical and often neglected step—the rational selection of reference and validation genes for RT-qPCR—based on the evidence contained within the researcher's own RNA-seq dataset. By providing a method that is both computationally robust and experimentally practical, GSV reduces the potential for error, saves time and resources, and ultimately fortifies the conclusions drawn from transcriptomic studies. Its successful application in model organisms and its ability to handle large datasets make it an indispensable tool for researchers and drug developers aiming to translate RNA-seq discoveries into validated biological insights.
Troubleshooting the Process: Overcoming Common Validation Pitfalls
The reproducibility of scientific findings is a cornerstone of biomedical research, yet it remains a significant challenge, particularly in preclinical life science research. Concerns about a "reproducibility crisis" have been raised following reports that in-house target validation reproduced only 20-25% of findings from 67 preclinical studies, with similar low success rates in validating preclinical cancer targets [43]. This crisis erodes public trust and wastes hundreds of millions of pounds in research funding [43].
Translating research discoveries into clinical applications requires ensuring that experimental results are reliable and consistent across different laboratories and technical platforms. This is especially critical for genomic technologies like RNA sequencing (RNA-seq), which are increasingly used in clinical diagnostics. Benchmarking studies that systematically evaluate inter-laboratory performance provide invaluable insights into the sources of variability and strategies for mitigation. This article explores lessons from large-scale benchmarking studies, specifically focusing on why validating RNA-seq with qPCR remains an essential practice in rigorous scientific research.
The Challenge of Inter-Laboratory Variability in RNA-Seq
RNA-seq has become a fundamental tool for exploring global gene expression patterns. However, when used to detect clinically relevant subtle differential expression—such as minor expression changes between different disease subtypes or stages—its technical limitations become apparent [44]. Subtle differential expression is particularly challenging to distinguish from the technical noise inherent to RNA-seq protocols [44].
A landmark multi-center RNA-seq benchmarking study across 45 laboratories revealed the extent of this challenge [44]. Using reference samples from the Quartet project, researchers systematically assessed performance across 26 experimental processes and 140 bioinformatics pipelines. The findings demonstrated "greater inter-laboratory variations in detecting subtle differential expressions" compared to samples with larger biological differences [44]. Primary sources of variation included:
- Experimental factors: mRNA enrichment protocols and library strandedness
- Bioinformatics steps: Each computational step in the analysis pipeline introduced variability
The study further highlighted the challenge of quality assessment, showing that quality metrics based on samples with large biological differences (like the MAQC samples) may not ensure accurate identification of clinically relevant subtle differential expression [44]. This underscores the necessity for more sensitive quality controls tailored to detecting minor expression changes.
qPCR as a Validation Standard: Comparative Performance
Quantitative PCR has traditionally served as the gold standard for gene expression quantification due to its sensitivity, reproducibility, and precision. The technical consistency of qPCR across laboratories is significantly enhanced through the use of standardized reference materials, as demonstrated by interlaboratory studies of Standard Reference Material 2917 (NIST SRM 2917) [45].
When 14 laboratories repeatedly measured 12 different qPCR assays using the same reference material, researchers found that "SRM 2917 allows for reproducible single-instrument run calibration models across laboratories, regardless of qPCR assay" [45]. The use of a reliable calibrant combined with protocol standardization improved qPCR measurement precision both within and between laboratories.
Table 1: Comparative Method Performance in Inter-Laboratory Studies
| Performance Metric | RNA-Seq (45 labs) | qPCR (14 labs) |
|---|---|---|
| Inter-lab variation in detecting subtle expression | Significant | Minimal with standardized reference materials |
| Primary variability sources | Experimental protocols (mRNA enrichment, strandedness) and bioinformatics pipelines | Assay design and calibration methods |
| Impact of standardization | Moderate (improves but doesn't eliminate variability) | High (dramatically improves inter-lab consistency) |
| Correlation with orthogonal methods | Variable correlation with qPCR (see Table 2) | High correlation with digital PCR [45] |
Direct comparisons between qPCR and RNA-seq reveal more complex relationships. A study analyzing HLA class I gene expression observed only "moderate correlation between expression estimates from qPCR and RNA-seq for HLA-A, -B, and -C (0.2 ≤ rho ≤ 0.53)" [32]. This discrepancy highlights the technical challenges specific to RNA-seq quantification of highly polymorphic genes and suggests that qPCR provides complementary rather than redundant information.
Table 2: RNA-seq and qPCR Correlation for HLA Class I Genes
| HLA Gene | Correlation Coefficient (rho) |
|---|---|
| HLA-A | 0.2 ≤ rho ≤ 0.53 |
| HLA-B | 0.2 ≤ rho ≤ 0.53 |
| HLA-C | 0.2 ≤ rho ≤ 0.53 |
Large-scale benchmarking studies have systematically identified the major contributors to inter-laboratory variability. The Quartet project's comprehensive analysis revealed that both experimental and computational factors significantly impact results [44].
Experimental Process Variability
The 45-laboratory study identified 26 different experimental processes contributing to variability. Key factors included:
- mRNA enrichment methods: Different protocols for RNA selection introduced systematic biases
- Library preparation protocols: Particularly stranded versus non-stranded methods
- Sequencing platforms: Different instruments and sequencing chemistries
- Batch effects: Sixteen laboratories introduced additional variability by distributing libraries across different flowcells or lanes [44]
Bioinformatics Pipeline Variability
The same study assessed 140 different analysis pipelines, finding that each computational step introduced variability [44]. Factors included:
- Gene annotation databases: Different reference annotations affected gene quantification
- Alignment algorithms: Three different genome alignment tools produced varying results
- Quantification methods: Eight different quantification tools with six normalization methods
- Differential analysis tools: Five different statistical packages for identifying differentially expressed genes
Similar variability has been observed in other omics fields. A large-scale benchmarking of circular RNA detection tools revealed that while "tool-specific precision is high and similar (median of 98.8%, 96.3% and 95.5% for qPCR, RNase R and amplicon sequencing, respectively)... the sensitivity and number of predicted circRNAs are the most significant differentiators" [46]. Different tools detected dramatically different numbers of circRNAs, ranging from 1,372 to 58,032 [46].
Best Practices for Minimizing Variability and Ensuring Reproducibility
Based on findings from large-scale benchmarking studies, researchers can adopt several strategies to minimize inter-laboratory variability and improve reproducibility.
Experimental Design Recommendations
- Implement reference materials: Use well-characterized reference samples like the Quartet or MAQC materials for quality control [44]
- Standardize protocols: Where possible, use consistent library preparation and sequencing protocols across collaborating laboratories
- Include technical replicates: Multiple replicates help distinguish technical variability from biological signals
- Control for batch effects: Process samples randomly and document potential batch effects
Bioinformatics Best Practices
- Pipeline transparency: Document all computational steps, parameters, and software versions
- Use established benchmarks: Select tools that perform well in benchmarking studies for specific applications
- Multiple normalization strategies: Compare results across different normalization methods
- Code and data sharing: Make analysis code and processed data available to enable reproducibility assessment
Quality Control Measures
- Orthogonal validation: Always validate key findings using orthogonal methods like qPCR [32]
- Signal-to-noise assessment: Use PCA-based signal-to-noise ratio to evaluate data quality [44]
- Data acceptance metrics: Establish minimum quality thresholds based on reference materials
The critical importance of orthogonal validation is further supported by circular RNA detection studies, which found that "precision values are lower when evaluating low-abundance circRNAs" [46]. This parallels the challenge of detecting subtle differential expression in RNA-seq data and underscores why qPCR validation remains essential, particularly for low-expression targets.
Table 3: Key Research Reagent Solutions for Minimizing Inter-Laboratory Variability
| Reagent/Resource | Function | Example/Benefit |
|---|---|---|
| Reference Materials | Quality control and calibration | Quartet project RNA samples, MAQC samples, NIST SRM 2917 for qPCR [44] [45] |
| Standardized Protocols | Experimental consistency | Identical library prep, sequencing, and analysis protocols across labs [44] |
| Cell Line Authentication | Ensuring biological material identity | STR profiling to confirm cell line identity [43] |
| ERCC Spike-in Controls | Technical control for RNA-seq | 92 synthetic RNA controls to monitor technical performance [44] |
| Quality-Checked Biologicals | Reproducible experimental materials | Certified cell lines with sterility, species ID, and mycoplasma testing [43] |
Large-scale benchmarking studies have unequivocally demonstrated that inter-laboratory variability represents a significant challenge in genomic research, particularly for sensitive techniques like RNA-seq. The 45-laboratory Quartet study revealed substantial variability in detecting subtle differential expression, stemming from both experimental and bioinformatics factors [44]. While standardization and improved computational methods can mitigate some variability, orthogonal validation using established methods like qPCR remains essential.
The consistent finding of only moderate correlation between RNA-seq and qPCR for challenging gene targets [32] underscores why qPCR validation should not be viewed as redundant but rather as a necessary component of rigorous study design. As the scientific community continues to address reproducibility challenges, the integration of standardized reference materials, transparent reporting, and orthogonal validation will be crucial for generating reliable, translatable research findings.
Future efforts should focus on developing more comprehensive reference materials, establishing field-specific best practices, and creating computational frameworks that explicitly account for technical variability. Through these coordinated efforts, the research community can enhance reproducibility and accelerate the translation of scientific discoveries into clinical applications.
The Challenge of Low-Expression Genes and Subtle Differential Expression
RNA sequencing (RNA-seq) has become the gold standard for whole-transcriptome gene expression quantification, offering an unbiased view of the transcriptome with a broad dynamic range [7]. However, the accurate identification of differentially expressed genes (DEGs), particularly those with low expression levels or subtle changes, remains technically challenging. The presence of noisy, low-expression genes can significantly decrease the sensitivity of DEG detection, potentially obscuring biologically relevant findings [47] [48]. These challenges necessitate rigorous validation strategies to ensure the reliability of RNA-seq results, especially in critical applications such as biomarker discovery and drug development.
Quantitative PCR (qPCR) retains its status as the gold standard for validating gene expression data due to its high sensitivity, specificity, and reproducibility [11]. This technical guide explores the specific challenges associated with low-expression genes and subtle differential expression in RNA-seq analysis and provides detailed methodologies for proper experimental design and validation protocols to ensure research robustness.
Technical Challenges in Detecting Subtle Expression Changes
The Impact of Low-Expression Genes on Analysis
Low-expression genes present a significant challenge in RNA-seq data analysis because their signal may be indistinguishable from sampling noise [47] [48]. Research has demonstrated that the presence of these noisy genes can decrease the sensitivity of detecting DEGs. Filtering of low-expression genes is often necessary to improve DEG detection sensitivity, but this process requires careful optimization [47].
The optimal threshold for filtering low-expression genes is not universal but depends on specific RNA-seq pipeline factors. Studies have shown that transcriptome reference annotation, expression quantification method, and DEG detection method are statistically significant factors that affect the optimal filtering threshold [47] [48]. The filtering threshold that maximizes the total number of DEGs closely corresponds to the threshold that maximizes DEG detection sensitivity [47].
Methodological Biases in Expression Quantification
Systematic discrepancies between quantification technologies can significantly impact the detection of subtle expression changes. A comprehensive benchmarking study revealed that a small but specific gene set consistently shows inconsistent expression measurements between RNA-seq and qPCR across different processing workflows [7]. These problematic genes are typically characterized by shorter length, fewer exons, and lower expression levels compared to genes with consistent expression measurements [7].
Table 1: Characteristics of Genes with Inconsistent Expression Measurements Between RNA-seq and qPCR
| Characteristic | Trend in Inconsistent Genes | Impact on Detection |
|---|---|---|
| Gene Length | Significantly shorter | Reduced read coverage |
| Exon Count | Fewer exons | Less efficient detection |
| Expression Level | Lower expressed | Higher technical variance |
| Dynamic Range | Limited fold changes | Challenging statistical detection |
Another significant challenge emerges in the analysis of highly polymorphic gene families, such as the human leukocyte antigen (HLA) genes. The extreme polymorphism at HLA genes creates technical difficulties for RNA-seq quantification due to challenges in aligning short reads to a reference genome that doesn't fully represent HLA allelic diversity [32]. This can result in moderate correlations between RNA-seq and qPCR (0.2 ≤ rho ≤ 0.53 for HLA class I genes), highlighting the necessity of validation for such genetically variable targets [32].
Experimental Design for Robust Detection
Replication Strategies and Sequencing Depth
Proper experimental design is paramount for reliably detecting subtle expression changes. The number of biological replicates significantly influences statistical power more than simply increasing sequencing depth. Studies have demonstrated that biological replicates are essential for accurate variance estimation and identifying genuine expression changes [49]. While pooled designs may reduce costs, maintaining separate biological replicates is ideal for detecting subtle changes [49].
Technical variation in RNA-seq experiments stems from multiple sources, with library preparation identified as the largest source of technical variation [49]. To mitigate these effects:
- Randomize samples during preparation
- Normalize concentrations across samples
- Utilize indexing and multiplexing to distribute samples across sequencing lanes
- Employ blocking designs when complete multiplexing isn't possible [49]
For sequencing parameters, both read length and sequencing depth must be optimized. While longer reads can improve mapping accuracy, sufficient depth is necessary to capture low-abundance transcripts. The optimal balance depends on the specific research goals and organism complexity.
Normalization and Statistical Methods
Normalization is a critical pre-processing step that serves to modulate values so they are directly comparable across samples [50]. The two most widely used normalization methods are:
- Trimmed Mean of M-values (TMM) : Used by edgeR, this method assumes most genes are not differentially expressed and estimates normalization factors to adjust for differences in library size and composition between samples [50].
- Geometric Mean : Used by DESeq2, this method involves calculating the geometric mean of expression values for each gene across all samples, adjusting for variations in sequencing depth and distributional differences [50].
For statistical analysis of differential expression, DESeq2 and edgeR are the most widely used tools, both utilizing negative binomial models to account for overdispersion in count data [50] [51]. DESeq2 is often preferred for its more robust handling of low-count genes and improved variability estimates, while edgeR uses empirical Bayes methods to stabilize variability estimates [51].
Validation Frameworks and Protocols
Reference Gene Selection for Accurate Normalization
The selection of appropriate reference genes is fundamental for reliable qPCR validation. Traditional housekeeping genes (e.g., actin and GAPDH) and ribosomal proteins are commonly used but may exhibit variable expression under different biological conditions [11]. Research has shown that these traditionally used genes can be modulated depending on the biological context, potentially leading to misinterpretation of results if used indiscriminately [11].
The GSV software tool provides a systematic approach for identifying optimal reference genes directly from RNA-seq data [11]. The algorithm applies stringent criteria to select stable, highly expressed genes suitable for qPCR normalization:
- Expression greater than zero in all libraries
- Low variability between libraries (standard deviation of log2(TPM) < 1)
- No exceptional expression in any library (within 2-fold of average)
- High expression level (average log2(TPM) > 5)
- Low coefficient of variation (< 0.2) [11]
This methodology was successfully applied to identify STAU1 as a stable reference gene for studies of endometrial decidualization, outperforming traditional reference genes [5].
Validation Workflow and Best Practices
A robust validation workflow begins with careful candidate gene selection from RNA-seq results, prioritizing genes based on both statistical significance and biological relevance. The following diagram illustrates the comprehensive validation workflow from RNA-seq analysis through qPCR confirmation:
When designing qPCR validation experiments, the following best practices are essential:
- Gene Selection : Include both variable genes of interest and stable reference genes identified from RNA-seq data [11]
- Replicate Strategy : Incorporate sufficient biological replicates (based on power analysis) and technical replicates to account for variability
- Assay Design : Design primers to span exon-exon junctions when possible to minimize genomic DNA amplification
- Normalization : Use multiple validated reference genes rather than a single housekeeping gene
- Data Analysis : Use established analysis methods such as the ΔΔCq method with proper efficiency correction
Essential Research Reagents and Tools
Table 2: Research Reagent Solutions for RNA-Seq Validation Studies
| Reagent/Tool | Function | Considerations for Low-Expression Genes |
|---|---|---|
| RNA Extraction Kits | Isolation of high-quality RNA | Prioritize kits with high recovery of low-abundance transcripts |
| * ribosomal Depletion Reagents* | Remove ribosomal RNA | Critical for non-polyA targets; improves detection of non-coding RNAs |
| Library Prep Kits | Prepare sequencing libraries | Select kits with low technical noise and minimal GC bias |
| qPCR Master Mixes | Amplify and detect targets | Choose mixes with high sensitivity and wide dynamic range |
| Reference Gene Panels | Normalize qPCR data | Validate stability across experimental conditions; use multiple genes |
| RNA Spike-In Controls | Monitor technical variation | Use synthetic controls for normalization, especially in single-cell studies |
Single-CRNA-Seq Considerations
Single-cell RNA sequencing (scRNA-seq) introduces additional complexities for detecting subtle expression changes due to increased technical noise and data sparsity. Benchmarking studies have revealed that analysis methods significantly impact results, with pseudobulk methods - which aggregate gene expression across cells within biological replicates - consistently outperforming single-cell methods that analyze individual cells [51].
Pseudobulk methods demonstrate higher concordance with bulk RNA-seq results, better prediction of protein abundance changes, and more accurate reflection of biological pathways in functional enrichment analyses [51]. Single-cell DE methods show a systematic bias toward identifying highly expressed genes as differentially expressed, even when their expression doesn't change, while pseudobulk methods reduce this bias by properly accounting for biological variability [51].
For scRNA-seq validation, researchers should:
- Increase biological replicates rather than simply increasing cells per sample
- Implement pseudobulk approaches for differential expression testing
- Validate findings with orthogonal methods such as qPCR or protein-level assays
- Exercise caution when interpreting results from single-cell methods alone
The challenges posed by low-expression genes and subtle differential expression in RNA-seq are significant but manageable through careful experimental design and rigorous validation. Filtering strategies must be optimized for specific analytical pipelines, and reference genes for qPCR validation should be selected based on stability within the experimental system rather than traditional assumptions. The integration of RNA-seq findings with qPCR validation remains essential, particularly for studies with potential translational impact. As sequencing technologies continue to evolve, maintaining this rigorous framework for validation will ensure the reliability and reproducibility of gene expression studies, ultimately strengthening the foundation upon which drug development and clinical applications are built.
The extreme polymorphism of genes within the Human Leukocyte Antigen (HLA) system presents unique computational and experimental challenges for RNA sequencing (RNA-Seq) analysis that are not encountered with typical human genes. These genes are not only essential for adaptive immune responses but also represent among the most polymorphic regions in the human genome, with over 25,000 known alleles reported in the IPD-IMGT/HLA database [52] [53]. Traditional RNA-Seq pipelines, which align short reads to a single reference genome, often fail to accurately quantify HLA gene expression due to the substantial sequence divergence between individual alleles and the reference sequence [32]. This technical limitation is particularly problematic given that HLA expression levels have been implicated in disease outcomes for HIV, autoimmune conditions, cancer, and transplantation success [32] [54]. Therefore, understanding these challenges and implementing specialized approaches is crucial for researchers validating RNA-Seq data with qPCR, as inaccuracies in transcript quantification can propagate errors throughout downstream analyses and biological interpretations.
The fundamental issue stems from the high sequence similarity among HLA alleles and between HLA paralogs. When short RNA-Seq reads are mapped to a standard reference genome, reads from polymorphic regions may fail to align altogether or may align incorrectly to similar but distinct HLA genes, leading to biased expression quantification [32] [55]. This problem is exacerbated by the fact that different HLA alleles can exhibit significantly different expression levels, which has functional consequences for immune responses [53] [54]. Consequently, specialized methodologies are required to overcome these limitations and generate reliable expression data for these critical immune genes.
Technical Obstacles in HLA Expression Quantification
Fundamental Limitations of Standard RNA-Seq Pipelines
Standard RNA-Seq analysis pipelines encounter several specific obstacles when applied to HLA genes:
Mapping Bias and Reference Divergence: The high polymorphism at HLA genes means that individual alleles often diverge significantly from the reference genome. This divergence causes mapping algorithms to either incorrectly assign reads to similar reference sequences or discard them entirely, leading to underestimation of true expression levels [32] [55]. One study noted that this mapping bias can substantially overestimate reference allele frequencies in population-level analyses [32].
Cross-Mapping Between Paralogs: The HLA gene family arose through successive duplication events, resulting in segments with high sequence similarity between paralogs. RNA-Seq reads from such regions cannot be uniquely mapped to their correct gene of origin, potentially inflating expression estimates for some genes while reducing those for others [32]. This cross-mapping particularly affects expression quantification for HLA-B and HLA-C, which share higher sequence similarity.
Ambiguous Allele Assignment: The combination of extreme polymorphism within each locus and sequence conservation between loci creates challenges for determining which specific alleles are present in a sample. Without knowing the exact alleles, expression quantification lacks precision and may not capture biologically relevant allele-specific expression differences [53] [54].
Comparative Performance: RNA-Seq Versus qPCR
The technical challenges of HLA expression quantification become evident when comparing RNA-Seq results to established qPCR methods. A direct comparison study analyzing HLA class I genes across the same set of individuals found only moderate correlation between expression estimates derived from qPCR and RNA-Seq:
Table 1: Correlation Between qPCR and RNA-Seq for HLA Class I Gene Expression
| HLA Locus | Correlation Coefficient (rho) |
|---|---|
| HLA-A | 0.2 ≤ rho ≤ 0.53 |
| HLA-B | 0.2 ≤ rho ≤ 0.53 |
| HLA-C | 0.2 ≤ rho ≤ 0.53 |
The relatively wide range of correlation coefficients (0.2-0.53) highlights the inconsistency between these methods and underscores the need for specialized approaches to RNA-Seq analysis for HLA genes. These discrepancies arise from fundamental differences in what each technique measures—qPCR typically targets a conserved region with locus-specific primers, while RNA-Seq attempts to capture overall transcript abundance—as well as the mapping issues inherent to standard RNA-Seq pipelines [32].
Specialized Methodologies for Accurate HLA Analysis
Computational Advancements for HLA-Specific Analysis
Several bioinformatic strategies have been developed to address the unique challenges of HLA gene analysis:
Personalized Reference Pipelines: These approaches incorporate known HLA allelic diversity into the alignment step, creating sample-specific references that minimize mapping errors. Tools such as seq2HLA [55], HLA-mapper [32], and other customized pipelines [53] [54] implement this strategy by building references that include all known HLA alleles or specifically the alleles identified through genotyping, thereby improving both HLA typing and expression quantification.
Allele-Specific Expression Estimation: Advanced computational pipelines can now provide expression estimates at both the locus level and allele level, capturing differences in expression between the two alleles of a gene [53]. This is particularly important for HLA genes, where allelic imbalance can have functional consequences. These methods typically use probabilistic assignment of multi-mapping reads based on known polymorphisms.
Targeted Enrichment Approaches: Methods like capture RNA-Seq use biotinylated oligonucleotide probes specific to HLA genes to enrich for target transcripts before sequencing [53]. This enrichment increases coverage of HLA genes, improving both genotyping accuracy and expression quantification by reducing competition from non-target transcripts.
Experimental Workflows for Enhanced Accuracy
Wet-lab methodologies have also evolved to address the technical challenges of HLA analysis:
The experimental workflow for HLA-focused RNA-Seq incorporates several key modifications to standard protocols:
UMI Integration: Unique Molecular Identifiers (UMIs) are short random nucleotide sequences added during reverse transcription that tag individual mRNA molecules. This enables bioinformatic correction of PCR amplification biases, which is particularly valuable for HLA genes where amplification efficiency may vary between alleles [54]. After sequencing, reads with the same UMI are identified as PCR duplicates originating from the same original molecule.
Long-Read Sequencing Technologies: Platforms such as Oxford Nanopore Technologies (ONT) and PacBio generate reads long enough to cover multiple polymorphic sites in a single read, greatly improving the accuracy of allele assignment [52] [58]. The extended read length helps distinguish between highly similar alleles by encompassing more distinguishing polymorphisms.
Template-Switching Reverse Transcription: This method, adapted from the STRT (Single-Cell Tagged Reverse Transcription) protocol, enables full-length cDNA synthesis while incorporating UMIs [54]. The template-switching mechanism provides more uniform coverage across transcripts, which is valuable for quantifying expression of different HLA alleles.
The Scientist's Toolkit: Essential Reagents and Platforms
Table 2: Key Research Reagent Solutions for HLA-Focused RNA-Seq Studies
| Reagent/Platform | Function in HLA Analysis |
|---|---|
| STRT-V3-T30-VN oligo | Reverse transcription primer for cDNA synthesis with template switching capability [54] |
| RNA-TSO with UMI | Template switching oligo with integrated Unique Molecular Identifier for PCR bias correction [54] |
| HLA-specific capture probes | Biotinylated oligonucleotides for targeted enrichment of HLA transcripts prior to sequencing [53] |
| Oxford Nanopore MinION | Portable long-read sequencer enabling real-time HLA analysis [52] [58] |
| SureSelect XTHS2 | Exome capture system adapted for HLA target enrichment in DNA and RNA sequencing [14] |
| KAPA HiFi HotStart ReadyMix | High-fidelity PCR enzyme for accurate amplification of HLA amplicons with minimal errors [54] |
Experimental Protocol: Capture RNA-Seq for HLA Genotyping and Expression
Sample Preparation and Library Construction
The capture RNA-Seq method enables simultaneous HLA genotyping and expression quantification through the following detailed protocol:
RNA Extraction and Quality Control:
- Isolate total RNA from peripheral blood mononuclear cells (PBMCs) using TRIzol or RNeasy Mini kits [53] [54].
- Assess RNA quality using Bioanalyzer to obtain RNA Integrity Number (RIN). Samples with RIN >8.0 are preferred for optimal results [54].
- Quantitate RNA using sensitive fluorescence-based methods (e.g., Qubit RNA HS Assay) rather than spectrophotometry for accurate concentration measurement.
Library Preparation and Target Enrichment:
- Convert total RNA to cDNA using reverse transcription with HLA-specific primers or random hexamers [53].
- Hybridize cDNA libraries to a complex pool of biotinylated HLA-specific RNA capture probes designed against all classical HLA loci (A, B, C, DPA1, DPB1, DQA1, DQB1, DRA, DRB1, DRB3, DRB4, DRB5) [53].
- Use streptavidin-coated magnetic beads to capture probe-bound HLA transcripts and remove non-target cDNA.
- Amplify enriched targets using PCR with 15-20 cycles to maintain representation while generating sufficient material for sequencing [53].
Sequencing and Data Analysis
Sequencing Parameters:
- Sequence enriched libraries on Illumina platforms (NovaSeq 6000) or long-read platforms (Oxford Nanopore MinION) [14] [52].
- For short-read platforms, aim for minimum 50 million paired-end reads (2×100 bp) per sample to ensure adequate coverage of HLA genes [55].
- For long-read platforms, use R9.4 or R10.3 flow cells with high-accuracy base calling enabled [58].
Bioinformatic Processing:
- For genotyping: Map reads to comprehensive HLA allele reference databases (IPD-IMGT/HLA) using specialized tools like OptiType or NGSengine [14] [58].
- For expression quantification: Use alignment-free methods like Kallisto or personalized genome alignment approaches that incorporate sample-specific HLA types [14] [53].
- For UMI-based data: Process raw reads to collapse PCR duplicates using the UMI information before expression quantification [54].
Validation Framework: Integrating RNA-Seq and qPCR Data
Strategic Validation Approaches
Given the technical challenges of HLA gene analysis, rigorous validation of RNA-Seq results is essential:
Targeted qPCR Assays: Design qPCR assays for conserved regions within each HLA locus to minimize amplification bias between alleles. Use these assays to validate expression trends observed in RNA-Seq data, particularly for alleles showing extreme expression values [32] [54].
Orthogonal Method Comparison: Where possible, compare RNA-Seq expression estimates with protein-level quantification using flow cytometry with HLA-specific antibodies. This provides validation across different molecular phenotypes (mRNA vs. protein) [32].
Spike-In Controls: Incorporate synthetic RNA spike-ins with known concentrations during library preparation to control for technical variation and enable more accurate cross-sample normalization [54].
Interpretation of Discrepant Results
When RNA-Seq and qPCR results show discrepancies in HLA expression quantification, consider these potential sources:
Primer/Probe Binding Efficiency: qPCR assays may show variable amplification efficiency between different HLA alleles due to polymorphisms in primer binding sites, potentially skewing results [32].
Multi-Mapping Read Exclusion: Standard RNA-Seq pipelines may discard reads that map equally well to multiple HLA loci, leading to underestimation of expression for certain genes [32] [55].
Reference Bias: Both methods may exhibit reference bias—qPCR through primer design and RNA-Seq through reference-based alignment—potentially favoring more common alleles over rare variants [32].
Understanding these methodological limitations is crucial for appropriate interpretation of conflicting data and highlights why a multi-technique approach provides the most comprehensive assessment of HLA expression.
The exceptional polymorphism of HLA genes demands specialized approaches throughout the RNA-Seq workflow, from experimental design through computational analysis. Standard RNA-Seq pipelines consistently underestimate the complexity of these loci, potentially leading to inaccurate biological conclusions. The methodologies outlined here—including personalized reference genomes, targeted enrichment strategies, UMI integration, and long-read sequencing—collectively address these challenges to generate more reliable HLA genotyping and expression data.
As research continues to illuminate the critical role of HLA expression levels in disease susceptibility and treatment outcomes, employing these optimized approaches becomes increasingly important. The validation of RNA-Seq findings with qPCR and other orthogonal methods remains essential, particularly for these complex loci where technical artifacts can easily mimic or obscure biologically significant patterns. By implementing the specialized considerations outlined in this technical guide, researchers can more accurately quantify expression for HLA and other highly polymorphic genes, leading to more robust findings in immunogenetics and personalized medicine.
Best Practices for Filtering, Normalization, and Data Interpretation
Within the framework of a broader thesis on the necessity of validating RNA-Seq data with qPCR, this guide details the critical steps of filtering, normalization, and data interpretation. RNA-Seq provides a comprehensive, genome-wide snapshot of the transcriptome, but its results are probabilistic and can be influenced by technical artifacts and complex computational pipelines. qPCR, with its superior sensitivity, precision, and direct quantification, serves as an essential orthogonal method to confirm key findings. The reliability of this validation hinges entirely on the rigorous application of best practices during the RNA-Seq data processing stage, which directly influences the selection of targets for qPCR and the interpretation of the correlative results. This document provides an in-depth technical guide for researchers and drug development professionals on these foundational steps.
RNA-Seq Normalization Methods
Normalization is a crucial first step in RNA-Seq data analysis to remove technical biases and enable accurate comparisons of gene expression levels between samples. These biases can include differences in library size, gene length, and sequencing depth. The choice of normalization method can significantly impact downstream analyses, such as the identification of differentially expressed genes (DEGs) and the subsequent selection of candidates for qPCR validation.
Table 1: Benchmarking of Common RNA-Seq Normalization Methods
| Normalization Method | Type | Key Principle | Impact on Model Variability (Based on Benchmarking Studies) | Best Use Cases |
|---|---|---|---|---|
| RLE (Relative Log Expression) | Between-sample | Calculates a correction factor as the median of the ratios of all genes in a sample to a pseudo-reference [59]. | Low variability in model content; high accuracy in capturing disease-associated genes [59]. | Differential expression analysis; creating consistent condition-specific models [59]. |
| TMM (Trimmed Mean of M-values) | Between-sample | Trims extreme log fold-changes and gene intensities to compute a scaling factor, assuming most genes are not differentially expressed [59]. | Low variability in model content; performance similar to RLE and GeTMM [59]. | General purpose differential expression; recommended when comparing between samples [59]. |
| GeTMM (Gene-length corrected TMM) | Between- & Within-sample | Combines the TMM method with gene length correction, reconciling both approaches [59]. | Low variability in model content; performance similar to RLE and TMM [59]. | When both within-sample and between-sample comparisons are needed [59]. |
| TPM (Transcripts Per Million) | Within-sample | Normalizes for both sequencing depth and gene length, with length correction performed first [59]. | High variability in personalized model content; can increase false positive predictions [59]. | Comparing expression levels across different genes within the same sample. |
| FPKM (Fragments Per Kilobase Million) | Within-sample | Similar to TPM but normalizes for sequencing depth before gene length, making it less comparable across samples [59] [60]. | High variability in personalized model content; can increase false positive predictions [59]. | Note: Largely superseded by TPM for within-sample comparisons. |
Benchmarking studies have shown that the choice of normalization method directly affects the outcomes of downstream analyses. For instance, when mapping normalized data to genome-scale metabolic models (GEMs) to create condition-specific models, between-sample normalization methods (RLE, TMM, GeTMM) produce models with considerably lower variability and more accurately capture disease-associated genes compared to within-sample methods (TPM, FPKM) [59]. This reduction in false positives is critical for prioritizing high-confidence candidates for qPCR validation.
Experimental Protocols and Workflows
A robust RNA-Seq workflow integrates best practices from data generation through validation. The following protocol outlines the key steps, with a focus on generating reliable data for downstream qPCR confirmation.
Protocol: An Integrated RNA-Seq to qPCR Validation Workflow
Part 1: RNA-Seq Data Preparation and Differential Expression Analysis
Experimental Design and Sequencing:
- Biological Replicates: Include a sufficient number of biological replicates (e.g., cells or tissues from different individuals) to account for biological variation. This is crucial for statistical power in later steps [61].
- Library Preparation: Use paired-end sequencing for more robust expression estimates. Strand-specific libraries are recommended as they provide information on the direction of transcription [60].
Quantification and Normalization:
- Pseudoalignment and Quantification: Process raw FASTQ files using tools like Salmon or kallisto. These tools rapidly quantify transcript abundance while modeling uncertainty in read assignments [60].
- Generate Count Matrix: Aggregate sample-level quantifications into a gene-level count matrix for differential expression analysis.
- Normalization: Apply a between-sample normalization method, such as RLE (used in DESeq2) or TMM (used in edgeR), to the count matrix to correct for library composition and other technical biases [59] [60].
Differential Expression Analysis:
- Statistical Testing: Use packages like
limma,DESeq2, oredgeRto identify statistically significant differentially expressed genes (DEGs). These tools use linear models or negative binomial distributions to test for expression changes between conditions [60]. - Covariate Adjustment: Account for known covariates (e.g., age, gender, batch effects) in the statistical model to prevent them from confounding the results [59].
- Statistical Testing: Use packages like
The following diagram illustrates the core computational workflow for RNA-Seq data processing:
Part 2: qPCR Experimental Validation
Target Selection and Primer Design:
- Select Candidate Genes: Choose a set of DEGs from the RNA-Seq analysis for validation. Include both significantly up-regulated and down-regulated genes.
- Reference Gene Selection: Select stable reference genes for normalization in qPCR. Critical: Do not use classic reference genes (e.g.,
β-actin) without validation. Ideally, identify stable genes from your RNA-Seq data using algorithms likegeNormorNormFinder, as demonstrated in studies where RNA-Seq identifiedSTAU1as a superior reference for decidualization studies [5]. - Primer Design: Design primers with high amplification efficiency (90–110%) and specificity. Ensure amplicons are unique and span an exon-exon junction to avoid genomic DNA amplification.
qPCR Setup and Execution:
- Reverse Transcription: Synthesize cDNA from the same RNA samples used for RNA-Seq.
- Replicates: Perform both technical replicates (repetitions of the same cDNA sample) to measure system precision and biological replicates to capture biological variation [61]. Triplicates are standard for technical replicates.
- Reaction Setup: Use a passive reference dye (e.g., ROX) and master mix to minimize pipetting error and well-to-well variation. Ensure good pipetting technique and centrifuge plates after sealing [61].
qPCR Data Analysis:
- Absolute vs. Relative Quantification: Use the relative quantification method (ΔΔCq method) to calculate fold-change in gene expression [61].
- Normalization: Normalize the Cq values of your target genes to the geometric mean of the stable reference genes selected in step 1 [5].
- Statistical Analysis: Perform appropriate statistical tests (e.g., t-test) to confirm that the fold-changes observed by qPCR are significant. A high correlation between RNA-Seq and qPCR fold-changes validates the initial transcriptomic findings [61].
The integrated validation pathway, from RNA-Seq to final confirmation, is summarized below:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for RNA-Seq and qPCR Studies
| Item | Function/Description | Example/Note |
|---|---|---|
| Stranded mRNA Library Prep Kit | Prepares sequencing libraries that preserve strand information, crucial for accurate transcript assignment. | Kits from Illumina, Thermo Fisher, or NEB. |
| Salmon | A software tool for fast and bias-aware quantification of transcript expression from RNA-Seq data. | Used in "STAR-salmon" workflows for alignment-based quantification [60]. |
| Reference Genes | Stable genes used for normalization in qPCR experiments to control for technical and biological variation. | Classic genes (e.g., β-actin, GAPDH) often require validation. New candidates (e.g., STAU1) can be identified from RNA-Seq data [5]. |
| Passive Reference Dye | A dye included in qPCR master mixes to normalize for non-PCR-related fluorescence fluctuations between wells. | ROX dye. Corrects for pipetting variations and optical anomalies, improving precision [61]. |
| Multiplex qPCR Assay | Allows amplification and detection of multiple gene targets (e.g., target and reference gene) in the same well. | Improves precision and throughput by normalizing target and reference data from the same well [61]. |
Data Interpretation and Integration
Interpreting data from RNA-Seq and qPCR requires an understanding of the strengths and limitations of each technology. A successful validation is not merely a significant p-value but a confirmation of the biological signal.
- Correlating Fold-Changes: The fold-changes obtained from RNA-Seq and qPCR should show a strong positive correlation. Discrepancies can arise from differences in the dynamic range of the techniques, the normalization methods used, or the specificity of the qPCR assays.
- Assessing Statistical vs. Biological Significance: A result can be statistically significant (e.g., a 1.2-fold change with a low p-value) but may not be biologically relevant. In eukaryotic gene expression, a two-fold change is often considered a minimum threshold for biological significance [61]. The context of the research question is paramount.
- Leveraging Integrated Data: Validated gene expression changes can be used to build more reliable biological models. For example, RNA-Seq data normalized with RLE or TMM has been shown to generate more accurate and less variable genome-scale metabolic models (GEMs), which better capture disease-associated metabolic perturbations [59].
Weighing the Evidence: A Balanced View on Validation Necessity
When is qPCR Validation Truly Needed? A Scenario-Based Analysis
In the landscape of modern molecular research, quantitative polymerase chain reaction (qPCR) remains a cornerstone technology for gene expression analysis, despite the rise of high-throughput sequencing methods like RNA-Seq. Its unparalleled sensitivity, specificity, and reproducibility make it the gold standard for validating transcriptomic data [11] [62]. However, the powerful exponential amplification that makes qPCR so sensitive also renders it vulnerable to methodological pitfalls that can compromise data integrity. The recent publication of the updated MIQE 2.0 guidelines underscores a persistent challenge in molecular biology: despite widespread awareness of quality standards, compliance remains patchy, and fundamental methodological failures continue to plague published literature [63]. This technical guide examines the specific scenarios where qPCR validation is not merely beneficial but essential, providing a framework for researchers and drug development professionals to ensure the reliability of their gene expression data within the context of a broader thesis on validating RNA-Seq findings.
The Foundation: Understanding qPCR Validation Parameters
qPCR validation establishes that an assay reliably detects and quantifies its intended target across the required range of conditions. Before examining specific scenarios, it is crucial to understand the core parameters that constitute a properly validated qPCR assay.
- Inclusivity and Exclusivity: Inclusivity measures the assay's ability to detect all intended target strains or variants, while exclusivity (or cross-reactivity) confirms that it does not amplify genetically similar non-targets [64]. This requires both in silico analysis (e.g., using BLAST to check primer specificity) and experimental confirmation [65] [64].
- Linear Dynamic Range and Efficiency: The linear dynamic range is the range of template concentrations over which the fluorescent signal is directly proportional to the input. It is typically assessed using a 7-point, 10-fold dilution series, with an R² value of ≥0.980 considered acceptable. The amplification efficiency, derived from the slope of the standard curve, should ideally be between 90% and 110% [64].
- Limit of Detection (LOD) and Limit of Quantification (LOQ): The LOD is the lowest concentration of target that can be detected in 95% of replicates. The LOQ is the lowest concentration that can be quantified with acceptable accuracy and precision [65] [64].
- Precision and Accuracy: Precision (repeatability and reproducibility) is measured by the percentage coefficient of variation (%CV) across replicates. Accuracy determines how close the measured value is to the true value, often assessed through spike-recovery experiments [66] [65].
The table below summarizes the key performance characteristics and their validation targets.
Table 1: Essential qPCR Validation Parameters and Their Targets
| Parameter | Definition | Validation Target |
|---|---|---|
| Inclusivity | Ability to detect all target variants/strains. | Detection of up to 50 certified target strains [64]. |
| Exclusivity (Cross-reactivity) | Ability to exclude non-targets. | No amplification of genetically similar non-target species [64]. |
| Linear Dynamic Range | Range where signal is proportional to input. | R² ≥ 0.980 over 6-8 orders of magnitude [64]. |
| Amplification Efficiency | Rate of target amplification per cycle. | 90-110% [64]. |
| Limit of Detection (LOD) | Lowest concentration that can be detected. | Concentration detectable in 95% of replicates [65] [64]. |
| Limit of Quantification (LOQ) | Lowest concentration that can be accurately quantified. | Quantifiable with defined accuracy and precision [65]. |
| Precision | Agreement between replicate measurements. | %CV specified and deemed acceptable for the application [66]. |
Key Scenarios Mandating qPCR Validation
Scenario 1: Validation of RNA-Sequencing Data
RNA-Seq is a powerful, hypothesis-free tool for transcriptome profiling, but it is not infallible. qPCR validation is crucial to confirm key findings before drawing major biological conclusions or investing in further research. This is especially true for genes with low expression levels or small fold-changes (e.g., 1.2- to 1.5-fold), which are prone to being exaggerated or misrepresented without orthogonal validation [63]. The process involves selecting appropriate candidate genes from the RNA-Seq data and designing specific qPCR assays.
- Experimental Protocol: A critical first step is the selection of stable, highly expressed reference genes. Traditionally, housekeeping genes like actin and GAPDH have been used, but their expression can vary significantly across biological conditions [11]. Software tools like Gene Selector for Validation (GSV) can identify optimal reference and variable candidate genes directly from RNA-Seq data using criteria such as high expression (average log₂TPM >5) and low variation (standard deviation of log₂TPM <1) across all samples [11]. Once candidates are selected, a validated qPCR assay is run, and the expression results are correlated with the normalized RNA-Seq data (e.g., TPM or FPKM values) to confirm the direction and magnitude of expression changes.
Scenario 2: Development of Diagnostic and Clinical Assays
In clinical diagnostics, the consequences of unreliable data are measured in patient lives, not just p-values [63]. qPCR is central to detecting pathogens, quantifying biomarkers, and monitoring treatment response. Any assay used in a clinical or regulatory context must undergo rigorous validation to ensure it is fit-for-purpose.
- Experimental Protocol: The development of a qPCR assay for detecting residual Vero cell DNA in rabies vaccines provides a robust template [66]. The process involves:
- Target Selection: Bioinformatic analysis to identify unique, highly repetitive genomic sequences (e.g., the "172 bp" tandem repeat or Alu repetitive sequence in Vero cells) to maximize sensitivity [66].
- Assay Optimization: Designing primers and probes with minimal dimer formation and secondary structure. A probe-based method (e.g., TaqMan) is often preferred over SYBR Green for its higher specificity and lower chance of false positives in a diagnostic setting [65].
- Full Validation: A comprehensive assessment of linearity, LOD, LOQ, precision (e.g., %CV from 12.4% to 18.3%), and accuracy (e.g., recovery rate from 87.7% to 98.5%) against certified reference standards [66]. Specificity must be tested against a panel of non-target organisms to exclude cross-reactivity [66] [64].
Scenario 3: Ensuring Reproducibility in Publications and Regulatory Submissions
Journals and regulatory agencies increasingly demand transparent and comprehensive methodological details to ensure the reproducibility of findings. The MIQE guidelines (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) were established for this purpose and have been recently updated to MIQE 2.0 [63] [67].
- Experimental Protocol: Adherence to MIQE 2.0 involves detailed reporting of the entire qPCR workflow [63] [67]:
- Sample Details: Nucleic acid extraction method, quality control (e.g., RIN, A260/280), and storage conditions.
- Assay Information: Primer and probe sequences, locations, and concentrations, along with evidence of their specificity.
- Validation Data: PCR efficiency, correlation coefficient (R²) of the standard curve, LOD, and dynamic range for each assay.
- Data Analysis: The method used for Cq determination and normalization, including the reference genes used and their stability values. Raw Cq values should be converted into efficiency-corrected target quantities [67].
Scenario 4: Detection of Low-Abundance Targets and Minimal Fold-Changes
qPCR's exceptional sensitivity makes it indispensable for applications requiring the detection of very few target molecules, such as in the early phases of infection, detection of residual DNA in biologics, or measuring subtle but biologically critical changes in gene expression.
- Experimental Protocol: For instance, a validated qPCR assay can detect residual Vero DNA at a limit of quantification (LOQ) of 0.03 pg/reaction, which is essential for ensuring vaccine safety per regulatory limits (e.g., ≤10 ng/dose) [66]. To achieve this:
- Maximize Sensitivity: Use assays targeting multi-copy genomic elements and probe-based chemistry.
- Empirically Determine LOD/LOQ: Perform a dilution series with a high number of replicates (e.g., n=24 or more) at concentrations near the expected limit to statistically determine the LOD (95% detection rate) and the LOQ (quantifiable with acceptable precision and accuracy) [65].
- Control Contamination: Perform nucleic acid extraction and PCR setup in physically separated, dedicated rooms or enclosures to prevent amplicon contamination, which is a significant risk when amplifying low-copy-number targets [65].
Diagram 1: A decision workflow outlining the primary scenarios that necessitate qPCR validation and linking them to their corresponding experimental protocols.
The Scientist's Toolkit: Essential Reagents and Materials
Successful qPCR validation relies on high-quality, purpose-selected reagents and materials. The following table details key components for developing and running a robust qPCR assay.
Table 2: Essential Research Reagent Solutions for qPCR Validation
| Item | Function/Description | Example Use Case |
|---|---|---|
| Probe-based Master Mix | Contains enzyme, dNTPs, and buffer. Probe-based chemistry (e.g., TaqMan) offers higher specificity than dye-based methods [65]. | Detecting residual host cell DNA in vaccines; clinical diagnostics [66] [65]. |
| SYBR Green Master Mix | Cost-effective intercalating dye that binds all double-stranded DNA. Requires melting curve analysis to verify specificity [68]. | Cost-sensitive large-scale screening, such as SARS-CoV-2 surveillance [68]. |
| Primers & Probes | Sequence-specific oligonucleotides for target amplification and detection. Must be designed for high specificity and efficiency. | Target-specific amplification, e.g., for Vero cell "172bp" sequence or SARS-CoV-2 N gene [66] [68]. |
| Nucleic Acid Extraction Kit | For purifying DNA or RNA from complex samples (e.g., stool, tissue, swabs). Method must be determined during development [65] [69]. | Isolving host DNA from vaccine samples; viral RNA from patient swabs [66] [68]. |
| Certified Reference Standards | Genomic DNA or RNA of known concentration and quality used to generate standard curves. | Determining assay linearity, efficiency, LOD, and LOQ [66] [64]. |
| No-Template Control (NTC) | Reaction mixture without template DNA to check for contamination. | Essential for every run to ensure no false positives from contaminating DNA [65]. |
qPCR validation is not an optional refinement but a fundamental requirement for generating trustworthy data. The scenarios outlined—RNA-Seq verification, clinical assay development, ensuring publication reproducibility, and low-abundance target detection—represent critical points where rigorous validation is non-negotiable. As emphasized by the updated MIQE 2.0 guidelines, the goal is a cultural shift toward transparency and rigor, treating qPCR not as a simple "black box" but as a technique demanding the same scrutiny as other molecular methods [63] [67]. By integrating the frameworks, protocols, and tools described in this guide, researchers and drug development professionals can ensure their qPCR results are not just publishable but are robust, reproducible, and reliable, thereby upholding the integrity of scientific research and the safety of clinical applications.
In the era of precision biology, RNA sequencing (RNA-seq) has become the gold standard for whole-transcriptome gene expression quantification, offering an unbiased view of the transcriptome [70]. Despite its widespread adoption, a critical question persists in molecular biology laboratories: how reliable are RNA-seq results, and do they require independent verification? The practice of validating RNA-seq findings with quantitative PCR (qPCR) is deeply ingrained, a tradition stemming from earlier microarray technologies that suffered from reproducibility and bias issues [9]. However, as we will explore, this practice remains relevant not because of fundamental flaws in RNA-seq technology, but because of specific technical and biological factors that create a "concordance spectrum" between these methodologies.
This guide examines the core reasons behind discrepancies between RNA-seq and qPCR, providing researchers and drug development professionals with a structured framework for understanding when and why orthogonal validation is scientifically warranted. Within a broader thesis on RNA-seq validation, we demonstrate that strategic qPCR verification adds rigor to transcriptomic studies, particularly for specific gene sets and in contexts where clinical or therapeutic decisions hinge on accurate gene expression measurement.
Performance Benchmarks: Quantifying Methodological Concordance
Large-scale benchmarking studies reveal both strong overall agreement and specific, reproducible discrepancies between RNA-seq and qPCR. Understanding the magnitude and nature of these differences is crucial for interpreting validation results.
A comprehensive benchmarking study using whole-transcriptome RT-qPCR data for over 18,000 protein-coding genes provides robust quantitative metrics on how these technologies compare [70]. The research evaluated five common RNA-seq workflows (Tophat-HTSeq, Tophat-Cufflinks, STAR-HTSeq, Kallisto, and Salmon) and found high expression correlations with qPCR data, with Pearson correlation coefficients (R²) ranging from 0.798 to 0.845 across workflows [70]. When comparing the capacity to detect differential expression, approximately 85% of genes showed consistent results between RNA-seq and qPCR when comparing fold changes between reference samples [70].
Table 1: Summary of RNA-seq and qPCR Concordance from Benchmarking Studies
| Metric | Alignment-Based Workflows | Pseudoalignment Workflows | Overall Range |
|---|---|---|---|
| Expression Correlation (R²) | 0.798 (Tophat-Cufflinks) to 0.827 (Tophat-HTSeq) [70] | 0.839 (Kallisto) to 0.845 (Salmon) [70] | 0.798 - 0.845 [70] |
| Non-Concordant Genes* | 15.1% (Tophat-HTSeq) [70] | 19.4% (Salmon) [70] | 15.1% - 19.4% [70] |
| Non-Concordant Genes with ΔFC >2 | 7.1% (Tophat-HTSeq) [70] | ~7.5% (average) [70] | 7.1% - 8.0% [70] |
Non-concordant genes defined as those with opposing differential expression status or direction between methods.
While these non-concordance rates might appear substantial, further analysis reveals that the majority of these discrepancies are relatively minor. Over 66% of non-concordant genes exhibit a fold change difference between methods (ΔFC) of less than 1, and 93% have a ΔFC < 2 [9]. This pattern indicates that most discrepancies occur in genes with small expression changes that hover near the threshold of statistical or biological significance, rather than in genes with large, unambiguous expression differences.
The Critical Gene Subset with Severe Discrepancies
A small but important subset of genes shows more substantial discrepancies. Approximately 1.8% of genes demonstrate severe non-concordance, characterized by large fold change differences (ΔFC > 2) between RNA-seq and qPCR methods [9]. These genes are not random outliers; they share common characteristics that make them particularly challenging for transcriptomic analysis. Problematic genes are typically shorter in transcript length, contain fewer exons, and are expressed at lower levels compared to genes with consistent expression measurements across platforms [70] [9].
This systematic pattern suggests the discrepancies are not due to random error but to specific technological limitations. For instance, the challenges with shorter transcripts may relate to the RNA-seq library preparation process, which often favors longer RNA species, while low expression levels push against the sensitivity limits of the technology.
The concordance spectrum between RNA-seq and qPCR arises from multiple factors spanning laboratory procedures, computational analysis, and molecular biology. Understanding these sources is key to designing effective validation strategies.
Technological and Analytical Workflow Differences
RNA-seq and qPCR measure gene expression through fundamentally different processes, each introducing specific biases that can affect the final results.
Table 2: Key Technical Sources of Discrepancy Between RNA-seq and qPCR
| Factor | Impact on RNA-seq | Impact on qPCR | Resulting Discrepancy |
|---|---|---|---|
| Transcript Length | Longer transcripts generate more reads, potentially overestimating expression [71]. | Amplification efficiency is generally independent of transcript length. | Bias for longer transcripts in RNA-seq compared to qPCR. |
| Expression Level | Low-abundance transcripts may be undersampled; high-abundance genes dominate sequencing capacity [71]. | High sensitivity even for low-copy transcripts with optimized assays. | Greater discrepancy for low-expression genes [70] [9]. |
| GC Content & Sequence | Extreme GC content can cause underrepresentation during library preparation and sequencing. | Optimized primer/probe design can mitigate GC content issues. | Differential performance for GC-rich or AT-rich transcripts. |
| Alignment to Reference | Polymorphic regions (especially in HLA genes) may align poorly, affecting quantification [32]. | Primer/probe sequences can be designed for specific alleles or variants. | Under-quantification of divergent alleles in RNA-seq [32]. |
| Normalization Methods | Relies on statistical normalization across entire transcriptome (e.g., TPM). | Typically normalized to one or a few reference genes. | Different assumptions about what constitutes "constant" expression. |
The extreme polymorphism of genes like those in the human leukocyte antigen (HLA) family presents particular challenges for RNA-seq. The short reads must be aligned to a reference genome, which cannot capture the full diversity of HLA alleles, leading to mapping errors and quantification bias [32]. One study comparing HLA class I expression between qPCR and RNA-seq found only moderate correlations (0.2 ≤ rho ≤ 0.53), highlighting the particular difficulty in accurately quantifying highly polymorphic genes with standard RNA-seq pipelines [32].
Molecular and Biological Considerations
Beyond technical factors, the biological nature of the transcript itself influences concordance. Genes with fewer exons consistently show higher discrepancy rates, potentially due to differences in how each technology captures and quantifies relatively simple transcript structures [70]. Additionally, the tissue source significantly impacts data quality and comparability, as different tissues present unique challenges in RNA quality, integrity, and composition [22].
For clinical applications, the dynamic range of detection is particularly important. While RNA-seq can identify novel transcripts and splicing events across the entire transcriptome, qPCR often provides superior sensitivity and dynamic range for quantifying specific, especially low-abundance, transcripts of interest [72]. This distinction makes qPCR particularly valuable for confirming expression changes in key target genes identified through RNA-seq screening.
Experimental Design for Effective Validation
Strategic validation of RNA-seq data requires careful planning in both wet-lab and computational approaches to ensure meaningful results.
RNA-seq Experimental Design and Analysis
Proper RNA-seq experimental design begins with sufficient biological replication, which is essential for robust differential expression analysis and reduces the need for downstream validation of every significant finding. During library preparation and sequencing, incorporating RNA quality controls and monitoring sequencing depth ensures that technical artifacts do not dominate the biological signal.
For data analysis, employing multiple RNA-seq quantification workflows can help identify genes whose reported expression is highly dependent on specific algorithms or alignment methods [70]. Studies have shown that while different computational workflows (e.g., Tophat-HTSeq, STAR-HTSeq, Kallisto, Salmon) generally show high agreement, each may produce a small set of unique outliers [70]. Additionally, for challenging gene families like HLA, using specialized alignment tools that account for known diversity rather than relying on a single reference genome can significantly improve quantification accuracy [32].
qPCR Validation Strategies
When designing qPCR validation experiments, the selection of reference genes is critical. Rather than relying on traditional "housekeeping" genes, researchers should use statistical approaches (such as NormFinder or GeNorm) to identify genes with stable expression in their specific experimental system [71]. Interestingly, one study demonstrated that with a robust statistical approach for reference gene selection, commonly used reference genes performed equally well as those pre-selected from RNA-seq data, potentially saving time and resources [71].
The following workflow diagram illustrates a recommended process for validating RNA-seq findings with qPCR:
For the actual validation experiments, researchers should validate all genes central to the study's conclusions, particularly those with low expression or small fold changes that fall into the more problematic regions of the concordance spectrum [9]. The number of biological replicates for qPCR should match or exceed those used in the RNA-seq experiment to ensure statistical rigor. Finally, when interpreting results, focus on the consistency of fold change direction and magnitude rather than expecting identical values, as absolute expression measures often differ between platforms due to their different normalization methods and dynamic ranges [70] [9].
Essential Reagents and Research Tools
Successful validation requires appropriate laboratory materials and computational resources. The following table outlines key solutions used in the featured studies:
Table 3: Essential Research Reagent Solutions for RNA-seq and qPCR Validation
| Reagent/Tool Category | Specific Examples | Function and Importance | Considerations for Selection |
|---|---|---|---|
| RNA Isolation Kits | RNeasy Mini Kit (Qiagen) [32] [22], AllPrep DNA/RNA Kit [14] | High-quality RNA with genomic DNA removal is critical for accurate quantification in both methods. | Choose based on sample type (e.g., FFPE vs. fresh frozen); include DNase treatment step. |
| RNA Quality Assessment | Qubit Fluorometer, TapeStation, Bioanalyzer [14] [22] | RNA integrity number (RIN) or similar metrics predict success in both RNA-seq and qPCR. | Essential QC step; poor RNA quality is a major source of technical variation. |
| Library Prep Kits | Illumina Stranded mRNA Prep [22], TruSeq Stranded mRNA [14], SureSelect XTHS2 [14] | Converts RNA to sequencing-ready libraries; choice affects coverage and bias. | Stranded protocols preferred for accurate transcript orientation; consider input RNA requirements. |
| qPCR Assays | TaqMan Gene Expression Assays [72], SYBR Green with validated primers | Target-specific detection with high sensitivity and specificity. | TaqMan assays offer better specificity; design assays to span exon-exon junctions. |
| Reference Gene Panels | Commercially available panels or laboratory-validated gene sets [71] [5] | Normalization of qPCR data using stably expressed genes. | Statistically validate stability in your specific experimental system [71]. |
| Alignment & Quantification Software | STAR [14], Kallisto [14], HTSeq [70] | Maps reads to genome/transcriptome and assigns to genes for expression quantification. | Pseudoaligners (Kallisto, Salmon) are faster; alignment-based may be more accurate for some applications. |
Clinical Applications and Special Considerations
In clinical diagnostics, the standards for analytical validation become more stringent, with implications for how RNA-seq and qPCR are utilized together.
Clinical Validation of RNA-seq Tests
The implementation of RNA-seq in clinical diagnostics requires rigorous validation frameworks. One approach for clinical RNA-seq tests involves establishing transcriptome-wide reference ranges for each reportable gene based on control data, against which patient samples are compared as outliers [22]. This differs from research applications where fold-change between conditions is typically the primary metric.
For Mendelian disease diagnostics, validation studies should include positive controls with known RNA-level changes (e.g., altered expression or splicing due to diagnostic DNA variants) to ensure the assay can detect clinically relevant abnormalities [22]. Additionally, tissue-specific validation is crucial, as gene expression and splicing patterns differ significantly between clinically accessible tissues like blood and fibroblasts [22].
When is qPCR Validation Essential?
While RNA-seq is generally reliable, specific scenarios warrant orthogonal verification with qPCR or other methods:
- Studying genes with inherent challenges: When focusing on genes prone to discrepancies (short transcripts, low expression, few exons, or high polymorphism), qPCR validation provides added confidence [70] [32].
- Critical findings supporting major conclusions: When the entire biological story hinges on differential expression of just a few genes, independent verification is prudent [9].
- Clinical or diagnostic applications: In contexts influencing patient care, the higher standard of evidence supports using multiple methodologies [22].
- Extending findings to new models or conditions: qPCR offers a cost-effective method to confirm key RNA-seq findings in additional sample types, time points, or genetic backgrounds [9].
The relationship between RNA-seq and qPCR is not competitive but complementary. While RNA-seq provides a powerful discovery platform for transcriptome-wide profiling, qPCR remains invaluable for targeted validation and specific applications where its sensitivity, reproducibility, and dynamic range are advantageous. The "concordance spectrum" between these technologies is influenced by specific technical and biological factors that systematically affect certain gene classes.
Strategic validation focuses resources on genes most likely to show discrepancies—those with low expression, short length, and few exons—or those most critical to research conclusions. By understanding the sources of discrepancy and implementing rigorous experimental designs, researchers can leverage the strengths of both technologies to produce robust, reproducible gene expression data that advances both basic research and clinical applications.
The prevailing paradigm in transcriptomics research often mandates quantitative PCR (qPCR) as an essential validation step for RNA sequencing (RNA-seq) results. This practice, inherited from the microarray era, is increasingly questioned as RNA-seq technologies mature. While qPCR validation provides orthogonal verification for specific targets, this approach has limitations—it is low-throughput, requires pre-selection of candidate genes, and may not capture transcriptome-wide complexities such as novel isoforms or global expression patterns. This article explores advanced strategies that move beyond qPCR, leveraging computational frameworks, integrated multi-omics, and emerging technologies to provide more comprehensive validation of RNA-seq findings. We examine how these approaches are reshaping validation standards while considering their applications within the broader context of establishing robust, reproducible transcriptomic insights.
Computational and Statistical Validation Frameworks
Computational methods provide powerful, scalable alternatives to wet-lab validation by assessing RNA-seq data reliability through statistical robustness measures and replicate consistency.
Statistical Robustness and Reproducibility: Properly designed RNA-seq experiments with adequate biological replication generate statistically robust data. Methods like NOISeq and GFOLD are particularly effective for identifying differentially expressed genes (DEGs) with high positive predictive value, especially when biological effect sizes are strong. With triplicate or larger replicate sizes, tools such as DESeq2 and edgeR demonstrate superior performance for system-level analyses, achieving over 90% and 60% mean positive predictive value respectively under optimal conditions [73].
Meta-analysis Across Studies: Combining results from multiple independent RNA-seq studies via meta-analysis significantly enhances the robustness of DEG identification. This approach increases statistical power and helps distinguish biologically consistent signals from study-specific artifacts. In livestock genomics, where individual studies often have small sample sizes, meta-analysis has successfully identified more reliable biomarkers for complex traits like feed efficiency and mastitis resistance that were not detectable in individual studies [74]. The key steps include:
- Dataset Identification: Apply stringent eligibility criteria based on biological and technical parameters
- Data Harmonization: Address inter-study variability from technical differences (library protocols, batch effects) and biological factors
- Jackknife Sensitivity Testing: Assess result stability by systematically excluding individual datasets to identify robust findings
Table 1: Statistical Methods for RNA-seq Validation
| Method Type | Examples | Best Use Cases | Key Advantages |
|---|---|---|---|
| Differential Expression Tools | NOISeq, GFOLD, DESeq2, edgeR | Strong effect sizes with adequate replicates | High positive predictive value, genome-wide coverage |
| Meta-analysis Approaches | P-value combination, effect size integration | Combining multiple independent studies | Increased statistical power, more robust biomarkers |
| Sample Size Assessment | Power analysis, replicate simulations | Experimental design optimization | Identifies required replicates for reliable detection |
Orthogonal Sequencing-Based Validation
Sequencing-based validation methods provide comprehensive alternatives to targeted qPCR by offering transcriptome-wide verification through technological diversification.
Integrated DNA-RNA Sequencing: Combining whole exome sequencing (WES) with RNA-seq from the same sample enables powerful internal validation. This integrated approach allows direct correlation of somatic DNA alterations with transcriptional consequences, improving variant interpretation and functional annotation. In clinical oncology, integrated assays have demonstrated enhanced detection of clinically actionable alterations—including gene fusions, allele-specific expression, and splicing variants—that would likely remain undetected with DNA-only testing [14]. The validation framework for combined assays includes:
- Analytical Validation: Using reference samples with known variants to establish accuracy metrics
- Orthogonal Testing: Comparing results with established methodologies on patient samples
- Clinical Utility Assessment: Demonstrating real-world impact on diagnostic interpretation
Long-Read RNA Sequencing: Long-read technologies (PacBio, Oxford Nanopore) provide orthogonal validation for transcriptome assembly and isoform detection by capturing full-length transcripts. According to the Long-read RNA-Seq Genome Annotation Assessment Project (LRGASP) consortium, libraries with longer, more accurate sequences produce more precise transcript models than short-read approaches with increased depth [75]. Key applications include:
- Transcript Isoform Validation: Resolving complete splice variants without assembly artifacts
- Novel Transcript Discovery: Identifying previously unannotated genes and isoforms
- Fusion Gene Verification: Characterizing chimeric transcripts with exact breakpoints
RNA-seq Validation Strategy Decision Tree
Multi-omics and Single-Cell Integration
Advanced integration approaches leverage complementary data types to provide biological validation across molecular layers and at single-cell resolution.
Multi-omics Correlation Analysis: Correlating RNA-seq findings with proteomic data provides strong biological validation at the functional level. While mRNA and protein levels don't always correlate perfectly due to post-transcriptional regulation, consistent patterns strengthen the biological significance of transcriptomic findings. This approach is particularly valuable for:
- Prioritizing Candidate Biomarkers: Genes with correlated RNA and protein expression changes have higher translational potential
- Pathway Activation Validation: Confirming signaling pathway alterations across molecular layers
- Drug Target Identification: Increasing confidence in target engagement assessments
Single-Cell RNA-seq Validation: Single-cell RNA sequencing (scRNA-seq) provides resolution unattainable with bulk sequencing or qPCR. While technically challenging due to low starting RNA, scRNA-seq enables validation of cell type-specific expression patterns in complex tissues [76]. Applications include:
- Cell Type-specific Expression: Verifying gene expression patterns in particular cell subpopulations
- Developmental Trajectories: Validating temporal expression patterns through pseudotime analysis
- Spatial Transcriptomic Correlation: Integrating with spatial transcriptomics to confirm localization patterns
Table 2: Research Reagent Solutions for Advanced Validation
| Reagent/Category | Specific Examples | Function in Validation |
|---|---|---|
| Reference Materials | SRSF2 NMD-sensitive transcripts | Internal control for NMD inhibition efficiency [15] |
| NMD Inhibitors | Cycloheximide (CHX), Puromycin (PUR) | Stabilize transcripts for detecting nonsense-mediated decay [15] |
| Library Prep Kits | TruSeq stranded mRNA, SureSelect XTHS2 | Ensure compatibility between RNA-seq and validation assays [14] |
| Single-cell Platforms | Fluidigm C1, 10X Genomics | Enable cell-type specific validation across technologies [76] |
| Orthogonal Sequencing | PacBio Iso-seq, Oxford Nanopore | Provide long-read verification of transcript isoforms [75] |
Reference Gene Selection from RNA-seq Data
Traditional validation practices often rely on presumed stable reference genes (e.g., ACTB, GAPDH), but evidence shows these can vary substantially across biological conditions. RNA-seq data itself enables data-driven selection of optimal reference genes specifically suited to experimental conditions.
The Gene Selector for Validation (GSV) software implements a systematic approach for identifying stable reference genes directly from RNA-seq data using TPM values and multiple filtering criteria [11]:
- Expression Threshold: TPM >0 across all samples
- Low Variability: Standard deviation of log2(TPM) <1
- Consistent Expression: No individual value >2-fold from mean log2 expression
- High Expression: Average log2(TPM) >5
- Low Coefficient of Variation: <0.2
This methodology identified STAU1 as a superior reference gene for endometrial decidualization studies, outperforming traditional choices like β-actin [5]. The data-driven approach eliminates guesswork and adapts reference selection to specific biological contexts.
Reference Gene Selection Workflow
Future Directions and Implementation Guidelines
The evolution of RNA-seq validation is progressing toward more integrated, standardized approaches that leverage technological advancements and collaborative science.
Emerging Technologies and Standards: Future validation frameworks will increasingly incorporate:
- Multi-omics Reference Materials: Commercially available standards with known DNA, RNA, and protein profiles
- Artificial Intelligence-Enhanced Quality Metrics: Machine learning algorithms to predict data quality and reproducibility
- Consortium-Led Benchmarking: Large-scale community efforts like LRGASP to establish best practices [75]
- Single-cell and Spatial Multi-omics: Integrated validation across cellular resolution and spatial context
Practical Implementation Guidelines: Researchers should consider these evidence-based recommendations:
For High-Quality RNA-seq Data: With sufficient biological replicates (n≥3) and strong effect sizes, extensive qPCR validation may offer limited added value [9] [17]
When qPCR Remains Appropriate: Target validation when RNA-seq uses minimal replicates, or when confirming specific genes crucial to the study conclusions [17]
Integrated Validation Framework: Combine computational checks, orthogonal sequencing, and biological replication for the most robust verification
Context-Dependent Strategy: Align validation intensity with study goals—hypothesis-generating screens require less validation than clinical biomarker studies
The transcriptomics field is transitioning from reflexive qPCR validation toward nuanced, multi-faceted validation strategies that better address the complexity of RNA-seq data. Computational robustness checks, orthogonal sequencing technologies, multi-omics integration, and data-driven reference standards collectively provide more comprehensive verification frameworks than targeted qPCR alone. While qPCR retains value for specific applications, particularly with low-replicate RNA-seq designs, these advanced approaches enable researchers to match validation strategies to their specific scientific goals and resource constraints. As RNA-seq technologies continue evolving toward long-read, single-cell, and spatial applications, validation practices must similarly advance—focusing not on ritualistic verification but on building robust, reproducible biological insights through appropriate methodological diversification.
Integrating RNA-Seq and qPCR in a Multi-Omics Framework for Drug Discovery
The integration of RNA sequencing (RNA-Seq) and quantitative PCR (qPCR) represents a methodological cornerstone within modern multi-omics frameworks for drug discovery. While RNA-Seq provides an unbiased, genome-wide view of the transcriptome, qPCR delivers a highly sensitive, specific, and quantitative method for validation, anchoring high-dimensional data in analytical robustness. This validation is not merely a procedural formality but a critical step that ensures the reliability of transcriptomic data used to inform decision-making in the drug development pipeline. The high sensitivity, specificity, and reproducibility of qPCR make it the gold standard for validating gene expression data obtained from transcriptome sequencing [11]. Within a multi-omics context, where data from genomics, transcriptomics, proteomics, and epigenomics are integrated to build a comprehensive model of biological systems and drug responses, the accuracy of each individual data layer is paramount [77]. Misinterpretation of RNA-Seq data due to a lack of proper validation can propagate errors through integrated models, leading to flawed biological conclusions and costly missteps in target identification and lead compound optimization.
The necessity of this integrated approach is further underscored by the growing application of RNA-Seq for discovering and profiling RNA-based drug response biomarkers, with the goal of improving the efficiency and success rate of the drug development process [78]. As next-generation sequencing (NGS) technologies become more accessible, a standardized workflow for cross-platform validation ensures that transcriptional signatures used to group compounds by mechanism of action (MoA) or to identify patient responders are analytically sound [79].
Analytical Foundations: Technical Comparison of RNA-Seq and qPCR
The strategic integration of RNA-Seq and qPCR is predicated on a clear understanding of their complementary technical profiles. RNA-Seq offers a discovery-oriented, hypothesis-generating capability, while qPCR provides a targeted, hypothesis-testing function with superior quantitative precision for specific genes.
Table 1: Comparative Analysis of RNA-Seq and qPCR Technologies
| Feature | RNA Sequencing (RNA-Seq) | Quantitative PCR (qPCR) |
|---|---|---|
| Throughput | High-throughput, whole transcriptome [78] | Low- to medium-throughput, targeted (dozens to hundreds of genes) |
| Dynamic Range | Broad (>10^5-fold) [70] | Very Broad (>10^6-fold) |
| Sensitivity | Moderate; depends on sequencing depth [79] | High; can detect single copies of RNA |
| Quantification | Relative (e.g., TPM, FPKM); can be influenced by transcriptome composition | Absolute or relative; relies on standard curves or comparative Cq method |
| Prior Knowledge Required | No prior knowledge needed; can discover novel transcripts, fusions, and SNPs [78] | Requires pre-defined sequence for primer/probe design |
| Primary Application in Drug Discovery | Biomarker discovery, MoA elucidation, transcriptional profiling [78] [79] | Validation of RNA-Seq findings, focused biomarker panels, high-confidence quantification |
| Cost per Sample | Moderate to high (though decreasing with methods like DRUG-seq) [79] | Low |
| Workflow & Turnaround Time | Complex, multi-day library prep and bioinformatics analysis | Simple, same-day results from purified RNA |
Benchmarking studies demonstrate that while RNA-Seq workflows show high gene expression correlations with qPCR data (with Pearson correlations, R², ranging from 0.798 to 0.845 for various workflows), a critical analysis of differential expression reveals that approximately 85% of genes show consistent fold-change results between RNA-Seq and qPCR [70]. This leaves a non-concordant fraction of genes whose expression changes are discrepant between the platforms, underscoring the need for validation rather than assuming equivalence.
Integrated Workflow: From Multi-Omics Discovery to qPCR Validation
A robust, integrated pipeline systematically transitions from broad-scale discovery to focused validation, ensuring that findings are both comprehensive and reliable.
Stage 1: Transcriptomic and Multi-Omics Profiling for Hypothesis Generation
The initial stage employs RNA-Seq to generate comprehensive transcriptional profiles from disease models or compound-treated samples. In drug discovery, this can be effectively applied using high-throughput transcriptomic platforms like DRUG-seq (Digital RNA with pertUrbation of Genes), which provides a cost-effective solution for profiling hundreds of compounds across multiple doses to group them by MoA based on transcriptional signatures [79]. The goal is to identify differentially expressed genes (DEGs), pathways, and gene signatures associated with a disease state or drug response.
The power of discovery is greatly enhanced by multi-omics integration. This involves combining RNA-Seq data with other data types, such as whole exome sequencing (WES) for somatic mutations, DNA methylation arrays for epigenomics, and proteomic profiles [77] [80] [14]. For instance, integrating transcriptomic, epigenetic, and somatic mutation data has been successfully used to classify molecular subtypes of gastric cancer with distinct prognostic and therapeutic implications [80]. Such integration provides a more holistic view of the biological system and can reveal regulatory mechanisms that would be missed by a single-omics approach.
Stage 2: Bioinformatics Analysis and Candidate Selection
The raw RNA-Seq data is processed through a bioinformatics pipeline, which typically includes alignment, quantification, and differential expression analysis. Benchmarking studies have shown that various processing workflows (e.g., Tophat-HTSeq, STAR-HTSeq, Kallisto, Salmon) perform with comparable accuracy for the majority of genes, though each may have a small, specific set of genes with inconsistent measurements [70]. The output is a list of candidate genes for validation, which may include:
- Potential Biomarkers: Genes whose expression strongly correlates with disease outcome or drug response [78].
- Key Drivers of Signatures: Genes that are central to a multi-omics-defined molecular subtype or a drug response signature [80] [81].
- Mechanistically Relevant Targets: Genes involved in pathways identified as critical through pathway enrichment analysis of the multi-omics data.
Stage 3: Strategic Selection of Genes for qPCR Validation
The selection of genes for qPCR validation is a critical step that should be guided by both statistical significance and biological rationale. Tools like GSV (Gene Selector for Validation) can objectively identify the most stable reference genes and the most variable target genes from RNA-seq data, thereby preventing the common pitfall of using inappropriate housekeeping genes [11].
Table 2: Key Reagent Solutions for Integrated RNA-Seq and qPCR Workflows
| Research Reagent Category | Example Products | Function in Workflow |
|---|---|---|
| RNA Library Prep Kits | TruSeq Stranded mRNA Kit (Illumina) [14], SMART-Seq v4 Ultra Low Input RNA Kit (Takara Bio) [82] | Converts purified RNA into sequencing-ready libraries for RNA-Seq. |
| Targeted RNA Panels | TruSight RNA Pan-Cancer Panel (Illumina) [78] | Focuses sequencing on a predefined set of genes of interest, reducing cost and data complexity. |
| Nucleic Acid Extraction Kits | AllPrep DNA/RNA Kit (Qiagen) [14], mirVana RNA Isolation Kit (Thermo Fisher) [82] | Simultaneously or separately isolates high-quality DNA and RNA from precious clinical samples (FFPE, fresh frozen). |
| One-Step qPCR Kits | Various one-step RT-qPCR kits | Integrates reverse transcription and quantitative PCR into a single reaction, reducing hands-on time and variability for validation. |
| Automated Library Prep Systems | Integrated with DRUG-seq workflow [79] | Enables high-throughput, automated RNA library construction in 384- and 1536-well formats for large-scale compound screening. |
The criteria for selecting a reference gene should include: expression greater than zero in all samples, low variability (standard deviation of log₂(TPM) < 1), no outlier expression (within 2x the average of log₂ expression), high average expression (log₂(TPM) > 5), and a low coefficient of variation (< 0.2) [11]. For target validation genes, the selection should focus on genes with high variability between conditions (standard deviation of log₂(TPM) > 1) and sufficient expression to be reliably detected by qPCR [11].
Stage 4: Experimental qPCR Validation and Data Integration
This final stage involves executing a rigorous qPCR experiment using the selected reference and target genes. The resulting Cq values are analyzed using software like GeNorm, NormFinder, or BestKeeper to confirm the stability of the chosen reference genes [11]. The expression levels of the target genes are then normalized accordingly. Successful validation is achieved when the qPCR data confirms the direction and significance of the expression changes observed in the RNA-Seq data. This validated dataset becomes a high-confidence component that can be integrated with other omics data layers to build reliable models for patient stratification, target identification, and MoA deconvolution.
Diagram 1: Integrated RNA-Seq and qPCR Workflow in Multi-Omics. This diagram outlines the sequential and synergistic process of using multi-omics discovery to inform targeted qPCR validation, leading to high-confidence models for drug discovery.
Advanced Applications in Drug Discovery and Development
The strategic integration of RNA-Seq and qPCR is leveraged across the entire drug discovery and development pipeline, providing critical insights that de-risk the process.
Mechanism of Action (MoA) Deconvolution: DRUG-seq enables the profiling of hundreds of compounds across multiple doses in a high-throughput manner. The resulting transcriptional signatures cluster compounds by their MoA [79]. For example, translation inhibitors like homoharringtonine and cycloheximide cluster together, while epigenetic regulators such as BET and HDAC inhibitors form another distinct cluster. qPCR can then be used to validate the expression of key genes within these signatures (e.g., specific cell cycle genes for kinase inhibitors) to confirm the MoA for novel compounds or to understand differential on- and off-target activities among similar compounds.
Biomarker Identification and Patient Stratification: In oncology, multi-omics studies integrate transcriptomic, epigenetic, and genomic data to define molecular subtypes of cancer with distinct prognoses and treatment responses [80] [81]. A resulting gene signature, such as a programmed cell death signature (GMPS) in gastric cancer, can be developed using machine learning and its hub genes validated with qPCR [80]. This validated signature can then be developed into a focused qPCR-based assay for clinical application, enabling the identification of patient populations most likely to respond to a specific therapy.
Microbiome-Informed Drug Response Analysis: The microbiome can significantly influence host gene expression, protein activity, and drug metabolism, contributing to variation in individual drug responses [77]. RNA-Seq can be used to profile host transcriptomic changes in response to a drug in the context of different microbial communities. qPCR assays targeting specific host genes or microbial pathways can then be used to validate these interactions and develop biomarkers that predict drug efficacy or toxicity based on the microbiome composition.
Validation and Quality Control Frameworks
Ensuring the analytical validity of the data generated from both RNA-Seq and qPCR is fundamental to the integrity of the entire research endeavor.
For RNA-Seq, comprehensive validation guidelines for clinical application include using custom reference samples with known mutations and expression profiles, orthogonal testing with patient samples, and demonstrating clinical utility in real-world cases [14]. Quality control metrics for RNA-Seq library preparation include RNA Integrity Number (RIN), library concentration, and average fragment size, while bioinformatics QC involves assessing alignment rates, read distribution across genomic features, and sample-level quality metrics [14] [82].
For qPCR, the validation process must include:
- Assay Specificity and Efficiency: Designing intron-spanning primers to avoid genomic DNA amplification and performing serial dilutions to generate a standard curve for determining amplification efficiency [82].
- Reference Gene Stability: Using software like GSV to select stable reference genes from the RNA-Seq dataset itself, rather than relying on traditional housekeeping genes which may vary under different biological conditions [11].
- Analysis of Discrepant Results: Investigating genes that show inconsistent results between RNA-Seq and qPCR. These discrepancies often involve genes that are smaller, have fewer exons, or are lowly expressed, and may require alternative validation methods [70].
Diagram 2: qPCR Experimental Validation Pathway. This flowchart details the critical steps for designing and executing a qPCR validation experiment, from candidate selection to resolving discrepant results.
The integration of RNA-Seq and qPCR within a multi-omics framework is not a linear process but a synergistic cycle that enhances the rigor and translational potential of drug discovery research. RNA-Seq provides the powerful, unbiased lens to observe the entire transcriptomic landscape, generating hypotheses about drug MoAs, disease mechanisms, and potential biomarkers. qPCR then provides the precise, reliable tool to validate and ground these hypotheses, ensuring that the key findings are robust and reproducible. This tandem approach transforms large, complex datasets into high-confidence, actionable knowledge, ultimately de-risking the drug development pipeline. As multi-omics integration and machine learning continue to advance, the role of qPCR as an essential validator will only become more critical, ensuring that the models and signatures which guide personalized medicine are built upon a foundation of analytically sound data.
Conclusion
qPCR validation remains a cornerstone of rigorous transcriptomic analysis, particularly when research conclusions hinge on a few key genes, involve subtle expression changes, or are destined to inform clinical and drug development decisions. However, it is not universally required; well-powered RNA-Seq studies serving as exploratory hypothesis generators may forgo this step. The future of validation lies in smarter, integrated approaches—leveraging bioinformatics tools for robust experimental design and reference gene selection, adopting combined RNA-DNA assays in clinical oncology, and establishing standardized guidelines based on large-scale benchmarking. By strategically applying qPCR validation, researchers can significantly enhance the credibility, reproducibility, and translational impact of their RNA-Seq findings, ultimately accelerating the pace of biomedical discovery.
References
- 1. Commentary: a review of technical considerations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12522984/]
- 2. The Importance of Quality Control in RNA-Seq Analysis [https://ngscloud.com/the-importance-of-quality-control-in-rna-seq-analysis/]
- 3. a practical guide to RNA sequencing data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599993/]
- 4. Long-read RNA sequencing: A transformative technology ... [https://www.sciencedirect.com/science/article/pii/S1525001624007524]
- 5. Selection and validation of reference genes for ... [https://www.sciencedirect.com/science/article/pii/S1642431X25000300]
- 6. RNA-Seq Experimental Design Guide for Drug Discovery [https://www.lexogen.com/blog/planning-for-success-a-strategic-design-guide-for-rna-seq-experiments-in-drug-discovery/]
- 7. Benchmarking of RNA-sequencing analysis workflows ... [https://www.nature.com/articles/s41598-017-01617-3]
- 8. An updated comparison of microarray and RNA-seq for ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11548-3]
- 9. Do results obtained with RNA-sequencing require ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7823214/]
- 10. Assessing concordance between RNA-Seq and ... [https://pubmed.ncbi.nlm.nih.gov/40211167/]
- 11. RNA-seq validation: software for selection of reference and ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10511-y]
- 12. Evaluation of normalisation strategies for qPCR data ... [https://www.nature.com/articles/s41598-025-06657-8]
- 13. Validation of RNA-Seq by qPCR [https://bio-protocol.org/exchange/minidetail?id=10617026&type=30]
- 14. Clinical and analytical validation of a combined RNA ... [https://www.nature.com/articles/s43856-025-00934-3]
- 15. Cracking rare disorders: a new minimally invasive RNA- ... [https://www.nature.com/articles/s41525-025-00502-7]
- 16. Experimental validation of methods for differential gene expression analysis and sample pooling in RNA-seq | BMC Genomics [https://link.springer.com/article/10.1186/s12864-015-1767-y]
- 17. Do I Need to Validate My RNA-Seq Results With qPCR? | RNA-Seq Blog [https://www.rna-seqblog.com/do-i-need-to-validate-my-rna-seq-results-with-qpcr/]
- 18. Validation of RNAseq Experiments by qPCR? [http://bridgeslab.sph.umich.edu/posts/validation-of-rnaseq-experiments-by-qpcr]
- 19. An approach for prioritizing candidate genes from RNA-seq ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10542560/]
- 20. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 21. Cracking rare disorders: a new minimally invasive RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12120114/]
- 22. Clinical validation of RNA sequencing for Mendelian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081282/]
- 23. Validation of Prognostic Circulating Cell-Free RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293443/]
- 24. RNA-seq analysis identifies key genes enhancing hoof ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11814-4]
- 25. Validation of RNA-Seq Data by Reverse Transcription ... [https://bio-protocol.org/exchange/minidetail?id=8461527&type=30]
- 26. RNA-Seq 5: Data Validation [https://nordicbiosite.com/blog/rna-seq-5-data-validation]
- 27. Evaluation and validation of reference genes for RT-qPCR ... [https://www.nature.com/articles/s41598-025-22650-7]
- 28. Identification and validation of stable reference genes for ... [https://www.sciencedirect.com/science/article/abs/pii/S092144882500224X]
- 29. Selection and validation of stable reference genes in ... [https://www.sciencedirect.com/science/article/pii/S0717345825000119]
- 30. Selection and validation of RT-qPCR reference genes for ... [https://pubmed.ncbi.nlm.nih.gov/41013193/]
- 31. Selection of Stable Reference Genes for Gene Expression ... [https://pubmed.ncbi.nlm.nih.gov/40725038/]
- 32. Comparison between qPCR and RNA-seq reveals challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9883133/]
- 33. Experimental Design – Sample size and Assigning Groups [https://lantsandlaminins.com/experimental-design-step-by-step-guide/experimental-design-preliminaries/correlative-or-manipulative/choice-of-model-system/experimental-design-sample-size-and-assigning-groups/]
- 34. Sample size, power and effect size revisited: simplified and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7745163/]
- 35. Efficient experimental design and analysis strategies for the ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-13-484]
- 36. Next-generation RNA sequencing of spatially mapped ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12381154/]
- 37. DV200 determination for FFPE RNA samples [https://www.agilent.com/en/promotions/dv200-determination?srsltid=AfmBOop6CDCskVHZXf-xFiEhQborCMWpaobjlKJLwgEC3rgFTa9Whk88]
- 38. RNA Sequencing [https://broadclinicallabs.org/rna-sequencing/]
- 39. RNA-seq validation: software for selection of reference and ... [https://pubmed.ncbi.nlm.nih.gov/39014352/]
- 40. RNA-seq validation – software for selection of reference ... [https://www.rna-seqblog.com/rna-seq-validation-software-for-selection-of-reference-and-variable-candidate-genes-for-rt-qpcr/]
- 41. rdmesquita/GSV: Gene Selector for Validation [https://github.com/rdmesquita/GSV]
- 42. The impact of PCR duplication on RNAseq data generated ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03613-7]
- 43. Reproducibility in pre-clinical life science research [https://www.culturecollections.org.uk/culture-collection-news/reproducibility-in-pre-clinical-life-science-research/]
- 44. A real-world multi-center RNA-seq benchmarking study ... [https://www.nature.com/articles/s41467-024-50420-y]
- 45. Interlaboratory performance and quantitative PCR data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9932931/]
- 46. Large-scale benchmarking of circRNA detection tools reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10870000/]
- 47. Effect of low-expression gene filtering on detection ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/26737772/]
- 48. Effect of low-expression gene filtering on detection ... [https://www.rna-seqblog.com/effect-of-low-expression-gene-filtering-on-detection-of-differentially-expressed-genes-in-rna-seq-data/]
- 49. RNA-seq Data: Challenges in and Recommendations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4230301/]
- 50. Differential gene expression analysis pipelines and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10951429/]
- 51. Decoding Differential Expression: Are Your Findings Really ... [https://bridgeinformatics.com/decoding-differential-expression-are-your-findings-really-true/]
- 52. Targeted RNA-Based Oxford Nanopore Sequencing for ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.635601/full]
- 53. Frontiers | Capturing Differential Allele-Level Expression and... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2020.00941/full]
- 54. HLA RNA Sequencing With Unique Molecular Identifiers ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.629059/full]
- 55. typing from HLA - RNA sequence reads | Genome Medicine Seq [https://genomemedicine.biomedcentral.com/articles/10.1186/gm403]
- 56. Comparison between qPCR and RNA-seq reveals ... [https://texasbiomedical.theopenscholar.com/kulkarni-lab/publications/comparison-between-qpcr-and-rna-seq-reveals-challenges-quantifying-hla-expression]
- 57. Comparison between qPCR and RNA - seq reveals challenges of... [https://link.springer.com/article/10.1007/s00251-023-01296-7]
- 58. A rapid high-resolution HLA typing assay using RNA-Seq [https://www.rna-seqblog.com/a-rapid-high-resolution-hla-typing-assay-using-rna-seq/]
- 59. A benchmark of RNA-seq data normalization methods for ... [https://www.nature.com/articles/s41540-024-00448-z]
- 60. [Tutorial] Bulk RNA-seq DE analysis [https://informatics.fas.harvard.edu/resources/tutorials/differential-expression-analysis/]
- 61. Quantitative PCR (qPCR) Data Analysis [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/precision-qpcr.html]
- 62. A practical guide to sequencing in neuropsychiatric research [https://pmc.ncbi.nlm.nih.gov/articles/PMC12331514/]
- 63. MIQE 2.0 and the Urgent Need to Rethink qPCR Standards [https://pmc.ncbi.nlm.nih.gov/articles/PMC12154065/]
- 64. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 65. Validation of qPCR Assays for Gene Therapy [https://www.bioagilytix.com/blog/validation-of-qpcr-assays-for-gene-therapy/]
- 66. Development and validation of a qPCR assay for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12536431/]
- 67. MIQE 2.0: Revision of the Minimum Information for ... [https://pubmed.ncbi.nlm.nih.gov/40272429/]
- 68. Development and validation of cost-effective SYBR Green ... [https://www.nature.com/articles/s41598-024-52250-w]
- 69. Development and validation of a high-throughput qPCR ... [https://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0012760]
- 70. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5431503/]
- 71. RNA-Seq is not required to determine stable reference genes for qPCR normalization | PLOS Computational Biology [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009868]
- 72. Real-Time PCR (qPCR) and Whole-Transcriptome NGS [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/ngs-comparison.html]
- 73. Getting the most out of RNA-seq data analysis [https://www.rna-seqblog.com/getting-the-most-out-of-rna-seq-data-analysis/]
- 74. Recent developments and future directions in meta ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9527320/]
- 75. Systematic assessment of long-read RNA-seq methods for ... [https://www.nature.com/articles/s41592-024-02298-3]
- 76. Single-cell RNA-seq: advances and future challenges - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4132710/]
- 77. Multi-omics data integration considerations and study ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8058243/]
- 78. Drug Response RNA Biomarker Discovery and Profiling [https://www.illumina.com/areas-of-interest/genomics-in-drug-development/ngs-for-drug-development/rna-biomarker-discovery-profiling.html]
- 79. DRUG-seq for miniaturized high-throughput transcriptome ... [https://www.nature.com/articles/s41467-018-06500-x]
- 80. Multiomics integration and machine learning reveal ... [https://www.nature.com/articles/s41598-024-82233-w]
- 81. Multi-omics integration and machine learning uncover ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06711-z]
- 82. Innovative qPCR Algorithm Using Platelet-Derived RNA for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11988175/]